

Abstract
Diabetic nephropathy is a serious complication of diabetes mellitus, and its prevalence has been increasing worldwide. Therefore, there is an urgent need to identify a new therapeutic target to prevent diabetic nephropathy. Autophagy is a major catabolic pathway involved in degrading and recycling macromolecules and damaged organelles to maintain intracellular homeostasis. The study of autophagy in mammalian systems is advancing rapidly and has revealed that it is involved in the pathogenesis of various metabolic or age-related diseases. The functional role of autophagy in the kidneys is also currently under intense investigation although, until recently, evidence showing the involvement of autophagy in the pathogenesis of diabetic nephropathy has been limited. We provide a systematic review of autophagy and discuss the therapeutic potential of autophagy in diabetic nephropathy to help future investigations in this field.
1. Introduction
The prevalence of diabetes mellitus has been increasing worldwide during recent years, and this is estimated to continue in the future [1, 2]. Diabetic nephropathy is a serious complication of diabetes mellitus and is the most common cause of end-stage renal disease [3, 4]. The increasing prevalence of diabetes mellitus and its complications, including diabetic nephropathy, has therefore become a major health problem worldwide. There is now an urgent need to identify new therapeutic target molecules or cellular processes that underlie the pathogenesis of diabetic nephropathy to establish an additional therapeutic option.
Hyperglycemia-mediated alteration of extra- and intracellular metabolism, such as advanced glycation end products [5], increased protein kinase C activity [6], and abnormal polyol metabolism [7], has been recognized as classical pathogenesis of diabetic nephropathy. In addition, intracellular stress associated with renal hypoxia [8, 9], mitochondrial reactive oxygen species (ROS) [10–13], and endoplasmic reticulum (ER) stress [14–16] has recently been proposed and focused as new pathogenesis of diabetic nephropathy. Thus, to maintain the cellular homeostasis against stress condition derived from organelle dysfunction or hypoxia may be a new therapeutic target of diabetic nephropathy.
Autophagy, a lysosomal protein degradation pathway in cells, plays a crucial role in removing protein aggregates as well as damaged or excess organelles to maintain intracellular homeostasis and cell integrity [17]. It has recently been highlighted because it can be stimulated by multiple types of cellular stressors including starvation, hypoxia, or ER stress. The study of autophagy in mammalian systems and in disease states is advancing rapidly, and many investigators are entering this new and exciting field (Figure 1). It has been revealed that autophagy plays a crucial role in several organs, especially in metabolic organs, and that its alteration is involved in the pathogenesis of metabolic [18–21] and age-related diseases [22–27]. The functional role of autophagy in the kidneys is currently under intense investigation (Figure 1), and it has been revealed that autophagy has a renoprotective role in several animal models including those used for aging and acute kidney injury [26–31]. However, the role of autophagy in diabetic nephropathy remains unclear.
Figure 1.
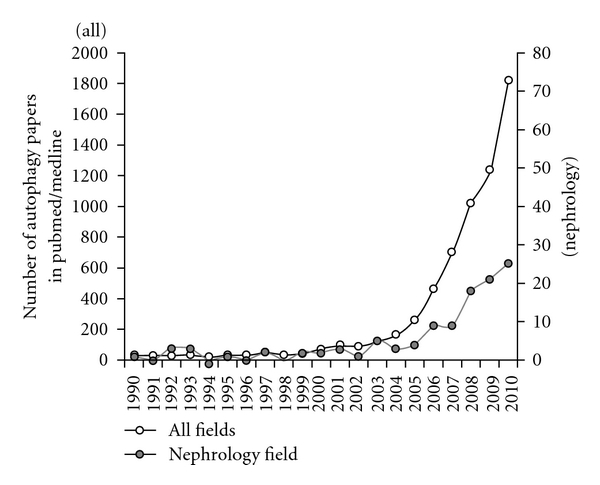
Substantial increases in the number of autophagy-related papers indexed in PubMed/Medline. The number of autophagy-related papers in all fields has increased remarkably over recent decades. Corresponding with the increase of autophagy-related papers in all fields, publication of autophagy-related papers in nephrology fields, also gradually increased.
Alteration of several nutrient-sensing pathways is related to the development of metabolic diseases, such as type 2 diabetes and its vascular complications. Major nutrient-sensing pathway involves the mammalian target of rapamycin (mTOR) [32–35], AMP-activated protein kinase (AMPK), [36–40] and oxidized NAD- (NAD+-) dependent histone deacetylase (Sirt1) [41–43], which are also recognized as the regulatory factors of autophagy under nutrient-depleted condition. As described above, autophagy can be induced by intracellular stresses that are involved in the pathogenesis of diabetic nephropathy [44]. Thus, alteration of these nutrient-sensing pathways under diabetic condition may impair the autophagic stress response stimulated by intracellular stress, which may lead to exacerbation of organelle dysfunction and subsequent diabetic nephropathy.
The above findings lead us to hypothesize that autophagy is involved in the pathogenesis of diabetic nephropathy and is a potential therapeutic option. Therefore, we provide a systematic review of autophagy and discuss its therapeutic potency in diabetic nephropathy to help future investigations in this field.
2. Autophagy
The term autophagy is derived from Greek and means self-eating. It is highly conserved from yeast to mammals and is a bulk degradation process that is involved in the clearance of long-lived proteins and organelles. Autophagy has two major roles in cells: to recycle intracellular energy resources in response to nutrient-depleted conditions and to remove cytotoxic proteins and organelles under stressful conditions. Autophagy works to maintain cell homeostasis under various stressful conditions. Several types of autophagy have been recognized in cells: macroautophagy, microautophagy, and chaperone-mediated autophagy; these differ in their mechanisms and functions [45, 46]. Of these three types of autophagy, macroautophagy is most prevalent and hereafter is referred to as autophagy. In this paper, we focus on the mechanisms and functions of autophagy.
3. Molecular Mechanisms of Autophagy
During macroautophagy, de novo isolation membranes (phagophores) elongate and fuse while engulfing a portion of the cytoplasm within double-membraned vesicles (autophagosomes) (Figure 2). Several origins of autophagosomes have been reported, including the ER [47–49], mitochondria [50], and plasma membrane [51]. Four major steps are involved in the formation of autophagosomes: initiation, nucleation, elongation, and closure. During these steps, autophagy-related proteins are involved (Figure 2). Autophagy is initiated by the unc-51-like kinase (Ulk) 1 (mammalian ortholog of the yeast autophagy-related genes (Atg)1) complex, which is composed of Ulk1 Ser/Thr protein kinase, Atg13, and FIP200 (mammalian homolog of the yeast Atg17) [52–54]. The phosphorylation of Atg13 and FIP200 by Ulk1 is essential for triggering autophagy. Phagophore nucleation is dependent on Beclin 1 (Atg6 in yeast)—an hVps34 or class III phosphatidylinositol 3-kinase (PI3K) complex, which consists of hVps34, hVps15, Beclin 1, and Atg14 [55, 56].
Figure 2.
