

Abstract
The protein arginine methyltransferases (PRMTs) are SAM-dependent enzymes that catalyze the mono- and di-methylation of peptidyl arginine residues. PRMT1 is the founding member of the PRMT family, and this isozyme is responsible for methylating ~85% of the arginine residues in mammalian cells. Additionally, PRMT1 activity is aberrantly upregulated in heart disease and cancer. As a part of a program to develop isozyme specific PRMT inhibitors, we recently described the design and synthesis of C21, a chloroacetamidine bearing histone H4 tail analog that acts as an irreversible PRMT1 inhibitor. Given the covalent nature of the interaction, we set out to develop Activity Based Probes (ABPs) that could be used to characterize the physiological roles of PRMT1. Herein, we report the design, synthesis, and characterization of fluorescein-conjugated C21 (F-C21) and biotin-conjugated C21 (B-C21) as PRMT1-specific ABPs. Additionally, we provide the first evidence that PRMT1 activity is negatively regulated in a spatial and temporal fashion.
Post-translational modifications (PTMs) are known to impact numerous cellular processes (e.g., cell growth, apoptosis, and transcriptional regulation) via their ability to alter protein activity and/or stability. One such modification is arginine methylation. This modification is catalyzed by the Protein Arginine Methyltransferases (PRMTs), a relatively small nine member family of enzymes that catalyze the transfer of a methyl group from S-adenosyl methionine (SAM) to the guanidinium nitrogens of arginine residues in proteins. Although the specific roles that the PRMTs play in controlling cellular physiology are only beginning to be understood, it is clear that these enzymes help to regulate a diverse array of cellular pathways including, cell growth, division, differentiation, gene transcription, and RNA splicing (1–4). In addition, the activity of several of these enzymes, most predominantly PRMTs 1 and 4, appears to be dysregulated and contribute to the pathophysiology of heart disease and cancer (5–7).
One of the most widely studied members of the PRMT family is PRMT1. This isozyme catalyzes the transfer of two methyl groups to the same terminal nitrogen atom of arginine to generate asymmetrically dimethylated arginine (ADMA) (Figure 1), and is responsible for generating ~85% of the methylated arginine residues in mammalian cells (8). PRMT1 is ubiquitously expressed and catalyzes the methylation of a diverse array of substrates (9). For instance, PRMT1 asymmetrically dimethylates Sam68 in proline-rich motifs and this modification decreases the ability of Sam68 to interact with SH3 domains present in other proteins (10). Additionally, PRMT1 has been shown to methylate Arg260 in estrogen receptor α (ERα). This modification is induced in the presence of estrogen, and ultimately triggers the formation of a macromolecular complex that activates protein kinase B (PKB/Akt) and promotes cell survival (11). PRMT1 is also known to catalyze the methylation of histone H4 at Arg3 and this modification is associated with increased transcription of genes under the control of ERα, the androgen receptor, and p53, among others (12–14).
Figure 1.

Structures of Activity-Based Probes (ABPs). The reporter tags, FITC and biotin, were coupled to the N-terminus of C21 to generate fluorescein-conjugated C21 (F-C21) and biotin-conjugated C21 (B-C21).
In order to guide the development of inhibitors/chemical probes targeting PRMT1, we initiated studies to characterize the mechanism of PRMT1 catalysis. Specifically, we showed that PRMT1 uses a rapid equilibrium random mechanism with dead-end EAP and EBQ complexes (15) to catalyze the dimethylation of at least a subset of its substrates in a partially processive fashion (16). Substrate specificity studies have also revealed that this enzyme preferentially methylates substrates that possess positively-charged residues distal to the site of methylation; these long range interactions stabilize the formation of the ternary complex (16). These observations aided our efforts to design and synthesize the most potent and selective PRMT1 inhibitor known to date (17). This compound, denoted C21, is a peptide-based inhibitor that contains a chloroacetamidine warhead in place of the target arginine residue (17). The fact that C21 irreversibly inactivates PRMT1 suggested that this compound could be used as the basis for generating PRMT1-targeted Activity-Based Probes (ABPs). ABPs targeting a variety of enzymes and enzyme families have been described (18–20), and these probes have been shown to have great utility in a number of areas, including inhibitor identification, the identification of novel enzymes, and the investigation of enzyme regulatory mechanisms (21–26). For PRMT1, we hypothesized that ABPs targeting this enzyme could be used to identify the PRMT Interacting Proteins (PIPs) and PTMs that regulate PRMT1 activity; because PRMT1 is constitutively active there must be a mechanism to regulate its activity. Herein we describe the design, synthesis, and characterization of biotin-conjugated C21 (B-C21) and fluorescein-conjugated C21 (F-C21) (Figure 1). Furthermore, we show for the first time that PRMT1 activity is regulated in response to estrogen.
Materials and Methods
Reagents and Chemicals
N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES), dithiothreitol (DTT), fluorescein isothiocyanate (FITC), and ethylenediamine tetraacetic acid (EDTA) were purchased from Sigma-Aldrich. N-α-Fmoc-protected amino acids and pre-loaded Wang based resins were obtained from Novabiochem. HOBt and HBTU were purchased from VWR. Anti-PRMT1 (ab7027), anti-PRMT3 (ab84202), anti-PRMT4 (ab50214), anti-PRMT6 (ab47244) and anti-PAD4 (ab50247) were purchased from Abcam. Radiolabelled reagents, i.e. 14C-labeled SAM and 14C-labeled bovine serum albumin (BSA), were purchased from Perkin-Elmer Life Sciences. The purification of recombinant human PRMT1 (hPRMT1) has previously been described (16).
