

Abstract
By using imaging spectrophotometry with paired images in the 200- to 280-nm wavelength range, we have directly mapped intracellular nucleic acid and protein distributions across a population of Chinese hamster ovary (CHO-K1) cells. A broadband 100× objective with a numerical aperture of 1.2NA (glycerin immersion) and a novel laser-induced-plasma point source generated high-contrast images with short (~100 ms) exposures and a lateral resolution nearing 200 nm that easily resolves internal organelles. In a population of 420 CHO-K1 cells and 477 nuclei, we found a G1 whole-cell nucleic acid peak at 26.6 pg, a nuclear-isolated total nucleic acid peak at 11.4 pg, and, as inferred by RNase treatment, a G1 total DNA mass of 7.4 pg. At the G1 peak we found a whole-cell protein mass of 95.6 pg, and a nuclear-isolated protein mass of 39.3 pg. An algorithm for protein quantification that senses peptide-bond (220-nm) absorbance was found to have a higher signal-to-noise ratio and to provide more reliable nucleic acid and protein determinations when compared to more classical 280-nm/260-nm algorithms when used for intracellular mass mapping. Using simultaneous imaging with common nuclear stains (Hoechst 33342, Syto-14, and Sytox Orange), we have compared staining patterns to deep-UV images of condensed chromatin and have confirmed bias of these common nuclear stains related to nuclear packaging. The approach allows absolute mass measurements with no special sample preparation or staining. It can be used in conjunction with normal fluorescence microscopy and with relatively modest modification of the microscope.
Keywords: deep ultraviolet, CHO cells, native contrast, Hoechst, nuclear stain
INTRODUCTION
Because of their size domain, intrinsic fragility, and heterogeneity it is surprisingly difficult to make absolute measurements on biological cells. Most of the characterization of cell heterogeneity is inferred indirectly through fluorescence with an unknown accuracy and only on accessible parameters. The values of mass during cell cycle, mass related to metabolism, content of intracellular compartments, distribution of nucleic acids, and even of genome size are often not well established. Deep-UV transmission imaging should be able to obtain fine intracellular structure tied directly to the fundamental optical properties of molecular composition (1). Self-calibrated images are generated via the native contrast encoded in spectral absorption. In principle, these methods should be universal and free of the unstable calibration. Biological imaging in this wavelength regime has antecedents dating from the 1950's (1 – 6), but becomes transformed by modern sources and detectors.
Although measurements using stains and extrinsic markers are the current standard, the absolute quantitative ability of fluorescence is only conveyed through rigorous external calibration to standards. Fluorescence-based methods for quantification of DNA and RNA are subject to sensitive chemical protocols (7 – 12). The choice and quality of dye is important. Attendant issues are access (membrane penetration and transport into histone-bound DNA), total binding site occupancy, stoichiometry, uniformity/stability of the light source, and photo-bleaching; all of which affect binding efficacy, excitation efficiency, and quantum yield of fluorescence (13 – 17). Buffer choice can also strongly influence the amplitude of the signal (18 – 19). Other approaches for intracellular quantification of nucleic acids include extractions for measurement from bulk heterogeneous samples. DNA and RNA can be measured from bulk solution with phosphate or nitrogen content or using UV spectroscopy (20 – 22). RNA can be measured using competitive or real-time RT-PCR methods (23). The situation for absolute determination of protein is nearly the same. Most standard methods use total extraction from a macroscopic sample, followed by reaction with a stain such as Coomassie Blue, to copper ions (Lowry reaction), or marking with dyes such as SYPRO (24). Extraction-based determination averages over the population, thereby making assumptions on the usually un-known distribution of heterogeneity, and is blind to the detailed phenotype. A great deal is gained by making measurements in specific phenotypes.
Apart from the assumption of averaged composition at the molecular level, UV absorption microscopy makes no such assumptions, does not rely on external standards, and provides cell-resolved cytometry measurements immune to errors from sample heterogeneity and unstable chemistry. For the most part, the underlying assumptions are identical to those applied in “gold standard” methods for quantifying protein and nucleic acids in bulk solution (3, 25).