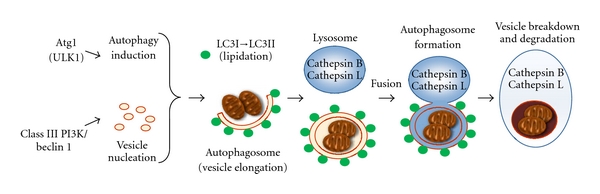
Scheme of autophagic pathways. Autophagic pathways consist of four steps: initiation, nucleation, elongation, and closure. Autophagy is initiated by the nucleation of an isolation membrane (phagophore). The phagophore elongates and closes on itself to form an autophagosome. Fusion of an autophagosome with a lysosome forms an autolysosome and, where the acid hydrolases in the lysosome, breaks down the inner membrane and cytoplasmic contents.
During autophagosome elongation/closure, two dependent ubiquitin-like conjugation systems are involved: Atg12 and LC3 (the mammalian ortholog of the yeast Atg8) [57]. The Atg12-Atg5 conjugate, which forms the Atg12-Atg5-Atg16 complex, contributes to the stimulation and localization of the LC3 conjugation reaction. The cytosolic isoform of LC3 (LC3-I) is conjugated to phosphatidylethanolamine through two consecutive ubiquitination-like reactions that are catalyzed by E1-like enzyme Atg7 and the E2-like enzyme Atg3 to form LC3-II [58]. Thus, LC3-II formation is recognized as a marker of existence of autophagosomes in cell or animal experiments [59–61]. After formation, the autophagosomes merge with the lysosomal compartment to form autolysosomes. The protein p62, also known as sequestosome 1 (SQSTM1), is known to localize to autophagosomes via LC3 interaction and to be constantly degraded by the autophagy-lysosome system [62, 63]. The accumulation of p62 is observed in autophagy-deficient cells [62, 63].
4. Methods Available for Monitoring Autophagy
It is necessary to keep in mind several important points as we monitor and assess the autophagy activity to prevent misconceptions. Some reviews about the methods for autophagy research have been published [59–61]. As briefly below summarized, some methods including electron microscopy (EM), detection of endogenous LC3 or green fluorescent protein (GFP)-LC3 by fluorescence microscopy, and detection of LC3-II by Western blotting are useful in monitoring the number of autophagosomes. However, these methods have some limitations. An accumulation of autophagosomes does not always mean increased formation of autophagosomes and may represent inhibited maturation of autolysosomes (or amphisomes). Simply counting the number of autophagosomes is insufficient for assessing autophagy activity. Autophagy flux is a term that represents a serial process of autophagy, including the synthesis of autophagosomes, the delivery of cargo to lysosomes, and the degradation of autolysosomes. To distinguish whether the accumulation of autophagosomes is caused by induction of autophagy or inhibition of autophagosome maturation and/or degradation of autophagic substrates in the lysosome, and then assess autophagy activity, an autophagy flux assay is more reliable than counting the number of autophagosomes. There are some useful assays to monitor autophagy flux. These include the LC3 turnover assay, or measurement of total levels of autophagic substrates such as LC3, GFP-LC3, and p62. Furthermore, several types of autophagy inhibitors and activators have recently become available to modulate the activity of autophagy processes. Pharmacological inhibitors of autophagy are PI3-kinase inhibitors such as wortmannin, LY294002, or 3-methladenine (3-MA) and inhibitors that block autophagosome-lysosome fusion or degradation of autophagic cargo in autolysosomes, such as E64d, pepstatin A, and bafilomycin A. However, a major problem is that there are no highly specific inhibitors or activators of autophagy. Thus, it is strongly recommended that pharmacological studies should be combined with studies that investigate deficiency/reduction of autophagy-related genes by genetic knockout/knockdown of ATG genes or dominant-negative mutant autophagy proteins, including Atg3, Atg5, Atg7, and Beclin 1.
5. Role of Nutrient Stress in Autophagy and Diabetic Nephropathy
The kidney is a structurally complex organ and is essential in several functions including excretion of the waste products of metabolism, regulation of body fluid volume, maintenance of appropriate acid balance, and secretion of a variety of hormones. The basic unit of the kidney is the nephron, which consists of a glomerulus and a series of tubules lined by a continuous layer of epithelial cells (Figure 3). The glomerulus consists of mesangial cells and a capillary wall with endothelial cells, glomerular basement membrane, and visceral epithelial cells (podocytes) (Figure 3). Among them, since podocytes play essential role to maintain glomerular filtration barrier, podocyte injury leads to proteinuria and glomerulosclerosis, which are major features of diabetic nephropathy. Podocytes are terminally differentiated cells with a limited proliferative capacity. Therefore, the fate of podocyte depends on its ability to cope with stress. Excess fluid filtered through glomerulus enters urinary space and is reabsorbed by the proximal tubular cells (Figure 3). The proximal tubular cells serve as a system to degrade several molecules reabsorbed from urinary space. Thus, autophagy may be essential to maintain their homeostasis and functions in both podocytes and proximal tubular cells, which might be altered in diabetic condition. If autophagy system is altered in diabetic condition, this alteration of autophagy may be involved in the pathogenesis of diabetic nephropathy.
Figure 3.
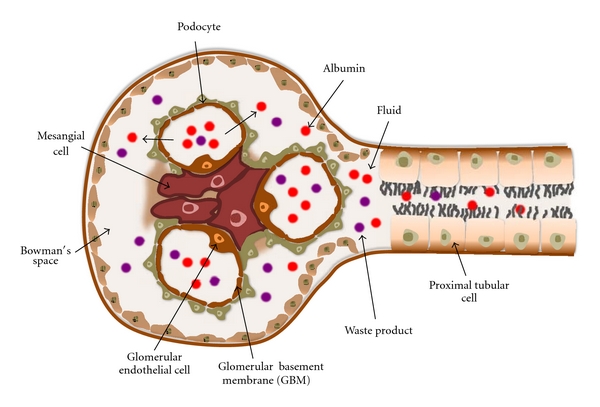
Schematic representation of nephron. The basic unit of the kidney is the nephron, which consists of a glomerulus and a series of tubules lined by a continuous layer of epithelial cells. The glomerulus consists of mesangial cells and a capillary wall with endothelial cells, glomerular basement membrane, and visceral epithelial cells (podocytes). Fluid containing albumins and waste products is filtered through glomerulus, enters urinary space, and is reabsorbed by the proximal tubular cells.
As expected, autophagy has been identified in both podocytes and proximal tubular cells and is regulated by a variety of stimuli including nutrient stress. Nutrient depletion is the most potent physiological inducer of autophagy, among several that have been reported to regulate autophagy. Here, we show the roles of mTOR, AMPK, and Sirt1, in the regulation of autophagy. The alteration of these pathways is involved in the pathogenesis of several kidney diseases including diabetic nephropathy.
5.1. mTOR
Several studies have shown that hyperactivation of the mTOR pathway in diabetic nephropathy plays a pivotal role in the hypertrophy of existing glomerular and tubular cells [64, 65] and is associated with podocyte injury and the progressive decline of glomerular filtration rates. Other studies have suggested that inhibition of the mTORC1 pathway with rapamycin has renoprotective effects on the progression of diabetic nephropathy in models of type 1 [33] and type 2 diabetes [32, 66–69]. Some reports have shown the additive renoprotective effects of rapamycin treatment including prevention of mesangial expansion and glomerular membrane thickness in type 1 diabetic rats [33] and attenuation of increased glomerular expression of laminin-β1 protein in type 2 diabetic mice [32, 67]. It has been reported that activation of the mTOR pathway is involved in the increased expression of profibrotic cytokines, such as TGF-β1 and connective tissue growth factor, and subsequent interstitial fibrosis in diabetic nephropathy [32–34]. Furthermore, more recent reports have shown that mTORC1 activity is essential to maintain podocyte homeostasis, but its hyperactivation is a causes of glomerular lesion of both type 1 and type 2 diabetic nephropathy [68, 69]. Complete deletion of podocyte mTORC1 activity in podocyte-specific Raptor-deficient mice causes podocyte injury [68]. In contrast, podocyte-specific mTORC1 hyperactivation by podocyte-specific tuberous sclerosis complex (TSC) 1-knockout mice show podocyte injury and glomerular lesion similar to diabetic nephropathy [69]. Finally, podocyte-specific Raptor-heterozygous mice show partial deletion of mTORC1 activity in podocyte and resistance to the development of diabetic nephropathy in both STZ-induced type 1 diabetic mice [68] and type 2 diabetic db/db mice [69].