Synthesis of F-C21
The H4-21(R3Orn(Dde)) peptide (NH2- SGXGKGGKGLGKGGAKRHRKV-COO−; where X is Dde-protected ornithine) was synthesized using standard Fmoc solid phase peptide chemistry on Wang resin using a Rainin PS3 automated peptide synthesizer. Fmoc-aminohexanoic acid (2 equiv) was then coupled to the N-terminus of the peptide in the presence of HOBt and HBTU. The peptide was deprotected with 20% piperidine/DMF and subsequently coupled, in an equimolar ratio, to FITC in the presence of Et3N (2 equiv) in DMF overnight, in the dark. Removal of the Dde-protecting group was accomplished by two 45 min incubations of the resin with 2% hydrazine in DMF. The resin containing free Orn was treated with ethylchloroacetimidate hydrochloride (4 equiv) and triethylamine (8 equiv) in DMF twice for 8 h. The resin was washed three times each with DMF, ethanol, and DCM. Cleavage and complete deprotection of F-C21 was accomplished by treatment with TFA/TIS/H2O (95/2.5/2.5) for 1 h. After the volatiles were removed, crude F- C21 was precipitated with ether, isolated by centrifugation, and purified by RP-HPLC using a H2O(0.05% TFA)/Acetonitrile (0.05% TFA) linear gradient. The identity of F-C21 was verified by MALDI-MS, calculated m/z is 2626, observed m/z is 2626.
Synthesis of B-C21
The H4-21(R3Orn(Dde)) peptide was synthesized as described above. Upon deprotection of the N-terminus, D-(+)-biotin (Alfa Aesar) (1 equiv) was coupled to the peptide, in the presence of HOBt and HBTU, twice for 3 h. The Dde protecting group was removed with two 45 min incubations of the resin with 2% hydrazine in DMF. Ethylchloroacetimidate was coupled to the peptide and the product was isolated and purified as described above. The identity of B-C21 was confirmed with MALDI-MS, calculated m/z is 2465, observed m/z is 2465.
Synthesis of F-F21
The H4-21(R3Orn(Dde)) peptide was synthesized as described above. Fmoc-aminohexanoic acid (2 equiv) was coupled to the N-terminus of the peptide in the presence of HOBt and HBTU. The peptide was deprotected with 20% piperidine/DMF and subsequently coupled, in an equimolar ratio, to FITC in the presence of Et3N (2 equiv) in DMF overnight, in the dark. Removal of the Dde-protecting group was accomplished by two 45 min incubations of the resin with 2% hydrazine in DMF. The resin containing free Orn was treated with ethylfluoroacetimidate hydrochloride (4 equiv) and triethylamine (8 equiv) in DMF twice for 8 h. The peptide was isolated and purified as described above. The identity of F-F21 was confirmed with MALDI-MS, calculated m/z is 2611, observed m/z is 2611.
IC50 assays
IC50 values for PRMT1 were determined as previously described (16, 27). The Assay Buffer consisted of 50 mM HEPES at pH 8.0, 50 mM NaCl, 1 mM EDTA, and 0.5 mM DTT. Briefly, various inhibitor concentrations were incubated with 200 nM PRMT1 and 15 μM 14C-methyl-SAM in Assay Buffer at 37 °C for 10 min. The reaction was initiated by the addition of peptide substrate (i.e., 25 μM AcH4-21) and quenched with tris-tricine gel loading dye after 15 min. Samples were run on 16.5% tris-tricine polyacrylamide gels and incorporated radioactivity was quantified by phosphorimage analysis (Molecular Dynamics). IC50 values were determined by fitting the data thus obtained to equation 2,
| (eq 2) |
using the GraFit version 5.0.11 software package (28), where [I] is the concentration of inhibitor and IC50 is the concentration of inhibitor that yields half-maximal activity. All assays were performed at least in duplicate and the standard deviation was typically ≤ 20 %.
Cell culture and extract preparation
MCF-7 cells were maintained at 37 °C and 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM) (VWR) supplemented with 10% Fetal Bovine Serum (FBS) (VWR). For estrogen stimulation experiments, cells were grown for 48 h in phenol-red free DMEM (VWR) with 10% charcoal-stripped FBS (Gemini Bio Products) at 37 °C and 5% CO2. Cells were stimulated with 10 nM E2 (Sigma) and incubated at 37 °C and 5% CO2 for the specified amount of time. Cells were harvested via scraping and incubated in RIPA buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% deoxycholate) for 30 min at 4 °C, with occasional agitation; following centrifugation, the supernatant was collected to generate whole cell extracts.
Cytoplasmic and nuclear extracts were obtained by incubating cells in cytoplasmic extract buffer (10 mM Tris-HCl pH 7.5, 137 mM NaCl, 1% Tween 20, 1 mM PMSF) for 30 min at 4 °C. Cells were lysed with a 25 gauge needle and the lysate was cleared by centrifugation. The supernatant was removed to afford the cytoplasmic extract. The pellet was then resuspended in nuclear extract buffer (20 mM HEPES pH 7.9, 25% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.5 mM DTT, 1 mM PMSF) and incubated at 4 °C for 30 min. Again, the lysate was centrifuged and the supernatant was removed to afford the nuclear extracts.
Time and Concentration Dependence of Labeling
2 μM PRMT1 was incubated with 2 μM F-C21 in Assay Buffer for 0–50 min at 37 °C. The reaction was quenched with SDS-PAGE loading dye and separated by SDS-PAGE. Labeled protein was visualized with a flatbed laser scanner. 2 μM PRMT1 was labeled with increasing concentrations of F-C21 (0 to 50 μM) or F-F21 (0 to 5000 μM) for 30 min at 37 °C. The samples were quenched and processed as described above.
Limits of Detection
2 μM B-C21 was reacted with 1 nM to 2 μM PRMT1 for 30 min in Assay Buffer at 37 °C. The reaction was quenched with SDS-PAGE loading dye and separated by SDS-PAGE. Labeled protein was visualized by western blot analysis using a streptavidin-HRP. 2 μM F-C21 was reacted with 1 nM to 2 μM PRMT1 for 30 min in Assay Buffer at 37 °C. The reaction was quenched with SDS-PAGE loading dye and separated by SDS-PAGE. Labeled protein was visualized by fluorescence imaging.