In the work described below, we generated high-resolution mass maps of intracellular protein and nucleic acid content in fixed CHO-K1 cells by applying mathematical operations to transmitted-light images at wavelengths from 200–280 nm. Mass maps were generated for several-hundred cells. Whole-cell and nuclear-isolated mass were measured. We also compared nucleic acid mass to the DNA-specific label Hoechst 33342 and to the general nucleic acid stains Syto-14 and Sytox Orange, and studied the correlation of these markers to the total protein and nucleic acid across the distribution of cells. The mass maps resolved the sub-compartments of the cell, including the nucleus, nucleoli, and endoplasmic reticulum. It was therefore possible to correlate intracellular mass data at the detail of some of these sub-compartments. We were able to show stain-neutral images of the nucleic acid distribution, and to place absolute numbers on these distributions in femtograms of nucleic acid and protein to the precision of a single pixel.
MATERIAL AND METHODS
UV microscope
The UV microscope is a Zeiss Axioskop modified to accept a fiber optic light guide. The light source is a CW laser-induced plasma in high-pressure Xe gas (EQ-99, Energetiq Inc., Woburn MA, details below). A shutter (Uniblitz, Rochester, NY) and a motorized filter wheel (#FW102, ThorLabs, Newton, NJ) were installed before the solarization-resistant fiber (450 μm core, 0.22 NA, #QP450-1-XSR, Ocean Optics, Dunedin, FL).
Five UV bandpass filters were used, 280, 265, 232, 220, and 200, all with 10-nm bandpass (Omega Optical, Brattleboro, VT). The fiber output was directed onto a collimator, passed through a second filter wheel and was focused onto the specimen by a UV-Kond condenser (Carl Zeiss AG, Oberkochen, Germany) adjustable between 0.4 and 0.8 NA. Transmitted light was captured in a straight-through configuration by either a 0.4 NA, Ultrafluar 32× (Carl Zeiss AG) or a 1.25 NA, Ultrafluar 100× (Carl Zeiss AG) glycerin-immersion objective. A post-specimen filter was placed between the objective and the EMCCD camera (PhotonMax 512, Princeton Instruments, Trenton, NJ). The camera was UV-sensitive, with a constant 35% quantum efficiency from 200 to 350 nm, rising to over 95% between 475 and 700 nm. Images were acquired with WinView (Princeton Instruments).
Fluorescence imaging was also conducted in this trans-illumination configuration. Filters (Chroma Technology Corp., Rockingham, VT) were used for Hoechst (ex: 350; em: 450), Syto-14 (ex: 480; em: 510LP), and Sytox Orange (ex: 525; 600).
The imaging resolution was limited by the image magnification at the CCD camera (80-nm pixel at 100× and 278-nm pixel at 32×) combined with the <120-nm diffraction limit of the 100× (1.2-NA) objective. From Ronchi-ruling and bead tests (data not shown) we estimate the effective resolution to be ~200-nm. It should be possible to improve image resolution somewhat by increasing the optical magnification at the CCD.
Laser-induced plasma deep-UV source
A prototype deep-UV light source based on laser-induced plasma point source (EQ-99, Energetiq Inc., Woburn MA) was coupled using a two-paraboloid mirror imaging system to a solarization-resistant, fused-silica fiber (above). This source produces a broad continuum from 170–750 nm with a near constant radiance of 10 mW/mm2/nm/sr and a source size of 100×100×200 μm. The fiber-coupled output at 250-nm wavelength is approximately 2.5 decades higher than for a 50W deuterium lamp.