In addition to the above-mentioned function, inhibition of autophagy is a main role of mTOR pathway [44, 58, 70]. Among the several signaling pathways that regulate autophagy in mammalian cells, the classical pathway of serine/threonine kinase, mTOR, plays a major role in the negative regulation of autophagy because it integrates signals that are emitted by growth factors, amino acids, glucose, and energy status [71]. Autophagy is inhibited by the activation of TOR under hypernutrient conditions [44, 58]. The mTOR pathway involves two functional complexes: mTOR complex 1 (mTORC1) and mTORC2. mTORC1, which consists of the mTOR catalytic subunit, regulatory associated protein of mTOR (Raptor), G protein β-subunit-like protein (GβL), proline-rich Akt substrate of 40 kDa (PRAS40), and DEP domain-containing mTOR-interacting protein (Deptor) [72], is sensitive to the immunosuppressant rapamycin [73, 74]. This complex regulates cell growth, metabolism (by integrating amino acid and growth factor signals), energy, and oxygen status [75]. The mTORC1 complex suppresses autophagy via phosphorylation and inactivation of Ulk1, an initiator of autophagosome formation [76]. Although no direct evidences have been provided, hyperactivation of mTOR pathway may suppress autophagy in podocyte and tubular cells in diabetic condition. Furthermore, enhanced activity of autophagy may be involved in the renoprotective effects of rapamycin treatment in diabetic nephropathy. The mTORC2 complex is less sensitive to rapamycin and includes mTOR, rapamycin-insensitive companion of mTOR (Rictor), GβL, stress-activated protein kinase-interacting protein 1 (Sin1), protein observed with Rictor (PROTOR), and Deptor [75, 77]. The mTORC2 complex regulates cytoskeletal organization, metabolism, and cell survival [75, 78, 79]. However, until now, the role of mTORC2 in regulation of autophagy has remained unclear.
5.2. AMPK
AMPK is activated under energy-depleted conditions and is likely to be suppressed in diabetic nephropathy. It has been reported that AMPK is inactivated (decreased phosphorylation of AMPK) in glomeruli and tubules in both type 1 and type 2 diabetic animal models [40, 81–84], which are reversed by agents such as metformin and resveratrol along with attenuation of diabetic glomerular and tubular injury [40, 85, 86]. This introduces the question of how decreases in AMPK activity can be involved in the pathogenesis of diabetic nephropathy. In type 1 and 2 diabetic kidneys, intrarenal lipid metabolism is altered, which is characterized by enhanced renal lipogenesis and suppressed lipolysis [87–90]. AMPK-mediated phosphorylation inactivates a lipogenic enzyme, acetyl-CoA carboxylase, which results in decreased lipogenesis and enhanced lipolysis [91]. Decreases in renal AMPK activity in these mouse models may be a mechanism of altered renal lipid metabolism and subsequent lipotoxicity-associated renal damage. Since AMPK can affect various cellular metabolism as wells as lipid metabolism [92, 93], the other molecular mechanism should be involved in AMPK-mediated renoprotection.
AMPK plays a central role in the integration of several stress stimuli and is a positive regulator of autophagy in response to nutrient-depleted conditions. AMPK monitors the energy status of the cell by sensing its AMP/ATP ratio [93]. Several upstream kinases, including liver kinase B1 (LKB1), calcium/calmodulin kinase kinase (CaMKII) β, and TGF-β-activated kinase-1 (TAK1), can activate AMPK by phosphorylating a threonine residue on its catalytic α subunit [93]. AMPK can crosstalk with the mTORC1 signal during multiple steps of autophagy regulation. AMPK induces autophagy by inhibiting mTORC1 activity via phosphorylation of its regulatory-associated proteins [44, 58, 94]. Recent studies have shown that AMPK-dependent phosphorylation of Ulk1 induces autophagy [94, 95]. A balance between mTORC1 and AMPK likely directly regulates Ulk1 activity and subsequent autophagy initiation [44]. Thus, in addition to the above-mentioned mechanism, AMPK-mediated induction of autophagy may be involved in its renoprotection. AMPK activation may be linked to autophagy for the maintenance of renal homeostasis in diabetic kidney.
5.3. Sirt1
Sirtuins, the silent information regulator 2 family, were originally identified as NAD+-dependent deacetylases in experiments in lower species and consist of seven members, Sirt1–Sirt7, in mammals [96, 97]. Sirtuins have been identified as antiaging molecules under calorie-restricted conditions and environmental stress. Some mammalian sirtuins, especially Sirt1, have been shown to play important roles in the regulation of aging, or in the pathogenesis of age-related metabolic diseases such as type 2 diabetes [41, 42, 96]. An increase in the intracellular concentration of NAD+ by caloric restriction can activate Sirt1. Results that demonstrate the role of Sirt1 in autophagy are still lacking compared with those for mTOR and AMPK, but they have recently been increasing. Sirt1 can deacetylate essential autophagic factors such as Atg5, Atg7, and LC3 [98] and has been shown to induce autophagy. Furthermore, Sirt1 deacetylates the transcription factor Forkhead box O3a (Foxo3a), which leads to enhanced expression of proautophagic BCL2/adenovirus E1V 19-kDa interacting protein 3 (Bnip3) [26].
Renal expression of Sirt1 decreases in type 1 diabetic animal models [99, 100]. Also, reduced forms of nicotinamide adenine dinucleotide (NADH) are metabolites of glucose and fatty acids. Thus, NAD+/NADH ratios are decreased in cells under conditions where nutrients are in excess, such as diabetes. Sirt1-deacetylase activity should decrease in diabetic nephropathy. Although direct renoprotective effects of Sirt1 in diabetic nephropathy have yet to be elucidated, Sirt1 has shown renoprotective activity in aging kidneys and fibrotic kidney diseases. The previously mentioned findings lead us to speculate that activation of Sirt1 should also have therapeutic efficacy in diabetic nephropathy. Furthermore, Sirt1-induced autophagy activation may contribute to Sirt1-mediated renoprotective effect in diabetic nephropathy.
6. Regulation of Autophagy by Intracellular Stress
Besides nutrient stress, autophagy is upregulated by several intracellular stresses, such as hypoxia, ROS, and ER stress [44]. Based on recent reports, this process is probably a compensatory response to maintain cell integrity. Furthermore, these intracellular stresses have recently been focused on as a pathogenesis of diabetic nephropathy, in addition to the classical pathogenesis of diabetic nephropathy.