Isolation of PRMT1 with B-C21
Extracts (100 μg total protein) were incubated with B-C21 for 30 min at 37 °C and then applied to streptavidin-agarose beads (Thermo Scientific) overnight at 4 °C. The beads were washed one time with 10X bead volume of 0.2% SDS/PBS, three times with PBS and three times with water. Bound proteins were eluted with 1 h incubation at 42 °C in elution buffer (2% SDS, 30 mM biotin, 6 M urea, 2 M thiourea). Eluent was concentrated by speed vacuum, reconstituted in SDS-PAGE loading dye and separated by SDS-PAGE. Proteins were then transferred to nitrocellulose and visualized by western blotting using an anti-PRMT1 antibody. A 10% loading control, representative of the extract samples prior to their incubation with the probe, was run on a separate gel and visualized by western blot analysis. Representative data from one of three experiments is depicted. The data were quantified using the image J software.
In-gel Tryptic Digestion
Excised gel bands were washed twice with Wash Solution (50% v/v methanol, 5% v/v acetic acid) for 4 h, then dehydrated with acetonitrile for 5 min and dried. The dried bands were incubated first with 10 mM DTT for 30 min, then with iodoacetamide for 30 min. After the bands were dehydrated and dried, they were incubated with trypsin (at a trypsin to substrate ratio of 1:20) in 50 mM ammonium bicarbonate overnight at 37 °C. Peptides were extracted with two 10 min incubations in Extraction Buffer (50% v/v acetonitrile, 5% v/v formic acid) and analyzed by MALDI-TOF mass spectrometry.
Western blotting
Proteins were separated on 12% SDS-PAGE gels and transferred to a nitrocellulose membrane. The blot was blocked for 1 h in 5% milk and then incubated overnight at 4 °C with the appropriate antibody in 2.5% milk. Following incubation with the secondary antibody, proteins were visualized with an enhanced chemiluminescence kit (Pierce Chemical).
Results and Discussion
Design and Synthesis of ABPPs
Based on our work demonstrating that haloacetamidine containing compounds act as irreversible inhibitors of the Protein Arginine Deiminases (PADs) and Agmatine Deiminases (29–34), we considered the possibility that such compounds might also inhibit other arginine modifying enzymes, including the PRMTs. To test this hypothesis, we replaced the guanidinium of Arg3 in the AcH4-21 peptide, which consists of the first 21 amino acids of histone H4 and is a highly efficient PRMT1 substrate (15–17) with both a fluoro- and chloroacetamidine warhead to generate F21 and C21, respectively (17). Inhibition studies with these compounds demonstrated that C21, but not F21, is a highly potent (kinact/KI = 4.6 × 106 min−1M−1) and selective irreversible PRMT1 inhibitor (17); F21 is a reversible competitive inhibitor (Ki = 1.8 ± 0.9 μM μM) (17). Given that irreversible inhibitors have been used to generate ABPs for a variety of enzymes (e.g. Ser Hydrolases, Cys Proteases, and PADs (18, 33, 34), we further hypothesized that C21 could be readily converted into ABPs, and that such compounds would be useful for identifying and studying the factors that regulate PRMT1 activity. For reporter tags, we chose to synthesize both fluorescein- and biotin-tagged derivatives of C21 because the former would allow for the facile labeling and detection of active PRMT1 whereas the latter would enable both the visualization and isolation of the enzyme. To generate these ABPs, the N-terminal acetyl group on C21 was replaced with either an aminohexanoic acid linker coupled to FITC or directly coupled to biotin to generate fluorescein-conjugated C21 (F-C21) or biotin-conjugated C21 (B-C21), respectively (Figure 1); the aminohexanoic acid linker was used to provide ample separation between fluorescein and the chloroacetamidine warhead. Note that we chose to modify the N-terminus of C21 because we have previously shown that this portion of the peptide contributes minimally to substrate recognition (16). Also note that we synthesized a fluorescein-derivatized version of F21 (i.e., F-F21), which does not covalently modify PRMT1, to serve as a negative control.
IC50 values and inhibitor selectivity
To evaluate whether the fluorescein and biotin reporter tags affected inhibitor potency or selectivity, IC50 values were determined for PRMT1, as well as PRMTs 3, 4 (also known as Coactivator associated Arginine Methyltransferase 1 or CARM1), 6, and PAD4. With respect to PRMT1, the results of these experiments indicate that F-C21 and B- C21, as well as F-F21, possess IC50 values that are comparable to the parent compounds C21 and F21 (Table 1), thereby indicating that the reporter tags do not impact inhibitor potency. With respect to PAD4 and PRMTs 3, 4, and 6, the IC50 values (Table 2) are also comparable to those obtained for the parent compounds, which indicates that the added reporter tags do not alter inhibitor selectivity, thereby demonstrating the potential utility of these compounds to specifically identify and characterize the factors that regulate PRMT1 activity.
Table 1.
IC50 values for PRMT1
Values from ref. 17.
Table 2.
ABP Selectivity: IC50 values for PRMTs 1, 3, 4, 6 and PAD4
| Enzyme | IC50 for C21 (μM) | IC50 for F-C21 (μM) | IC50 for B-C21 (μM) |
|---|---|---|---|
| PRMT1 | 1.8 ± 0.1a | 3.4 ± 0.4 | 1.2 ± 0.2 |
| PRMT3 | > 500a | > 500 | > 500 |
| PRMT4/CARM1 | > 500a | 280 ± 100 | > 500 |
| PRMT6 | 8.8 ± 0.5a | 14 ± 3 | 13 ± 4 |
| PAD4 | 145 ± 20a | 120 ± 20 | 235 ± 10 |
Values from ref. 17
Time and concentration dependence and limits of detection
Having established that F-C21 and B-C21 inhibit PRMT1 as effectively as C21, their ability to label purified recombinant PRMT1 in a time and concentration dependent manner was assessed. For these experiments, PRMT1 was first incubated with F-C21 (2 μM final) in Assay Buffer for 0 to 50 min. The reaction was then quenched with 6 x SDS-PAGE loading buffer, and the reaction components separated by SDS-PAGE. Labeled PRMT1 was then visualized using a flatbed laser scanner. The results of these experiments (Figure 2A) indicate that F-C21 labels PRMT1 in a time dependent fashion with maximum labeling occurring within the first 30 min. Note that the enzyme was efficiently labeled in both the presence and absence of SAM, thereby indicating that the cofactor does not need to be bound to the enzyme prior to peptide binding. This observation is consistent with our observation that PRMT1 utilizes a rapid equilibrium random mechanism, with dead-end EAP and EBQ complexes, because this mechanism dictates that the order of substrate binding is not obligatory (15).