Cell Culture and sample preparation
CHO-K1 cells were grown in 25-ml flasks in complete HyClone Dulbecco's Modified Eagles Medium (#SH30285, Fisher Scientific, Pittsburgh, PA), with 10% fetal bovine serum (#SH30070.02HI, Fisher Scientific), 2 mM Glutamax (#35050-079, Invitrogen, San Diego, CA), and 100 I.U. penicillin/100 μg/ml streptomycin (#30-002-CI, Cellgro, Herndon, VA) at 37°C in 5% CO2, and were passed three times a week. For imaging experiments, cells were transferred to a fused-silica coverslip during passage and incubated overnight in complete medium. Cells were then washed with filtered (0.22 μm, #430758, Corning, Corning, NY) PBS and stained live with either Syto-14 (#S7576, Invitrogen) or Sytox Orange (#S11368, Invitrogen), fixed with 3.7% formaldehyde, and then stained with 1:1000 dilution of Hoechst 33342 (#S3570, Invitrogen). Coverslips were then washed with filtered PBS.
Determination of optical density, mass quantitation, cell segmentation and analysis
UV images were collected at all five UV bands for a given cell field, along with trans-illuminated fluorescence images for the same field. Background fields were obtained in cell-free region. Z-axis focus was registered with a MAC2000 motor (Ludl, Hawthorne, NY). All images were stored and processed off-line with MatLab (The Mathworks, Natick, MA) programs designed for batch processing of files.
Mass maps were generated as follows. Absorption and transmission based on each pixel (100×: 80 × 80 nm, 32×: 278 × 278 nm). Beer's law is given by
| (1) |
where transmission (I) through a specimen is given by the amount of UV light passing through a blank field that is attenuated by a specimen's attributes: (ε decadic molar extinction coefficient, L cm−1 mol−1), c (concentration, mol L−1), and 1 (path length, cm−1). The relationship between optical density and concentration is given by solving equation 1 to give
| (2) |
The leak term is a correction factor added to account for the imperfect block of visible light by the UV filters used in the study. Visible light accounted for, at most, 20% of the total light transmitted in a blank field. To generate an optical density (OD) image, a probe image and a leak image were captured for every field and later processed to calculate content maps.
To a good approximation, at wavelengths between 220 and 280 nm, the total absorption at a given UV band is the linear sum of the absorption of protein and nucleic acid components with negligible contribution from other cell components such as salts, fatty acids, esters and anhydrides (26). Thus,
| (3) |
This formula gives the OD at each pixel. We treated the concentration (c) and path length (l) product as a single unknown factor. With two unknown factors (one each for protein and nucleic acid concentrations), a system of equations was established using OD280 nm and OD260 nm, as previously described (1). This approach using two wavelengths (260 nm and 280 nm), produces two equations and two unknowns that is solved for the classical “gold-standard” determination of protein and nucleic acid in solution. We developed an alternative algorithm, which we term “220/260-nm”, that uses the Eqn. 3 couplet with the 220-nm and 260-nm image pair. At 260 and 280 nm, the protein-related absorption is strongly dominated by the aromatic residues tryptophan, tyrosine, and phenylalanine. At the 220 nm (and 200 nm), all amino acids contribute through absorption in the peptide bond. Furthermore, the relative contribution from nucleic acid is much-reduced, typically the to a few percent of the protein absorption at 220 nm.
Molar extinction coefficients for protein and nucleic acid for 260 and 280 nm were the same as in a previous study (1). For the 220-nm absorption coefficient, we used the recent method provided by Kuipers and Gruppen (27) combined with scaling from the classical measurements of Rosenheck and Doty (29) for random-coil averaged protein. The Kuipers and Gruppen model presents whole protein absorption, at 214 nm, as the summation of the absorption of individual amino acids and peptide bonds. An “average” protein was composed from the frequency of amino acid distribution in mammalian cells (30) over an “average” protein of length 443 residues (calculated using a mean protein molecular weight of 53 kDa and a mean peptide mass of 120 Da.) The protein epsilon (ε) value at 220 nm (432500 M−1 cm−1) was derived as a 50% reduction (29) from the ε value at 214-nm (27, 31). See Table 1 for ε-values used. However we accept the conclusion of Kuipers and Gruppen (27) and have calibrated for hyperchomic effects at 220 nm using an empirical method (see Results.)