6.1. Oxidative Stress
Under conditions where nutrients are in excess, such as diabetes and obesity, the production of ROS in the kidneys is enhanced by high glucose concentrations [101, 102]. Furthermore, high levels of free fatty acids, especially polysaturated fatty acids, also induce ROS production in the kidneys [88, 103]. Oxidative stress is a by-product of mitochondrial respiration and is associated with cell dysfunction. Actually, a recent report has shown abnormal mitochondrial morphology in diabetic kidney [95, 104], suggesting that diabetic kidney fails to remove damaged mitochondria. Thus, restoring the ability to control mitochondria homeostasis should be a therapeutic target of diabetic nephropathy.
Mitochondrial quality control is mediated by mitochondrial autophagy (mitophagy) [105]. Similarly, oxidative stress can induce autophagy to remove damaged mitochondria to protect cells. Thus, autophagy-mediated quality control of mitochondria and subsequent reduction of ROS should be essential to protect kidney in diabetic condition. It has been reported that exogenous hydrogen peroxide activates protein kinase RNA-like ER kinase (PERK), which subsequently phosphorylates eukaryotic initiation factor-2α, activates Atg4, and inhibits mTOR [106]. In response to cellular stress or damage, mitochondrial membranes can be permeabilized. The autophagic recognition of depolarized mitochondria is mediated by a refined voltage sensor, which involves the mitochondrial kinase, phosphatase, and tensin homolog-induced putative kinase 1.
6.2. Hypoxia
In early-stage diabetic nephropathy, hypoxia is aggravated by manifestations of chronic hyperglycemic abnormalities of red blood cells [107, 108], oxidative stress [109], and diabetes mellitus-induced tubular apoptosis; as such, tubulointerstitial hypoxia in diabetes mellitus might be an important early event.
Hypoxia is also a stimulatory factor of autophagy. Hypoxia-induced autophagy largely depends on hypoxia-inducible factor-1α (HIF-1α), which is a transcription factor that is activated and stabilized under hypoxic conditions [110, 111]. HIF-1α activates transcription of Bnip3 and Bnip3L and subsequently induces autophagy. Normally, Beclin 1 interacts with Bcl-2 proteins. Bnip3 can disrupt this interaction, liberating Beclin 1 from Bcl-2 in cells and leading to autophagy. Thus, HIF1α-induced Bnip3 overexpression promotes autophagy [112]. The transcription of Bnip3 is also upregulated by the transcription factor FOXO3, which is deacetylated and positively regulated by Sirt1 [26]. Hypoxia causes damage to the mitochondria and intracellular accumulation of ROS [113]. Removing the damaged mitochondria under hypoxic conditions is also an important role of Bnip3-mediated autophagy. Thus, to investigate whether hypoxia-induced and Sirt1-mediated autophagy is altered in diabetic kidney is interesting. If it is altered, to restore autophagy activity even under diabetic condition should be important to protect kidney form hypoxia.
6.3. ER Stress
ER stress has recently been focused as a pathogenesis of diabetic nephropathy. The induction of ER stress and subsequent apoptosis by hyperglycemia and high levels of free fatty acids (polysaturated fatty acids) are observed in podocytes [114]. Additionally, in proteinuric kidney diseases, including diabetic nephropathy, massive proteinuria filtered from glomeruli causes ER stress responses and subsequent apoptosis in renal tubular cells [14, 115]. Thus, to suppress inadequate ER stress is thought as a therapeutic strategy of diabetic nephropathy.
It is known that ER stress as well as hypoxia and ROS also cause autophagy. The ER is not only involved in protein synthesis and maturation but may also constitute a major source/scaffold for the autophagic isolation membrane [47]. When misfolded proteins are not exported efficiently to the cytoplasm and accumulate in the ER, the unfolded protein response (UPR) is often induced [116–118]. The UPR consists of three main branches that are controlled by the ER membrane proteins: PERK; activating transcription factor-6 (ATF6); inositol requiring enzyme 1 (IRE1) [116–118]. Among these UPR-related proteins, PERK and ATF6 have been reported to induce autophagy [44]. PERK induces the transcriptional activation of LC3 and Atg5 through the action of the transcription factors ATF4 and CCAAT-enhancer-binding protein homologous protein, respectively [119]. It has been suggested that IRE1 is also involved in the induction of autophagy by phosphorylation of Beclin 1 via c-Jun NH2-terminal kinase-1 [44]. Enhanced and prolonged ER stress causes several pathogenic features such as apoptosis and inflammation [117, 118]. Thus, autophagy-mediated ER degradation (ERphagy) may be required for cell protection from prolonged cytotoxic ER stress shown in diabetic kidney.
7. Autophagy in the Kidneys
The study of autophagy has previously been undertaken in lower species. The study of autophagy in mammalian systems is advancing rapidly and has revealed that mammalian autophagy is involved in the pathogenesis of various metabolic or age-related diseases [18–27].
Recently, nephrologists have also entered this exciting field of study. In this section, we review recent studies on the pathophysiology of autophagy in the kidneys. Autophagy has been observed in various parts of the kidneys, including proximal tubules, and thick ascending limbs. In particular, in podocytes, higher levels of constitutive autophagy have been observed using GFP-LC3 transgenic mice even under normal conditions [27]. As for the role of autophagy in renal pathophysiology, several researchers have reported the significance of autophagy in experimental renal injury models. In several experimental animal models of glomerulonephritis, including puromycin aminonucleoside and adriamycin-induced proteinuria, autophagy has been identified and shown to play renoprotective and antiproteinuric roles in podocytes through the use of podocyte-specific Atg5 knockout mice [27]. It has been recently reported that the normal aging process suppresses autophagy in podocytes, and that podocyte-specific deletion of Atg5 leads to glomerulopathy in aging mice that is accompanied by accumulation of oxidized and ubiquitinated proteins, ER stress, and proteinuria [27].
In renal tubules as well as in podocytes, autophagy has been reported to play a renoprotective role under several pathological conditions. In renal ischemia-reperfusion injury models, the upregulation of autophagy to protect the kidneys was observed using 3-MA, chloroquine [28], and proximal tubular epithelial cell-specific Atg5 knockout mice [29]. Additionally, in cisplatin-induced acute kidney injury models, the increase of autophagosomes was observed by EM, LC3-II Western blotting [30], and GFP-LC3 transgenic mice [31]. Hypoxia is one of the causes of renal tubular damage in aged kidney [120]. We have previously shown that hypoxia-induced autophagy activity declined with age, which led to accumulations of damaged mitochondria and mitochondrial ROS in the kidney [26]. Interestingly, long-term calorie restriction (CR) restored autophagy activity even in aged kidney [26]. As a mechanism, Sirt1-mediated autophagy was essential in CR-mediated renoprotection in aged kidney [26]. Bnip3 expression is essential to induce autophagy under hypoxic condition [121] and is positively regulated by a transcriptional factor Foxo3a [122]. This regulation was altered in aged kidney. On the other hand, CR-mediated Sirt1 activation deacetylated and activated Foxo3a transcriptional activity and subsequent Bnip3-mediated autophagy even in aged kidney [26]. Furthermore, the kidney of heterozygous Sirt1-knockout mice showed lower autophagy activity along with the decrease in Bnip3 expression, and thus they were resistant to CR-mediated antiaging effects [26]. This finding suggests that Sirt1 is essential for CR-mediated renoprotection.