Figure 2.

F-C21 Labels PRMT1 in a Time and Concentration Dependent Manner. A. Time Course with 2 μM PRMT1 and 2 μM F-C21. B. Concentration dependence of F-C21with 2 μM PRMT1 and 30 minute reaction time. C. Concentration dependence of F-F21 with 2 μM PRMT1 and 30 minute reaction time. D. F-C21 limit of detection was determined to be 0.5 μM, which corresponds to 250 ng PRMT1. E. B-C21 limit of detection was determined to be 0.05 μM, which corresponds to 25 ng PRMT1.
To evaluate the concentration dependence of labeling, PRMT1 was incubated with increasing concentrations of F-C21 (0 to 50 μM) or F-F21 (0 to 5000 μM), as a negative control for 30 min. The samples were then processed as described above, and the results (Figure 2B and 2C) indicate that F-C21 labels PRMT1 in a concentration dependent manner and that near maximum labeling occurs at 2 μM F-C21. The fact that F-F21 does not label PRMT1 even at concentrations as high as 5,000 μM, which represents a 2,500-fold higher concentration than the ideal concentration of F-C21, is consistent with the fact that F21 is a reversible PRMT1 inhibitor (17), and suggests that the leaving group potential of the fluoride is too low to effect reaction with the active site nucleophile in PRMT1. In Figure 2, it is also noteworthy that despite an excess of probe, two bands, corresponding to unmodified and modified PRMT1, are clearly visible in the Coomassie stained gels (Figure 2A-E). This result, which is most apparent in Figure 2B, causes the reduction in the intensity of PRMT1 in the Coomassie stained gels. Given that dimerization of PRMT1 is required for its activity (35), this result suggests that inactivation of PRMT1 by C21 may be due to the modification of a single PRMT1 monomer present in the dimer. It is also noteworthy that at concentrations of F-C21 in excess of 5 μM (Figure 2B), two bands are clearly visible in both the fluorescent images. This result suggests that, at higher concentrations, PRMT1 is subject to multiple modifications and indicates that care must be taken to choose the appropriate ABP concentration to prevent multiple labeling events. Note that the time and concentration dependence of B-C21 labeling was similar to that obtained for F-C21 (not shown).
To evaluate the sensitivity of F-C21 and B-C21, limit of detection assays were performed. Briefly, increasing concentrations of PRMT1 (0 to 2 μM or 0 to 2 μg) were incubated with fixed concentrations of F-C21 (2 μM, final) and B-C21 (2 μM, final) for 30 min. The samples were then processed as described above and the results (Figure 2D and 2E) indicate that F-C21 and B-C21 can detect as little as 250 ng and 25 ng of PRMT1, respectively. The higher sensitivity of B-C21 likely reflects the use of streptavidin-conjugated HRP to amplify the signal.
F-C21 Labeling of PRMT1 in MCF-7 cell lysates
Having established the ability of the probes to label recombinant PRMT1, we were interested in evaluating whether they could be used to label PRMT1 in a complex protein mixture. Given that PRMT1 is highly expressed in MCF-7 human breast cancer cells (11), this cell line was used to demonstrate the ability of the probes to label PRMT1 amongst a myriad of other proteins. To serve as a control, purified recombinant PRMT1 was incubated without (Figure 3; Lane 1) or with F-C21 (2 μM final; Figure 3; Lane 2), and as expected this led to the robust labeling of PRMT1 only in the presence of F-C21. To establish our ability to label PRMT1 in MCF7 cell extracts, purified recombinant PRMT1 (2 μM) was either added to MCF7 whole cell extracts (100 μg; Figure 3; Lane 3) or omitted (Figure 3; Lane 4), and then incubated with F-C21 (2 μM final) for 30 min. The results of these experiments indicate that F-C21 can readily label PRMT1 even in complex proteomes, as is apparent by the presence of a fluorescent band corresponding to the size of PRMT1. To probe the selectivity of labeling, 1 mM C21 was added to F-C21 treated cell extracts, and this treatment effectively blocked PRMT1 labeling. Interestingly, in the presence of the extracts the extent of labeling of the exogenously added purified PRMT1 is greatly diminished (compare Lanes 2 and 3). This result may suggest that either PRMT1 is rapidly degraded in the cell extracts or that factors within those extracts modulate the activity of the enzyme such that it is no longer capable of being modified by F-C21. To control for the former possibility, recombinant PRMT1 was pre-labeled with F-C21 for 30 min at 37 °C, and then the labeled enzyme was added MCF-7 whole cell extracts and incubated at 37 °C for 0 to 60 min. The results of these studies (Figure S1) indicate that labeled PRMT1 is not rapidly degraded by the cell extracts. Although an unknown constituent in the cell extracts may preferentially bind and sequester F-C21, which would prevent labeling, we favor the notion that factors (e.g. PRMT1 modifying enzymes, binding proteins, or substrates) within the extracts must alter recombinant PRMT1 activity so that it can no longer be modified by F-C21.
Figure 3.