Table 1.
Molar extinction coefficients by UV wavelength
| Wavelength (nm) | nucleic acid (M−1 cm−1) | protein (M−1 cm−1) |
|---|---|---|
| 280 | 3500# | 54129# |
| 260 | 7000# | 36057# |
| 220 | 4000#* | 432500*,**,346000## |
| 200 | 10500#* | 768000*,** |
Taken from (1), values for hybridized averaged nucleic acid.
Taken from (32)
calculated using formula from (27)
Table 1 provides the calculated value of the molar extinction coefficient for average protein in a random coil conformation.
The molar extinction coefficient at 220-nm was calibrated empirically; see Figure 3.
For conversion of concentration maps to mass maps, an area factor was used, as determined by the pixel resolution of the system with either the 32× (77284 nm2) or the 100× (6400 nm2) objective. The pixel resolutions were determined by use of a fused silica Ronchi ruling (Edmund Optics, Barrington, NJ) and beads (0.5, 1.1, and 15 μm diameter, Polysciences, Inc., Warrington, PA). Optical aberration and correction factors were also determined using beads and then applied to all images. Images were resized (using `imresize' function in MatLab) and then aligned with predetermined offsets before processing.
Analysis of images was performed in MatLab. For whole-cell content, cells were first outlined manually. Each region of interest (ROI) was then used to read out pixel values; mean mass per pixel, total mass, and cell size could all be determined in this way. Histograms and distribution fits were calculated in MatLab (function: `polyfit'). Two-dimensional plots of distributions and linear regression were performed in MatLab. For determination of nuclear-isolated mass, a custom-written automated image segmentation algorithm was used (morphological watershed), based on a mask generated by Hoechst 33342 fluorescence using a disk-shaped structuring element (radius = 3). Objects smaller or larger than 1 standard deviation of mean nucleus size were excluded from analysis. Remaining objects were converted into ROIs, allowing for automated determination of nuclear content and Hoechst fluorescence.
RESULTS
We began by first developing the image algorithms for extracting high-resolution mass maps from the deep-UV images. We tested the speculation that the better isolation of the nucleic acid and protein absorptions with the 220/260-nm image pair, combined with the stronger absolute protein absorption at the shorter wavelength, would lead to a more reliable measurement when compared to the classical 260/280-nm method. We then did a series of population-level comparisons on CHO-K1 and cross-correlated mass measurements for nucleic acid and protein. After this we describe measurements at progressively higher resolution and compare the UV mass maps to imaging with common nuclear stains.
Comparison of UV-derived cell images
The primary data generated by mass mapping in the deep UV is illustrated in Figures 1 and 2. The OD values range between 0 and 1, within the nearly linear portion of Beer's Law. The pixel values are integrations in the Z direction calibrated in absolute units of femtogram (fg) per X,Y pixel and derived by the specific algorithms discussed more below. Condensed DNA is clearly visible as high density for 235, 260, and 280 nm UV bands (Fig. 1A–C), but appears in reverse contrast for the 220- and 200-nm images (Fig. 1D–E) where the nucleic acids are nearly transparent. The 220-nm and 200-nm images represent largely protein (27 – 28, 33) and, with appropriate normalization, these shortest wavelength images closely approximate the protein mass map (Fig. D, E, and G). A large amount of fine structure is visible under 100× magnification, particularly in the nucleic acid map (Fig. 2B), some of which is not apparent with either the RNA selective nucleic acid stain Syto-14 (Fig. 2A) or the widely-used DNA stain Hoechst 33342 (see below). A few salient observations are:
-
(i)
the total nucleic acid content in the cytoplasm is nearly as high as it is in the nucleus (e.g., Fig. 2B),
-
(ii)
there is a region of low protein density surrounding the nucleolus (Fig. 1D, 1G and Fig. 2C).