Thus, accumulative evidence has demonstrated the pathophysiological importance of autophagy in the kidneys (Table 1). However, the role and existence of autophagy in other types of renal cells besides podocytes and proximal tubular cells is not known.
Table 1.
Autophagy-related kidney diseases.
| Species and methods to monitor autophagy | Disease model | Effects of autophagy | Reference |
|---|---|---|---|
| Sprague-Dawley rats, immunohistochemistry of LC3 and Western blotting of LC3-II | Cyclosporine A-induced nephrotoxicity | Protection against tubular cell death | [80] |
|
| |||
| C57BL/6 mice, EM, and Western blotting of LC3-II | Cisplatin injury | Protection against tubular cell death | [30] |
|
| |||
| C57BL/6 mice, EM, immunofluorescence of LC3, and Western blotting of LC3-II | Aging | Protection against aging and hypoxia-related tubular damage | [26] |
|
| |||
| GFP-LC3 mice | Cisplatin injury | Protection against tubular cell death | [31] |
|
| |||
| C57BL/6 mice, EM, and Western blotting of LC3-II with 3-MA and chloroquine | Ischemia reperfusion | Protection against tubular cell death | [28] |
|
| |||
| Proximal tubular epithelial cell-specific Atg5-deficient mice | Ischemia reperfusion | Protection against tubular cell death | [29] |
|
| |||
| Podocyte-specific Atg5-deficient mice | Aging, protein overload-, LPS-, PAN-, and adriamycin-induced glomerular injury | Protection against podocyte injury | [27] |
EM: electron microscopy; GFP: green fluorescent protein; 3-MA: 3-methladenine; Atg: autophagy-related genes; LPS: lipopolysaccharide; PAN: puromycin aminonucleoside.
8. Perspective
It is evident that the above-mentioned nutrient-sensing signals exist in the kidneys. However, what are their physiological roles in this organ? The kidneys require sufficient amounts of ATP for maintenance of their functions and avidly consume oxygen to drive mitochondrial oxidative phosphorylation among major organs. A small percentage of oxygen consumed by mitochondria is incompletely reduced to ROS, and this unremitting generation of oxidants during mitochondrial respiration, albeit in small amounts, may cumulatively damage the kidneys, which are heavily dependent on mitochondrial metabolism. Regulating mitochondrial metabolism in response to nutrient conditions via regulation of autophagy that can remove damaged mitochondria and subsequent ROS may be a physiological role of renal nutrient-sensing signals.
Autophagy is regulated by nutrient conditions, and its alteration associates with various metabolic and age-associated diseases. Although studies on autophagy have methodological limitations, as outlined above, it is evident that autophagy deficiency is associated with podocyte and tubular cell injuries from the studies using Atg5-knockout mice [27, 29]. These findings lead us to hypothesize that autophagy is altered in diabetic kidneys, and autophagy deficiency should contribute to the pathogenesis of diabetic nephropathy. Altered nutrient-sensing signals in diabetic kidneys may contribute to accumulation of mitochondrial ROS via suppression of autophagy, which may be associated with initiation of the early stages of diabetic nephropathy. Both hypoxia and proteinuria-induced ER stress contribute to proximal tubular cell damage in the progressive and overt stages of diabetic nephropathy. Why do diabetic kidneys show a weakness against these stresses? How can we protect the kidneys from these stresses even under diabetic conditions? One answer may be derived from autophagy studies. Autophagy deficiency in diabetic kidneys may make tubular cells fragile under hypoxic and ER stress and possibly lead to progression of diabetic nephropathy (Figure 4). Activation of autophagy may be a therapeutic option for the advanced stages of diabetic nephropathy.
Figure 4.
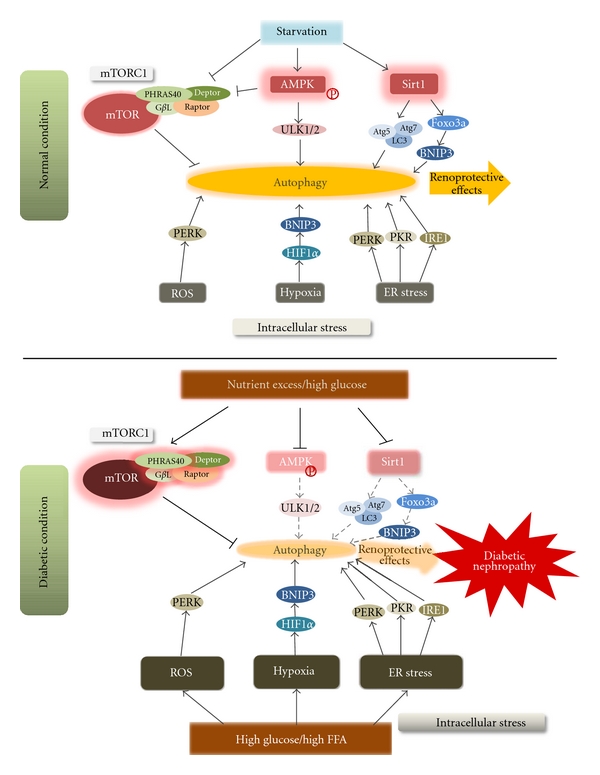
Regulation of autophagy by nutrient and intracellular stresses and the relationship between autophagy and the progression of diabetic nephropathy. Under normal conditions, intracellular stresses such as hypoxia, mitochondrial ROS, and ER stress induce autophagy. Nutrient depletion enhances autophagy by inhibiting mTORC1 and by activating AMPK and Sirt1. This activation of autophagy helps to maintain intracellular homeostasis and may have renoprotective effects. In contrast, under diabetic conditions, high glucose or FFA levels increase intracellular stresses, leading to the progression of diabetic nephropathy. Furthermore, nutrient excess and high glucose levels under diabetic conditions inhibit autophagy by inhibiting AMPK and Sirt1, and by activating mTORC1. This inactivation of autophagy may reinforce the progression of diabetic nephropathy. ROS: reactive oxygen species; ER: endoplasmic reticulum; mTORC1: mammalian target of rapamycin (mTOR) complex 1 (mTORC1); AMPK: AMP-activated protein kinase; FFA: free fatty acid.
9. Concluding Comments
In recent decades, numerous investigators have been making efforts to identify the molecular mechanisms involved in the initiation and progression of diabetic nephropathy to develop new therapeutic strategies. However, end-stage renal disease due to diabetic nephropathy continues to increase worldwide. There is an urgent need to identify additional new therapeutic targets for prevention of diabetic nephropathy. We have provided a perspective on whether autophagy is involved in the pathogenesis of diabetic nephropathy and whether it is an acceptable new therapeutic target. Unfortunately, there have still not been many studies that have focused on autophagy in diabetic nephropathy. In the next few years, studies using Atg-gene knockout/knockdown mice combined with different methodologies will elucidate this possibility. Finally, these studies will ultimately give us a clearer perspective as to whether autophagy should be considered as a novel therapeutic target to halt the progression of diabetic nephropathy.
Acknowledgment
This study was supported in part by a Grant-in-Aid For Diabetic Nephropathy Research, from the Ministry of Health, Labour and Welfare of Japan.