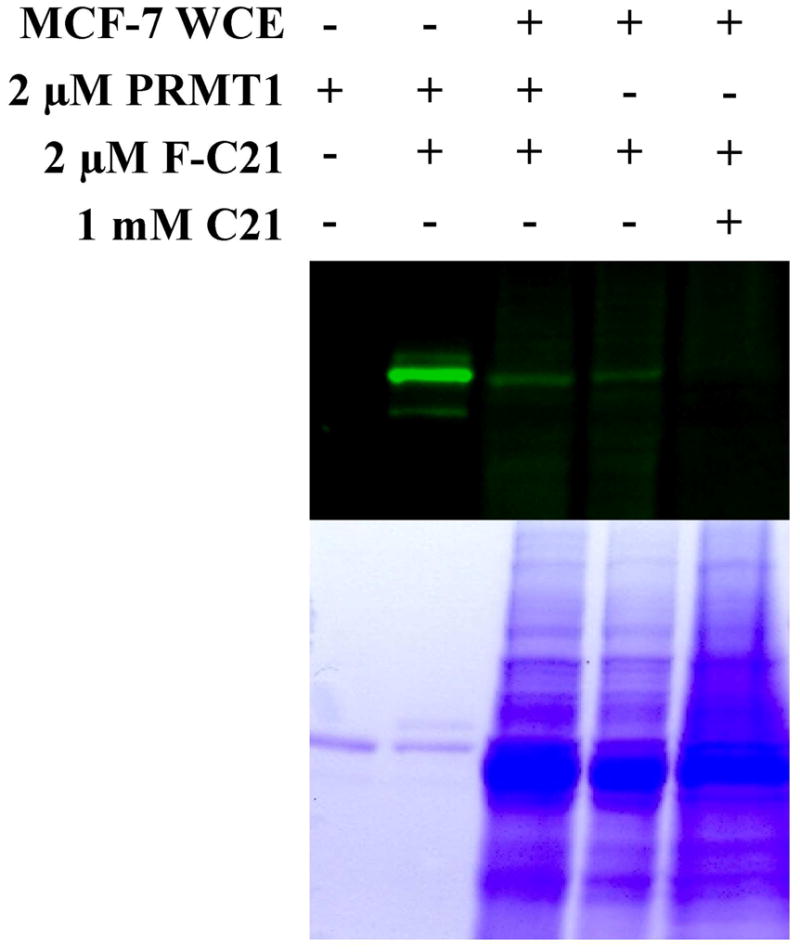
Labeling PRMT1 in MCF-7 Whole Cell Extracts with F-C21. MCF-7 whole cell extracts (WCE) were incubated in the prescence or absence of recombinant PRMT1 and F-C21. F-C21 labeling was outcompeted with C21.
B-C21 Labeling of PRMT1 in MCF-7 cell lysates
Having established the methodology to label PRMT1 in MCF7 cell extracts, we moved forward to examine the ability of B-C21 to label the enzyme under similar conditions. For these experiments, increasing concentrations of BC21 were used to label endogenous PRMT1 present in MCF7 whole cell extracts (Figure 4A). The results of these experiments indicate that B-C21 can also effectively label a protein with a molecular weight consistent with endogenous PRMT1. Note that while the molecular weight of PRMT6 is similar to that of PRMT1 (41.9 kDa versus 42.4 kDa), which could suggest that the protein highlighted is a mixture of PRMT1 and PRMT6, the data in Figure 4B (see below) indicates that B-C21 selectively isolates PRMT1 over PRMT6. Thus, this band most likely corresponds to PRMT1. Although the probe is more selective at lower concentrations (e.g., 0.5 μM) versus higher concentrations (e.g., 5.0 μM), this observation is not unexpected as most chemical probes, and even antibodies (18, 23, 24), lose selectivity at higher concentrations. Nevertheless, the results indicate that care must be taken in using an appropriate amount of this ABP to prevent the non-specific modification of bystander proteins. Note that protein bands denoted by an asterisk correspond to biotinylated proteins. Although these bands appear to get darker as the concentration of B-C21 is increased above 1 μM, the bands are clearly present at equal intensity in the 0 μM B-C21 control lane and the 0.1 and 0.5 μM lanes. The increased intensity is most likely due to a loss of specificity at higher probe concentrations, which results in the labeling of proteins with similar molecular weights.
Figure 4.

Labeling PRMT1 in MCF-7 Cell Lysates with B-C21. A. MCF-7 breast cancer whole cell extracts were labeled with increasing amounts of B-C21 for 30 minutes and subject to western blot analysis using a streptavidin-HRP conjugate. Proteins marked with (*) appear to be biotinylated proteins since they are present in the absence of B-C21. B. Selective Isolation of PRMT1 from MCF-7 Whole Cell Extracts with B-C21. MCF-7 WCE were incubated with the specified amount of B-C21 for 30 minutes, then bound to streptavidin agarose beads overnight at 4 °C. Beads were washed several times before bound proteins were eluted, separated by SDS-PAGE, and subject to western blot analysis with the appropriate antibody.
To probe the selectivity of B-C21 in a proteome context and demonstrate our ability to isolate and enrich for PRMT1, the ability of B-C21 to isolate PRMTs 1, 3, 4, and 6, as well as PAD4, was evaluated. For these experiments, MCF-7 whole cell extracts were incubated with either 5 or 50 μM B-C21 for 30 min. Streptavidin-conjugated agarose beads were then added to the reaction mixtures and incubated overnight at 4 °C. Non-specific proteins were washed away and bound proteins were eluted from the beads and subjected to western blot analysis, using antibodies for PRMTs 1, 3, 4, and 6 and PAD4. As depicted in Figure 4B, PRMT1 can be selectively isolated from MCF-7 whole cell extracts. Note that the amount of each isozyme in the input lane is consistent with previous reports of the relative expression levels of the isozymes in vivo (36). PRMTs 3 and 8 are considered to be minor PRMTs due to their low cellular expression level in most tissues, while PRMT1 is the most abundant family member and is responsible for ~85% of protein arginine methylation in mammalian cells (8, 37). Although we do see cross reactivity with PRMT6 when we use a 10-fold higher concentration of B-C21, it is noteworthy that at this concentration this compound isolates ≥10% of total PRMT1 versus less than 1% of PRMT6 (compare the intensities of the bands 50 μM lanes to the 10% input controls (Figure 4B)). This is particularly impressive when one considers that the difference in the IC50 values for F-C21 and B-C21 between PRMT1 and PRMT6 are only 4.1- and 10.8-fold respectively.
To demonstrate the ability to isolate and enrich for PRMT1, MCF7 whole cell extracts (200 μg) were incubated with B-C21 (25 μM) and the samples were processed as described above. Coommassie staining of the SDS-PAGE gels identified a band corresponding to the correct molecular weight for PRMT1 (Figure 5A). The band was excised from the gel and subjected to in gel tryptic digestion. MALDI MS and MS/MS analyses of the tryptic digest identified a number of peptides corresponding to the sequence of human PRMT1 (sequence coverage was 36%; Figure 5B and 5C), thereby demonstrating that B-C21 can be used to isolate and enrich for PRMT1.
Figure 5.