-
(iii)
there is a large protein concentration present within the nucleolus, and, in particular, the nucleolus and a region near the nuclear membrane are also seen as dense UV-absorbent structures (UV-opaque at 200 and 220 nm. Fig. 1),
-
(iv)
Syto-14 staining (e.g., Fig. 2A) shows dense punctuate structures in the perinuclear region, indicating localized, elevated RNA. However these same areas correspond to regions with low (or background) total nucleic acid in the UV mass map.
Figure 1. UV absorption images and mass maps.

UV images at (A) 280 nm, (B) 260 nm, (C) 235 nm, (D) 220 nm, and (E) 200 nm. Pixel values are expressed as optical density (OD) values, calculated as the logarithm of the ratio of the background and cell images. Images of the calculated nucleic acid (F) and protein mass (G), based on 260-nm and 280-nm UV images. The mass maps (F,G) are normalized to a maximum of 1.1×10−14 and 2.0×10−13 pg per 0.077-μm2 pixel (140 fg and 2.6 pg per square micrometer peak nucleic acid and protein density, respectively). NA: nucleic acid.
Figure 2. High-resolution, images of CHO-K1 cells using a 100×, 1.2 NA objective.
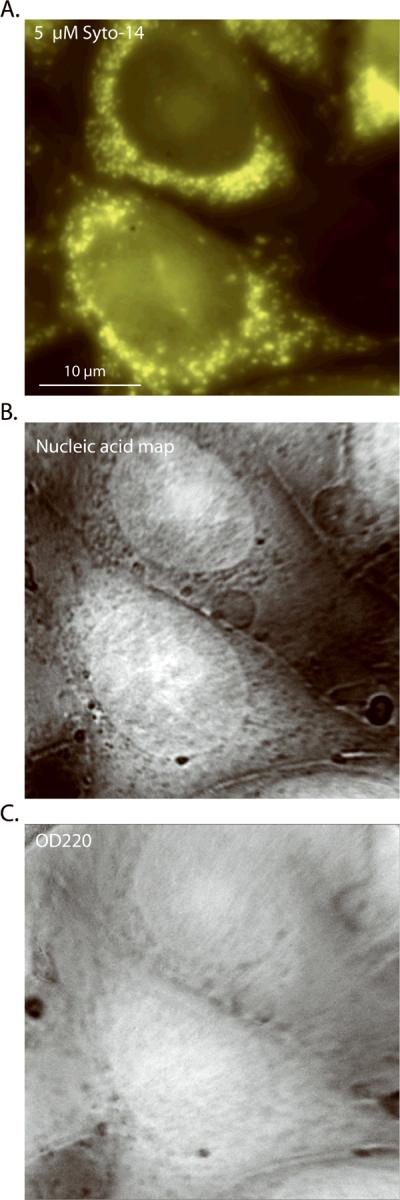
(A) 5 μM Syto-14 stain, (B) nucleic acid mass map deduced from the 260-nm and 280-nm images, (C) OD220 nm image, which closely approximates the protein mass map (see text).
Histogram population-level comparisons of whole-cell mass
To compare UV mass mapping to flow cytometry, and to assess different methods of data reduction, we collected several hundred images in all five UV bands then reduced them using the various mass algorithms to produce histogram distributions of whole-cell mass. We derived nucleic acid and protein maps using the 260/280 nm and 220/260-nm algorithms (See Methods). We took a series of full-field images using the 32× objective, segmented these fields into cells (n = 420 cells, manual segmentation) and measured the mass values within each whole-cell perimeter. The distributions of total masses are presented in Fig. 3A–D. The histograms were fitted with first-degree polynomial functions (black lines).
Figure 3. Distribution and comparison of whole-cell nucleic acid and protein mass calculated by deep-UV algorithms.