References
- 1.Amos AF, McCarty DJ, Zimmet P. The rising global burden of diabetes and its complications: estimates and projections to the year 2010. Diabetic Medicine. 1997;14, supplement 5:S1–S85. [PubMed] [Google Scholar]
- 2.Mokdad AH, Ford ES, Bowman BA, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. Journal of the American Medical Association. 2003;289(1):76–79. doi: 10.1001/jama.289.1.76. [DOI] [PubMed] [Google Scholar]
- 3.Lasaridis AN, Sarafidis PA. Diabetic nephropathy and antihypertensive treatment: what are the lessons from clinical trials? American Journal of Hypertension. 2003;16(8):689–697. doi: 10.1016/s0895-7061(03)00864-1. [DOI] [PubMed] [Google Scholar]
- 4.Molitch ME, DeFronzo RA, Franz MJ, et al. Nephropathy in diabetes. Diabetes Care. 2004;27, supplement 1:S79–S83. doi: 10.2337/diacare.27.2007.s79. [DOI] [PubMed] [Google Scholar]
- 5.Forbes JM, Thallas V, Thomas MC, et al. The breakdown of preexisting advanced glycation end products is associated with reduced renal fibrosis in experimental diabetes. FASEB Journal. 2003;17(12):1762–1764. doi: 10.1096/fj.02-1102fje. [DOI] [PubMed] [Google Scholar]
- 6.Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K, King GL. Characterization of protein kinase C β isoform activation on the gene expression of transforming growth factor-β, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. Journal of Clinical Investigation. 1997;100(1):115–126. doi: 10.1172/JCI119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunlop M. Aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney International, Supplement. 2000;58(77):S3–S12. doi: 10.1046/j.1523-1755.2000.07702.x. [DOI] [PubMed] [Google Scholar]
- 8.Miyata T, de Strihou CVY. Diabetic nephropathy: a disorder of oxygen metabolism? Nature Reviews Nephrology. 2010;6(2):83–95. doi: 10.1038/nrneph.2009.211. [DOI] [PubMed] [Google Scholar]
- 9.Singh DK, Winocour P, Farrington K. Mechanisms of disease: the hypoxic tubular hypothesis of diabetic nephropathy. Nature Clinical Practice Nephrology. 2008;4(4):216–226. doi: 10.1038/ncpneph0757. [DOI] [PubMed] [Google Scholar]
- 10.Ha H, Hwang IA, Park JH, Lee HB. Role of reactive oxygen species in the pathogenesis of diabetic nephropathy. Diabetes Research and Clinical Practice. 2008;82, supplement 1:S42–S45. doi: 10.1016/j.diabres.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 11.Hakim FA, Pflueger A. Role of oxidative stress in diabetic kidney disease. Medical Science Monitor. 2010;16(2):RA37–RA48. [PubMed] [Google Scholar]
- 12.Kashihara N, Haruna Y, Kondeti VK, Kanwar YS. Oxidative stress in diabetic nephropathy. Current Medicinal Chemistry. 2010;17(34):4256–4269. doi: 10.2174/092986710793348581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circulation Research. 2010;107(9):1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cybulsky AV. Endoplasmic reticulum stressin proteinuric kidney disease. Kidney International. 2010;77(3):187–193. doi: 10.1038/ki.2009.389. [DOI] [PubMed] [Google Scholar]
- 15.Qi W, Mu J, Luo ZF, et al. Attenuation of diabetic nephropathy in diabetes rats induced by streptozotocin by regulating the endoplasmic reticulum stress inflammatory response. Metabolism. 2011;60(5):594–603. doi: 10.1016/j.metabol.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 16.Wu J, Zhang R, Torreggiani M, et al. Induction of diabetes in aged C57B6 mice results in severe nephropathy: an association with oxidative stress, endoplasmic reticulum stress, and inflammation. American Journal of Pathology. 2010;176(5):2163–2176. doi: 10.2353/ajpath.2010.090386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ebato C, Uchida T, Arakawa M, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metabolism. 2008;8(4):325–332. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 19.Masini M, Bugliani M, Lupi R, et al. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia. 2009;52(6):1083–1086. doi: 10.1007/s00125-009-1347-2. [DOI] [PubMed] [Google Scholar]
- 20.Fujitani Y, Kawamori R, Watada H. The role of autophagy in pancreatic β-cell and diabetes. Autophagy. 2009;5(2):280–282. doi: 10.4161/auto.5.2.7656. [DOI] [PubMed] [Google Scholar]
- 21.Jung HS, Chung KW, Kim JW, et al. Loss of autophagy diminishes pancreatic β cell mass and function with resultant hyperglycemia. Cell Metabolism. 2008;8(4):318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 22.Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. Journal of Biological Chemistry. 2000;275(40):31505–31513. doi: 10.1074/jbc.M002102200. [DOI] [PubMed] [Google Scholar]
- 23.Donati A, Cavallini G, Paradiso C, et al. Age-related changes in the regulation of autophagic proteolysis in rat isolated hepatocytes. Journals of Gerontology—Series A. 2001;56(7):B288–B293. doi: 10.1093/gerona/56.7.b288. [DOI] [PubMed] [Google Scholar]
- 24.Terman A. The effect of age on formation and elimination of autophagic vacuoles in mouse hepatocytes. Gerontology. 1995;41, supplement 2:319–326. doi: 10.1159/000213753. [DOI] [PubMed] [Google Scholar]
- 25.Vittorini S, Paradiso C, Donati A, et al. The age-related accumulation of protein carbonyl in rat liver correlates with the age-related decline in liver proteolytic activities. Journals of Gerontology—Series A. 1999;54(8):B318–B323. doi: 10.1093/gerona/54.8.b318. [DOI] [PubMed] [Google Scholar]
- 26.Kume S, Uzu T, Horiike K, et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. Journal of Clinical Investigation. 2010;120(4):1043–1055. doi: 10.1172/JCI41376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartleben B, Gödel M, Meyer-Schwesinger C, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. Journal of Clinical Investigation. 2010;120(4):1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. American Journal of Pathology. 2010;176(3):1181–1192. doi: 10.2353/ajpath.2010.090594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimura T, Takabatake Y, Takahashi A, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. Journal of the American Society of Nephrology. 2011;22(5):902–913. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney International. 2008;74(5):631–640. doi: 10.1038/ki.2008.214. [DOI] [PubMed] [Google Scholar]
- 31.Inoue K, Kuwana H, Shimamura Y, et al. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clinical and Experimental Nephrology. 2010;14(2):112–122. doi: 10.1007/s10157-009-0254-7. [DOI] [PubMed] [Google Scholar]
- 32.Sakaguchi M, Isono M, Isshiki K, Sugimoto T, Koya D, Kashiwagi A. Inhibition of mTOR signaling with rapamycin attenuates renal hypertrophy in the early diabetic mice. Biochemical and Biophysical Research Communications. 2006;340(1):296–301. doi: 10.1016/j.bbrc.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 33.Yang Y, Wang J, Qin L, et al. Rapamycin prevents early steps of the development of diabetic nephropathy in rats. American Journal of Nephrology. 2007;27(5):495–502. doi: 10.1159/000106782. [DOI] [PubMed] [Google Scholar]
- 34.Lloberas N, Cruzado JM, Franquesa M, et al. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. Journal of the American Society of Nephrology. 2006;17(5):1395–1404. doi: 10.1681/ASN.2005050549. [DOI] [PubMed] [Google Scholar]
- 35.Inoki K. Role of TSC-mTOR pathway in diabetic nephropathy. Diabetes Research and Clinical Practice. 2008;82, supplement 1:S59–S62. doi: 10.1016/j.diabres.2008.09.028. [DOI] [PubMed] [Google Scholar]