A. PRMT1 was isolated from MCF-7 WCE and its identity was confirmed via western blot analysis (top) and Coomassie staining (bottom). B. B-C21 labeled enzyme was excised from the gel, subjected to tryptic digestion and the resulting peptides were observed and analyzed using MALDI-TOF MS and MS/MS analyses. The identities of the peptides and the sequence coverage is depicted. C. The expected and observed y and b ions for a representative tryptic peptide (i.e., 325DLDFTIDLDFK335) that was subjected to MS/MS analysis is depicted; the peptide is composed of amino acids 325 to 335 of human PRMT1 and the observed ions are highlighted in red.
In total, the results of the above validation experiments highlight both the utility and drawbacks of both F-C21 and B-C21. For example, while F-C21 enables the rapid detection/visualization of PRMT1 in vitro, its ability to act as an ABP in cell extracts is somewhat limited in comparison to B-C21 because higher concentrations of F-C21 are required to detect PRMT1; at these concentrations the level of non-specific labeling is relatively high. Although not as rapid as F-C21, B-C21 readily detects low ng amounts of PRMT1 and can be used to both visualize and isolate active PRMT1. These characteristics indicate that the greatest utility of F-C21 is likely to be as a tool for drug discovery/screening similarly to other systems (38–40). In contrast, the high sensitivity of B-C21 highlights its utility in isolating and/or visualizing PRMT1, thereby suggesting that this compound will be an excellent tool for the discovery of the factors that regulate PRMT1 activity.
B-C21 preferentially labels active PRMT1
Although the experiments described above demonstrate the ability of our PRMT1-targeted probes to label and isolate the enzyme, it is unclear whether they truly represent ABPs because PRMT1 is constitutively active in vitro, and modifications that down regulate its activity have not been discovered. Nevertheless, when one considers that PRMT1 is a key cell signaling enzyme that plays roles in a variety of important processes, including transcriptional regulation and cell survival (3, 13), it is clear that this enzyme is likely to be regulated similarly to other enzymes, e.g. the protein kinases, in a temporal and/or spatial fashion. Therefore, to demonstrate that our probes are in fact ABPs, we examined whether estrogen could modulate PRMT1 activity in nuclear and cytosolic extracts as a function of time. This model system was chosen because previous work by LeRomancer et al had demonstrated that PRMT1 methylates cytosolic ERα at Arg260 in response to estrogen treatment in MCF-7 breast adenocarcinoma cells, and that the presence of this is modification occurred in a time-dependent manner (11). Specifically, the methylated receptor was most abundant in the cytosol 5 min after estrogen stimulation and was no longer present after 15 minutes, suggesting that estrogen initiates a mechanism by which PRMT1 activity is directed to ERα for a short period of time (11). Since PRMT1 also resides in the nucleus of cells, where it acts to methylate histone H4, we also examined the temporal control of PRMT1 activity in nuclear extracts. For these experiments, MCF-7 cells were incubated with estrogen for a specified amount of time and subsequently the cells were lysed and the cytosolic and nuclear extracts prepared. These lysates were then incubated with B-C21, and the proteins isolated on streptavidin agarose as described above. Bound proteins were eluted and the relative presence of PRMT1 detected by western blot analysis (Figure 6). As a control, extracts were loaded onto a separate gel and the amount of PRMT1 present in each sample shown to be equal (Figure 6A, lower panel). The results of these experiments demonstrate that basal levels of active PRMT1 are present in both the cytoplasm and in the nucleus. However, following 5 minutes of estrogen stimulation, the levels of active PRMT1 in the nucleus begin to decline, reaching a maximum 3-fold reduction at the 5 minute time point. In contrast, the levels of active PRMT1 in the cytoplasm increase by 2.5-fold, 5 minutes after the addition of estrogen, reach a maximum at 10 minutes, and return to basal levels after ~15 minutes. Since PRMT1 levels in the cytosolic and nuclear extracts remain constant (Figure 6A lower panel), it is unlikely that the observed changes are due to a redistribution of bulk PRMT1 between the nucleus and cytosol. Although we cannot completely rule out the possibility that the subset of active PRMT1 is redistributed between these two compartments, the observed changes in the amount of active PRMT1 isolated from both the cytosolic and nuclear extracts suggest that PRMT1 activity is negatively regulated in a manner that ultimately precludes the enzyme from interacting with B-C21. Although the specific regulatory mechanism is unknown, it is likely that the enzyme is post-translationally modified, as has been observed for PRMTs 4 and 5; these isozymes are known to be negatively regulated by phosphorylation (41–43). Alternative explanations also consistent with the data include (1) the possibility that estrogen stimulates the assembly and disassembly of PRMT1 oligomers in the cytosol and nucleus, respectively; or (2) that estrogen may stimulate an unknown constituent in the cell extracts to adopt a conformation that is preferentially bound by B-C21; the sequestration of the probe would cause less PRMT1 to be isolated in the presence of estrogen. As B-C21 is a substrate-based probe, this phenomenon may mimic an estrogen-induced alteration of the substrate specificity of PRMT1. Although the latter mechanism is possible, the simplest explanation for the observed temporal changes in the amount of active PRMT1 is that the enzyme is negatively regulated.
Figure 6.

Labeling MCF-7 Cytoplasmic and Nuclear Extracts Following Estrogen Stimulation. A. MCF-7 cells were stimulated with estrogen for the indicated amount of time and then the cytoplasmic and nuclear extracts were incubated with B-C21 and bound to streptavidin-agarose beads overnight. Bound proteins were eluted from the beads, separated by SDS-PAGE and visualized by western blot analysis using an anti-PRMT1 antibody (upper panel). To demonstrate equal protein loading, a 10% loading control was loaded onto a separate gel and visualized by western blot analysis using the same antibody (lower panel). Representative data from one of three experiments is depicted. B. Quantification of the western blots in panel A.
Given that B-C21 is a substrate analog, this result suggests that this mode of negative regulation prevents substrate binding, and is consistent with the fact that PRMT1 methylates cytosolic ERα for a short time following estrogen treatment. The mechanism by which PRMT1 is directed to ERα is unknown; though it is possible that estrogen activates dormant cytosolic PRMT1 by stimulating post-translational modification of the enzyme or interaction with other proteins that upregulate its activity. Further investigation of this phenomenon with B-C21 is ongoing.