Histograms (n = 420 cells, bins: 50) of nucleic acid (A) and protein mass (B) calculated using 260/280-nm and nucleic acid (C) and protein (D) by 220/260-nm algorithms. The same manually defined radius of interest (ROI) was used, for each cell, for all histograms. Black lines are the fitted distributions for the histogram (single-degree polynomial with `polyfit' function in MatLab.) Gray lines (C and D) denote the distribution of protein mass calculated using an ε-220 value derived using the work of Kuipers and Gruppen (28) and Rosenheck and Doty (29), without hypochromic correction (see text). Black lines (C and D) show distribution calculated using an ε-220 value corrected for the hypochromic shift (see text). (E) Two-dimensional scatter plot showing the distribution of OD280 and OD260 across all cells. The lower line (black) denotes the OD combinations that produce zero protein mass; OD combinations below this line result in non-physiological, “negative” protein mass (circles). Note the proximity of OD values for the 260/280-nm algorithm to the zero-mass line for protein by the 260/280-nm algorithm, as is consistent with the negative and near-zero values observed in Fig. 3B. The upper line (E, gray) denotes the OD combinations that would yield negative nucleic acid mass. (F) A similar scatter plot and noise analysis for the 220/260-nm algorithm. Note that OD values in Fig. 3E fall within a range halfway between the two zero-mass lines. Therefore the 220/260-nm calculation is optimal for maximizing signal-to-noise ratio for both nucleic acid and protein mass. (G) The 220/260-nm calculation shows nucleic acid and protein as correlated (R=0.79, cells = 420). (H) However, whole-cell protein mass calculated by 260/280-nm shows little correlation to the nucleic acid mass (R = 0.149). The different result is attributed to the higher noise of the 260/280-nm algorithm, which hides the real relationship (see text). Regression fit was implemented using least squares method in MatLab.
While the qualitative features of the histograms are quite similar to cytometry data for both algorithms (20 – 21, 34), some problems in terms of statistical broadening and absolute calibration appear for the classical 260/280-nm data reduction. For the nucleic acid by this classical algorithm, there is a well-shaped histogram (Fig. 3A) but the G1 peak is at 14.0 pg (μ = 17.4 pg, σ= 6.9 pg, CV = 0.40) for total nucleic acid (DNA and RNA). As confirmed with the RNase treatments (data below), the entire nucleic acid histogram is shifted to unphysiologically low values. This problem is more extreme for protein distribution, which is broadened to the point that the 260/280-nm algorithm calculates negative protein mass for nearly 23% (93) of the cells (Fig. 3B). Neither of these artifacts occurs with the 220/260-nm algorithm. For nucleic acid (Fig. 3C) the G1 peak lies at 26.6 pg (μ = 33.3 pg, σ= 13.3 pg, CV = 0.40), with a G2 shoulder at nearly twice the G1 mass (Fig. 3C, arrow). While the optical properties of intracellular protein at 220-nm remain under-characterized, our two algorithms agree quite well without correction on the location of the protein peak value (i.e. most probably G1 mass). The 260/280-nm algorithm gives a peak of 91.7 pg (Fig. 3B; μ = 72.4 pg, σ = 108.1 pg, CV = 1.49). Using the 220/260-nm couplet and the most straightforward method of estimating ε-220 (27, 29), and before accounting for a hypochromic shift, the 220/260-nm algorithm places the G1 protein peak at 76.2 pg (Fig. 3D, gray line), within 17% of the classical 260/280-nm data reduction.
To add the hypochromic correction, we accepted the recent analysis of Kuipers and Gruppen (27) and determined ε-220 empirically from ensemble averaged 280-nm protein absorption. We calculated ε-220 by iterating the 220-nm/260-nm equation couplet using the data extracted from the 25–75 percentile of the 260-nm/280-nm protein histogram. By matching the means of these reduced data sets, we optimized for an ε-220 value of 346000 M−1 cm−1. We adopt this value for the effective extinction coefficient of intracellular protein in CHO cells.
Using this value, we then recalibrated the protein distribution (Fig. 3D, black line), and place the best value for the G1 peak at 95.6 pg (n = 420 cells, μ = 116.2 pg, σ = 53.3 pg, CV = 0.46). It is important to point out that the nucleic acid histograms are insensitive; by correcting the protein histogram with a hypochromic shift the nucleic acid histograms move by less than 1% (Fig. 3C, gray dashed line).