- 36.Sharma K, RamachandraRao S, Qiu G, et al. Adiponectin regulates albuminuria and podocyte function in mice. Journal of Clinical Investigation. 2008;118(5):1645–1656. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hwi JK, Zhang Z, Dae YJ, et al. Nutrient stress activates inflammation and reduces glucose metabolism by suppressing AMP-activated protein kinase in the heart. Diabetes. 2009;58(11):2536–2546. doi: 10.2337/db08-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kusmic C, L’Abbate A, Sambuceti G, et al. Improved myocardial perfusion in chronic diabetic mice by the up-regulation of pLKB1 and AMPK signaling. Journal of Cellular Biochemistry. 2010;109(5):1033–1044. doi: 10.1002/jcb.22486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo Z, Zheng C, Qin Z, Wei P. Effect of telmisartan on the expression of cardiac adiponectin and its receptor 1 in type 2 diabetic rats. Journal of Pharmacy and Pharmacology. 2011;63(1):87–94. doi: 10.1111/j.2042-7158.2010.01157.x. [DOI] [PubMed] [Google Scholar]
- 40.Ding DF, You N, Wu XM, et al. Resveratrol attenuates renal hypertrophy in early-stage diabetes by activating AMPK. American Journal of Nephrology. 2010;31(4):363–374. doi: 10.1159/000300388. [DOI] [PubMed] [Google Scholar]
- 41.Liang F, Kume S, Koya D. SIRT1 and insulin resistance. Nature Reviews Endocrinology. 2009;5(7):367–373. doi: 10.1038/nrendo.2009.101. [DOI] [PubMed] [Google Scholar]
- 42.Kume S, Takashi U, Atsunori K, Daisuke K. SIRT1, A calorie restriction mimetic, in a new therapeutic approach for type 2 diabetes mellitus and diabetic vascular complications. Endocrine, Metabolic and Immune Disorders—Drug Targets. 2010;10(1):16–24. doi: 10.2174/187153010790827957. [DOI] [PubMed] [Google Scholar]
- 43.Maeda S, Koya D, Araki SI, et al. Association between single nucleotide polymorphisms within genes encoding sirtuin families and diabetic nephropathy in Japanese subjects with type 2 diabetes. Clinical and Experimental Nephrology. 2011;15(3):381–390. doi: 10.1007/s10157-011-0418-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Molecular Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klionsky DJ. The molecular machinery of autophagy: unanswered questions. Journal of Cell Science. 2005;118, part 1:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Massey AC, Zhang C, Cuervo AM. Chaperone-mediated autophagy in aging and disease. Current Topics in Developmental Biology. 2006;73:205–235. doi: 10.1016/S0070-2153(05)73007-6. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nature Cell Biology. 2009;11(12):1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 48.Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5(8):1180–1185. doi: 10.4161/auto.5.8.10274. [DOI] [PubMed] [Google Scholar]
- 49.Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. Journal of Cell Biology. 2008;182(4):685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hailey DW, Rambold AS, Satpute-Krishnan P, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141(4):656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nature Cell Biology. 2010;12(8):747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ganley IG, Lam DH, Wang J, Ding X, Chen S, Jiang X. ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. Journal of Biological Chemistry. 2009;284(18):12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORCl association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular Biology of the Cell. 2009;20(7):1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Molecular Biology of the Cell. 2009;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 56.Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and beclin 1 complexes. Biochimie. 2008;90(2):313–323. doi: 10.1016/j.biochi.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 57.Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nature Reviews Molecular Cell Biology. 2009;10(7):458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- 58.Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiological Reviews. 2010;90(4):1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 59.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nature Cell Biology. 2010;12(9):823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 63.Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. Journal of Cell Biology. 2005;171(4):603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee CH, Inoki K, Guan KL. mTOR pathway as a target in tissue hypertrophy. Annual Review of Pharmacology and Toxicology. 2007;47:443–467. doi: 10.1146/annurev.pharmtox.47.120505.105359. [DOI] [PubMed] [Google Scholar]
- 65.Chen JK, Chen J, Neilson EG, Harris RC. Role of mammalian target of rapamycin signaling in compensatory renal hypertrophy. Journal of the American Society of Nephrology. 2005;16(5):1384–1391. doi: 10.1681/ASN.2004100894. [DOI] [PubMed] [Google Scholar]
- 66.Sataranatarajan K, Mariappan MM, Myung JL, et al. Regulation of elongation phase of mRNA translation in diabetic nephropathy: amelioration by rapamycin. American Journal of Pathology. 2007;171(6):1733–1742. doi: 10.2353/ajpath.2007.070412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mori H, Inoki K, Masutani K, et al. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochemical and Biophysical Research Communications. 2009;384(4):471–475. doi: 10.1016/j.bbrc.2009.04.136. [DOI] [PubMed] [Google Scholar]
- 68.Gödel M, Hartleben B, Herbach N, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. Journal of Clinical Investigation. 2011;121(6):2197–2209. doi: 10.1172/JCI44774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Inoki K, Mori H, Wang J, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. Journal of Clinical Investigation. 2011;121(6):2181–2196. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Research. 2010;20(7):748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 71.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Current Opinion in Cell Biology. 2005;17(6):596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 72.Efeyan A, Sabatini DM. MTOR and cancer: many loops in one pathway. Current Opinion in Cell Biology. 2010;22(2):169–176. doi: 10.1016/j.ceb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang Q, Guan KL. Expanding mTOR signaling. Cell Research. 2007;17(8):666–681. doi: 10.1038/cr.2007.64. [DOI] [PubMed] [Google Scholar]
- 74.Guertin DA, Stevens DM, Saitoh M, et al. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15(2):148–159. doi: 10.1016/j.ccr.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dos DS, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current Biology. 2004;14(14):1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 76.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Current Opinion in Cell Biology. 2010;22(2):132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 77.Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature Cell Biology. 2004;6(11):1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 78.Kim JE, Chen J. Cytoplasmic-nuclear shuttling of FKBP12-rapamycin-associated protein is involved in rapamycin-sensitive signaling and translation initiation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(26):14340–14345. doi: 10.1073/pnas.011511898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 80.Pallet N, Bouvier N, Legendre C, et al. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy. 2008;4(6):783–791. doi: 10.4161/auto.6477. [DOI] [PubMed] [Google Scholar]
- 81.Cammisotto PG, Londono I, Gingras D, Bendayan M. Control of glycogen synthase through ADIPOR1-AMPK pathway in renal distal tubules of normal and diabetic rats. American Journal of Physiology—Renal Physiology. 2008;294(4):F881–F889. doi: 10.1152/ajprenal.00373.2007. [DOI] [PubMed] [Google Scholar]
- 82.Kitada M, Kume S, Imaizumi N, Koya D. Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1- independent pathway. Diabetes. 2011;60(2):634–643. doi: 10.2337/db10-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sokolovska J, Isajevs S, Sugoka O, et al. Influence of metformin on GLUT1 gene and protein expression in rat streptozotocin diabetes mellitus model. Archives of Physiology and Biochemistry. 2010;116(3):137–145. doi: 10.3109/13813455.2010.494672. [DOI] [PubMed] [Google Scholar]