Conclusions
Although protein arginine methylation is critical for properly maintaining normal physiological functions, its dysregulation has been found to contribute to a variety of disease states, including heart disease and cancer. As such, inhibitors targeting this enzyme represent potential therapeutics. However, the specific roles of the PRMTs in these various different pathways remain incompletely understood. Thus, the overall benefit of PRMT inhibitors is unclear because such compounds could interfere with normal cellular processes and as a result cause a number of undesired side effects. Given these issues, chemical probes that can be used to better discern the physiological roles of the PRMTs and their mechanisms of regulation are clearly needed. Towards this goal, we have developed F-C21 and B-C21 as PRMT1-specific ABPs that can be used to labels as little as 250 ng and 25 ng of PRMT1, respectively. Additionally, these studies have shown that B-C21 can be used to selectively isolate PRMT1 from cell extracts and monitor PRMT1 activity in response to external stimuli. In fact, we have used B-C21 to show for the first time that PRMT1 activity is regulated, both temporally and spatially, in response to estrogen. In total, these ABPs will undoubtedly prove to be powerful chemical probes that can be used to investigate PRMT1 activity in a variety of cellular contexts, and thereby increase our understanding of its physiological roles.
Supplementary Material
ABBREVIATIONS
- PRMT
protein arginine methyltransferase
- SAM
S-adenosyl-L-methionine
- CARM1
coactivator-associated methyltransferase 1
- SAH
S-adenosyl-L-homocysteine
- ω-MMA
omega-monomethylarginine
- ADMA
asymmetric dimethylarginine
- SDMA
symmetric dimethylarginine
- HEPES
N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)
- EDTA
ethylenediamine tetraacetic acid
- MALDI
matrix-assisted laser desorption/ionization
- DTT
dithiothreitol
Footnotes
This work was supported in part by the University Of South Carolina Research Foundation (P.R.T), The Scripps Research Institute, and, in part, by NIH grant GM079357 to PRT.
Supporting Information Available
Supplementary Figure S1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lin WJ, Gary JD, Yang MC, Clarke S, Herschman HR. The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J Biol Chem. 1996;271:15034–15044. doi: 10.1074/jbc.271.25.15034. [DOI] [PubMed] [Google Scholar]
- 2.McBride AE, Silver PA. State of the arg: protein methylation at arginine comes of age. Cell. 2001;106:5–8. doi: 10.1016/s0092-8674(01)00423-8. [DOI] [PubMed] [Google Scholar]
- 3.Torres-Padilla ME, Parfitt DE, Kouzarides T, Zernicka-Goetz M. Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature. 2007;445:214–218. doi: 10.1038/nature05458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friesen WJ, Massenet S, Paushkin S, Wyce A, Dreyfuss G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol Cell. 2001;7:1111–1117. doi: 10.1016/s1097-2765(01)00244-1. [DOI] [PubMed] [Google Scholar]
- 5.Tran CT, Leiper JM, Vallance P. The DDAH/ADMA/NOS pathway. Atheroscler. 2003;(Suppl 4):33–40. doi: 10.1016/s1567-5688(03)00032-1. [DOI] [PubMed] [Google Scholar]
- 6.Vallance P, Leiper J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler Thromb Vasc Biol. 2004;24:1023–1030. doi: 10.1161/01.ATV.0000128897.54893.26. [DOI] [PubMed] [Google Scholar]
- 7.Yoshimatsu M, Toyokawa G, Hayami S, Unoki M, Tsunoda T, Field HI, Kelly JD, Neal DE, Maehara Y, Ponder BA, Nakamura Y, Hamamoto R. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer. 2010 doi: 10.1002/ijc.25366. [DOI] [PubMed] [Google Scholar]
- 8.Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, Clarke S, Herschman HR. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J Biol Chem. 2000;275:7723–7730. doi: 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- 9.Gary JD, Clarke S. RNA and protein interactions modulated by protein arginine methylation. Prog Nucleic Acid Res Mol Biol. 1998;61:65–131. doi: 10.1016/s0079-6603(08)60825-9. [DOI] [PubMed] [Google Scholar]
- 10.Bedford MT, Frankel A, Yaffe MB, Clarke S, Leder P, Richard S. Arginine methylation inhibits the binding of proline-rich ligands to Src homology 3, but not WW, domains. J Biol Chem. 2000;275:16030–16036. doi: 10.1074/jbc.M909368199. [DOI] [PubMed] [Google Scholar]
- 11.Le Romancer M, Treilleux I, Leconte N, Robin-Lespinasse Y, Sentis S, Bouchekioua-Bouzaghou K, Goddard S, Gobert-Gosse S, Corbo L. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell. 2008;31:212–221. doi: 10.1016/j.molcel.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 12.Herrmann F, Pably P, Eckerich C, Bedford MT, Fackelmayer FO. Human protein arginine methyltransferases in vivo--distinct properties of eight canonical members of the PRMT family. J Cell Sci. 2009;122:667–677. doi: 10.1242/jcs.039933. [DOI] [PubMed] [Google Scholar]
- 13.Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR. Regulation of transcription by a protein methyltransferase. Science. 1999;284:2174–2177. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- 14.Zhao X, Jankovic V, Gural A, Huang G, Pardanani A, Menendez S, Zhang J, Dunne R, Xiao A, Erdjument-Bromage H, Allis CD, Tempst P, Nimer SD. Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev. 2008;22:640–653. doi: 10.1101/gad.1632608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obianyo O, Osborne TC, Thompson PR. Kinetic mechanism of protein arginine methyltransferase 1. Biochemistry. 2008;47:10420–10427. doi: 10.1021/bi800904m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osborne TC, Obianyo O, Zhang X, Cheng X, Thompson PR. Protein arginine methyltransferase 1: positively charged residues in substrate peptides distal to the site of methylation are important for substrate binding and catalysis. Biochemistry. 2007;46:13370–13381. doi: 10.1021/bi701558t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obianyo O, Causey CP, Osborne TC, Jones JE, Lee YH, Stallcup MR, Thompson PR. A chloroacetamidine-based inactivator of protein arginine methyltransferase 1: design, synthesis, and in vitro and in vivo evaluation. Chembiochem. 2010;11:1219–1223. doi: 10.1002/cbic.201000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 19.Jones JE, Causey CP, Knuckley B, Slack-Noyes JL, Thompson PR. Protein arginine deiminase 4 (PAD4): Current understanding and future therapeutic potential. Curr Opin Drug Discov Devel. 2009;12:616–627. [PMC free article] [PubMed] [Google Scholar]