We next investigated the source of the error for the 260/280 nm for total nucleic acid at low OD values. To explore this failure, we examined the noise sensitivity of each of our algorithms at the OD values typical of the experiment (Fig. 3E). The zero-mass boundary lines were calculated by determining the mass values using different combinations of OD values, ranging from 0 to 1.0 and regressing across points with zero and near-zero values. The OD combinations falling below the zero-mass protein line will generate negative values and, in our sample, 92 cells (circled points) fall in this region. In contrast, the same OD values generate all positive nucleic acid masses with the 260/280-nm algorithm, with the cells distributed far from the nucleic acid zero line. A ratio of the standard deviation to the average distance from the zero-mass line is a measure of the signal-to-noise ratio of each algorithm. This analysis explains the greater distortion of protein histogram in comparison to nucleic acid by the classical 260/280-nm algorithm in our application. Based on this analysis we chose the 220/260-nm algorithm as more reliable, particularly for single-cell (un-averaged) measurements, and adopted it for the absolute calibration of the results discussed below.
Although images at 200 nm were also taken, due to absorption in the optical system the exposure times needed were 10–30 times greater than the 220-nm wavelength. These images also exhibited significant out-of-band leakage through the 200-nm bandpass filter (exhibited by apparent OD values in a limited dynamic range between 0.005 and 0.1, peak at 0.05); they were, therefore, deemed unreliable for absolute quantification of protein without optics improvements.
Cell-by-cell correlation of whole-cell nucleic acid to whole-cell protein
Next we tested whether UV-based mass measurements reproduce correlated increases in nucleic acid and protein depending on cell cycle, as has been inferred from previous flow cytometry measurements using stains (35). With the 220/260-nm algorithm we reproduced a clear linear monotonic relationship (R = 0.79, slope=3.18) suggesting that protein mass tends to be approximately three times that of the nucleic acid (Fig. 3G). In contrast, the systematic relationship between nucleic acid and protein is nearly hidden by the high noise of the 260/280-nm algorithm; we observed a very weak relationship (R = 0.15) using the classical data reduction (Fig. 3H).
Mass in the nuclear compartment: Nuclear protein and nuclear nucleic acid content
We then analyzed the mass in the nuclear compartment by using Hoechst-stained cells and automated segmentation. A mask was generated from the Hoechst image using standard fill and dilate operations and this was used to define the nuclear area. Total nucleic acid, protein and integrated Hoechst brightness were then measured for each nucleus (n = 477 nuclei, Figure 4A–D).
Figure 4. The nuclear compartment during cell cycle - distribution and comparison of total nucleic acid, protein, and Hoechst 33342 signal.

Values in the nuclear compartment: (A) Nucleic-acid and (B) protein mass by 220/260-nm are shown for 477 nuclei (automated watershed segmentation based on Hoechst 33342 fluorescence). (C) Integrated Hoechst signal derived across the same CHO-K1 cell population. The panel (D) shows the correlation (R = 0.87) of Hoechst fluorescence and nucleic acid mass (regression calculated in MatLab) across all nuclei. (E) Distribution of nucleic acid mass in nuclei following DNA isolation by treatment with RNase (see text). All histograms were binned to 100 bins and fitted to single-degree polynomial (black line, `polyfit' function in MatLab). (F) Images of 4 different cells in various stages of mitosis, identified by Hoechst fluorescence (top row). Pseudo-colored mass maps of the same cells are shown below. Each mass map is scaled to its own maximum. Whole-cell nucleic acid and protein masses are shown for the ROI (white) indicated in the image.
We used the 220/260-nm algorithm (ε-220 protein: 346000 M−1 cm−1) for the analysis exclusively. The G1 peak for CHO-K1 (Fig. 4A) falls at 11.38 pg nucleic acid (RNA plus DNA in the nucleus, μ= 16.5 pg, σ = 9.2 pg, CV = 0.55). For total protein in the nuclear compartment, the G1 peak (Fig. 4B) lies at 39.3 pg (μ = 51.3 pg, σ = 28.3 pg, CV = 0.55).