- 84.Yamazaki T, Tanimoto M, Gohda T, et al. Combination effects of enalapril and losartan on lipid peroxidation in the kidneys of KK-Ay/Ta mice. Nephron—Experimental Nephrology. 2009;113(2):e66–e76. doi: 10.1159/000228714. [DOI] [PubMed] [Google Scholar]
- 85.Chang CC, Chang CY, Wu YT, Huang JP, Yen TH, Hung LM. Resveratrol retards progression of diabetic nephropathy through modulations of oxidative stress, proinflammatory cytokines, and AMP-activated protein kinase. Journal of Biomedical Science. 2011;18(1):p. 47. doi: 10.1186/1423-0127-18-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee MJ, Feliers D, Mariappan MM, et al. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. American Journal of Physiology—Renal Physiology. 2007;292(2):F617–F627. doi: 10.1152/ajprenal.00278.2006. [DOI] [PubMed] [Google Scholar]
- 87.Kume S, Uzu T, Araki SI, et al. Role of altered renal lipid metabolism in the development of renal injury induced by a high-fat diet. Journal of the American Society of Nephrology. 2007;18(10):2715–2723. doi: 10.1681/ASN.2007010089. [DOI] [PubMed] [Google Scholar]
- 88.Tanaka Y, Kume S, Araki SI, et al. Fenofibrate, a PPARα agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney International. 2011;79(8):871–882. doi: 10.1038/ki.2010.530. [DOI] [PubMed] [Google Scholar]
- 89.Jiang T, Wang Z, Proctor G, et al. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. Journal of Biological Chemistry. 2005;280(37):32317–32325. doi: 10.1074/jbc.M500801200. [DOI] [PubMed] [Google Scholar]
- 90.Wang Z, Jiang T, Li J, et al. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes. 2005;54(8):2328–2335. doi: 10.2337/diabetes.54.8.2328. [DOI] [PubMed] [Google Scholar]
- 91.Saha AK, Ruderman NB. Malonyl-CoA and AMP-activated protein kinase: an expanding partnership. Molecular and Cellular Biochemistry. 2003;253(1-2):65–70. doi: 10.1023/a:1026053302036. [DOI] [PubMed] [Google Scholar]
- 92.Cantó C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cellular and Molecular Life Sciences. 2010;67(20):3407–3423. doi: 10.1007/s00018-010-0454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Steinberg GR, Kemp BE. AMPK in health and disease. Physiological Reviews. 2009;89(3):1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 94.Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS ONE. 2010;5(11) doi: 10.1371/journal.pone.0015394. Article ID e15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466(7302):68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Imai SI, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends in Pharmacological Sciences. 2010;31(5):212–220. doi: 10.1016/j.tips.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annual Review of Pathology. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.In HL, Cao L, Mostoslavsky R, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(9):3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tikoo K, Tripathi DN, Kabra DG, Sharma V, Gaikwad AB. Intermittent fasting prevents the progression of type I diabetic nephropathy in rats and changes the expression of Sir2 and p53. FEBS Letters. 2007;581(5):1071–1078. doi: 10.1016/j.febslet.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 100.Tikoo K, Singh K, Kabra D, Sharma V, Gaikwad A. Change in histone H3 phosphorylation, MAP kinase p38, SIR 2 and p53 expression by resveratrol in preventing streptozotocin induced type I diabetic nephropathy. Free Radical Research. 2008;42(4):397–404. doi: 10.1080/10715760801998646. [DOI] [PubMed] [Google Scholar]
- 101.Koya D, Hayashi K, Kitada M, Kashiwagi A, Kikkawa R, Haneda M. Effects of antioxidants in diabetes-induced oxidative stress in the glomeruli of diabetic rats. Journal of the American Society of Nephrology. 2003;14(8, supplement 3):S250–S253. doi: 10.1097/01.asn.0000077412.07578.44. [DOI] [PubMed] [Google Scholar]
- 102.Brezniceanu ML, Liu F, Wei CC, et al. Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney International. 2007;71(9):912–923. doi: 10.1038/sj.ki.5002188. [DOI] [PubMed] [Google Scholar]
- 103.Morse E, Schroth J, You NH, et al. TRB3 is stimulated in diabetic kidneys, regulated by the ER stress marker CHOP, and is a suppressor of podocyte MCP-1. American Journal of Physiology—Renal Physiology. 2010;299(5):F965–F972. doi: 10.1152/ajprenal.00236.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kitada M, Takeda A, Nagai T, et al. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in Diabetic Wistar fatty (fa/fa) rats, a model of type 2 diabetes. Experimental Diabetes Research. 2011;2011:11 pages. doi: 10.1155/2011/908185. Article ID 908185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Archives of Biochemistry and Biophysics. 2007;462(2):245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu L, Wise DR, Diehl JA, Simon MC. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. Journal of Biological Chemistry. 2008;283(45):31153–31162. doi: 10.1074/jbc.M805056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Peterson CM, Jones RL, Koenig RJ. Reversible hematologic sequelae of diabetes mellitus. Annals of Internal Medicine. 1977;86(4):425–429. doi: 10.7326/0003-4819-86-4-425. [DOI] [PubMed] [Google Scholar]
- 108.Jones RL, Peterson CM. Hematologic alterations in diabetes mellitus. American Journal of Medicine. 1981;70(2):339–352. doi: 10.1016/0002-9343(81)90771-3. [DOI] [PubMed] [Google Scholar]
- 109.Katavetin P, Miyata T, Inagi R, et al. High glucose blunts vascular endothelial growth factor response to hypoxia via the oxidative stress-regulated hypoxia-inducible factor/hypoxia-responsible element pathway. Journal of the American Society of Nephrology. 2006;17(5):1405–1413. doi: 10.1681/ASN.2005090918. [DOI] [PubMed] [Google Scholar]
- 110.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Molecular Cell. 2010;40(2):294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mazure NM, Pouysségur J. Hypoxia-induced autophagy: cell death or cell survival? Current Opinion in Cell Biology. 2010;22(2):177–180. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 112.Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Molecular and Cellular Biology. 2009;29(10):2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JGN, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. American Journal of Physiology—Heart and Circulatory Physiology. 2008;294(2):H570–H578. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 114.Sieber J, Lindenmeyer MT, Kampe K, et al. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. American Journal of Physiology—Renal Physiology. 2010;299(4):F821–F829. doi: 10.1152/ajprenal.00196.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lindenmeyer MT, Rastaldi MP, Ikehata M, et al. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. Journal of the American Society of Nephrology. 2008;19(11):2225–2236. doi: 10.1681/ASN.2007121313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circulation Research. 2010;107(5):579–591. doi: 10.1161/CIRCRESAHA.110.225698. [DOI] [PubMed] [Google Scholar]
- 117.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 118.Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends in Biochemical Sciences. 2007;32(10):469–476. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 119.Rouschop KMA, Beucken TVD, Dubois L, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. Journal of Clinical Investigation. 2010;120(1):127–141. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tanaka T, Kato H, Kojima I, et al. Hypoxia and expression of hypoxia-inducible factor in the aging kidney. Journals of Gerontology—Series A. 2006;61(8):795–805. doi: 10.1093/gerona/61.8.795. [DOI] [PubMed] [Google Scholar]
- 121.Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. Journal of Biological Chemistry. 2008;283(16):10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 122.Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metabolism. 2007;6(6):458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]