- 20.Knuckley B, Jones JE, Bachovchin DA, Slack J, Causey CP, Brown SJ, Rosen H, Cravatt BF, Thompson PR. A fluopol-ABPP HTS assay to identify PAD inhibitors. Chem Commun (Camb) 2010;46:7175–7177. doi: 10.1039/c0cc02634d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenbaum D, Baruch A, Hayrapetian L, Darula Z, Burlingame A, Medzihradszky KF, Bogyo M. Chemical approaches for functionally probing the proteome. Mol Cell Proteomics. 2002;1:60–68. doi: 10.1074/mcp.t100003-mcp200. [DOI] [PubMed] [Google Scholar]
- 22.Berger AB, Vitorino PM, Bogyo M. Activity-based protein profiling: applications to biomarker discovery, in vivo imaging and drug discovery. Am J Pharmacogenomics. 2004;4:371–381. doi: 10.2165/00129785-200404060-00004. [DOI] [PubMed] [Google Scholar]
- 23.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 24.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010 doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leung D, Hardouin C, Boger DL, Cravatt BF. Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat Biotechnol. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 26.Adam GC, Cravatt BF, Sorensen EJ. Profiling the specific reactivity of the proteome with non-directed activity-based probes. Chem Biol. 2001;8:81–95. doi: 10.1016/s1074-5521(00)90060-7. [DOI] [PubMed] [Google Scholar]
- 27.Osborne T, Roska RL, Rajski SR, Thompson PR. In situ generation of a bisubstrate analogue for protein arginine methyltransferase 1. J Am Chem Soc. 2008;130:4574–4575. doi: 10.1021/ja077104v. [DOI] [PubMed] [Google Scholar]
- 28.Leatherbarrow RJ. Grafit Ver 5.0. Erathicus Software; Staines, UK: 2004. [Google Scholar]
- 29.Jones JE, Causey CP, Lovelace L, Knuckley B, Flick H, Lebioda L, Thompson PR. Characterization and inactivation of an agmatine deiminase from Helicobacter pylori. Bioorg Chem. 2010;38:62–73. doi: 10.1016/j.bioorg.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knuckley B, Causey CP, Jones JE, Bhatia M, Dreyton CJ, Osborne TC, Takahara H, Thompson PR. Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deiminase 3. Biochemistry. 2010;49:4852–4863. doi: 10.1021/bi100363t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knuckley B, Causey CP, Pellechia PJ, Cook PF, Thompson PR. Haloacetamidine-based inactivators of protein arginine deiminase 4 (PAD4): evidence that general acid catalysis promotes efficient inactivation. Chembiochem. 2010;11:161–165. doi: 10.1002/cbic.200900698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo Y, Arita K, Bhatia M, Knuckley B, Lee YH, Stallcup MR, Thompson PR. Inhibitors and Inactivators of Protein Arginine Deiminase 4: Functional and structural characterization. Biochemistry. 2006;45:11727–11736. doi: 10.1021/bi061180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo Y, Knuckley B, Bhatia M, Pellechia PJ, Thompson PR. Activity-based protein profiling reagents for protein arginine deiminase 4 (PAD4): synthesis and in vitro evaluation of a fluorescently labeled probe. J Am Chem Soc. 2006;128:14468–14469. doi: 10.1021/ja0656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo Y, Knuckley B, Lee YH, Stallcup MR, Thompson PR. A fluoroacetamidine-based inactivator of protein arginine deiminase 4: design, synthesis, and in vitro and in vivo evaluation. J Am Chem Soc. 2006;128:1092–1093. doi: 10.1021/ja0576233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, Cheng X. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure. 2003;11:509–520. doi: 10.1016/s0969-2126(03)00071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frankel A, Clarke S. PRMT3 is a distinct member of the protein arginine N-methyltransferase family. Conferral of substrate specificity by a zinc-finger domain. J Biol Chem. 2000;275:32974–32982. doi: 10.1074/jbc.M006445200. [DOI] [PubMed] [Google Scholar]
- 38.Knuckley B, Jones JE, Bachovchin DA, Slack J, Causey CP, Brown SJ, Rosen H, Cravatt BF, Thompson PR. A fluopol-ABPP HTS assay to identify PAD inhibitors. Chem Commun (Camb) 2010 doi: 10.1039/c0cc02634d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knuckley B, Luo Y, Thompson PR. Profiling Protein Arginine Deiminase 4 (PAD4): a novel screen to identify PAD4 inhibitors. Bioorg Med Chem. 2008;16:739–745. doi: 10.1016/j.bmc.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bachovchin DA, Brown SJ, Rosen H, Cravatt BF. Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes. Nat Biotechnol. 2009;27:387–394. doi: 10.1038/nbt.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng Q, He B, Jung SY, Song Y, Qin J, Tsai SY, Tsai MJ, O’Malley BW. Biochemical control of CARM1 enzymatic activity by phosphorylation. J Biol Chem. 2009;284:36167–36174. doi: 10.1074/jbc.M109.065524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higashimoto K, Kuhn P, Desai D, Cheng X, Xu W. Phosphorylation-mediated inactivation of coactivator-associated arginine methyltransferase 1. Proc Natl Acad Sci U S A. 2007;104:12318–12323. doi: 10.1073/pnas.0610792104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu F, Zhao X, Perna F, Wang L, Koppikar P, Abdel-Wahab O, Harr MW, Levine RL, Xu H, Tefferi A, Deblasio A, Hatlen M, Menendez S, Nimer SD. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell. 2011;19:283–294. doi: 10.1016/j.ccr.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.