We then looked for systematic differences within the nucleus between UV-derived mass and the nuclear stain. The Hoechst fluorescence in our microscope exhibited a classical histogram (Fig. 4C) (15, 35 – 36). Plotting this fluorescence versus UV-derived nucleic acid mass for each nucleus revealed a linear relationship with high correlation (R = 0.87) (Fig. 4D). This linear relationship persisted despite significant known differences in the two measurements. Packaging is known to strongly affect Hoechst staining; a factor of 88% increase in staining brightness has been observed under some conditions for Hoechst 33342 (and greater increases for many other nuclear stains) when nuclear proteins are extracted with HCl (15). In particular, there is negligible staining of the nucleolus and RNA by Hoechst (13 – 17, 35 – 36), while the nucleolus is strongly represented in the UV-derived nucleic acid mass map. However, despite these differences, the nearly linear relationship of total nucleic acid to Hoechst staining is confirmed to a reasonable accuracy in our experiments.
Despite much effort over a period of several decades, quantifying genome size remains an area of active interest (7, 19, 31 – 38). The UV mass mapping method has the ability to separate nucleic acid and protein contribution, although it cannot, by itself, distinguish between RNA and DNA. We therefore attempted to isolate the DNA component. We treated a population of CHO-K1 cells with DNase-free RNase (25 μg / ml) after fixation with 4% formaldehyde and permeabilization with methanol (Fig. 4E). In these treated nuclei, we observed a (presumptive) DNA-only, G1 peak mass of 7.35 pg (n = 403 nuclei, μ = 10.0 pg, σ = 4.8 pg, CV = 0.48). Furthermore, as expected, the RNase treatment affected nucleic acids only; the protein histogram is unchanged with a G1 peak value at 42.0 pg (μ = 53.6 pg, σ = 28.5 pg, CV = 0.53), matching the untreated cells (Fig. 4B).
As a cross check, we also took data by the 260/280-nm method for the same RNase-treated cells. We found consistently low values (most probable DNA mass = 3.8 pg, μ = 5.5 pg, σ = 3.8 pg, CV = 0.69), that is, a G1 peak even below the known mass of the hamster genome. The protein determination repeated the whole-cell results, with a large variance (μ = 25.2 pg, σ = 184.6 pg, CV = 7.3). This corroborates our earlier analysis and confirms our preference for using the 220/260-nm algorithm to avoid noise related to low optical densities for 260/280-nm.
After the RNase treatment the total nucleic acids may include oligonucleotides as well as genomic DNA, however the haploid number from our measurement for CHO-K1 after RNase, i.e., half the total remaining nucleic acids is 3.67 pg. This should be compared to three existing literature C-values by Feulgen densitometry namely; 3.10 pg (39), 3.20 pg (40) and, 3.9 pg (41). Our approach handles the main issues of genome size measurement. It provides considerable population averaging, handles the issue of cell cycle effectively, isolates DNA content, and uses an established computation for determining nucleic acid. Some further testing by enzymatic methods would be needed to confirm the absolute calibration, especially given the known hyperchromic shift in UV absorption by nucleic acids upon denaturing.
Mitotic Cells
Of the 420 cells in the full data set, we isolated 28 cells with circular morphology and either dense Hoechst labeling or with apposed Hoechst clusters indicating immediately post-mitotic pairs. We sorted these according to the phase of mitosis and measured the level of Hoechst fluorescence, protein mass, and nucleic acid mass (Fig. 4F). Hoechst image and nucleic acid mass maps are presented with the same coloring scheme to facilitate comparison. Much of the fine structure is duplicated between the nucleic acid mass map (second panel) and the Hoechst image. The total nucleic acid mass from the 28 mitotic cells averages 38.4 pg (σ = 12.2 pg, CV = 0.32), which is 44% larger than the mass at the G1 peak for the full distribution of cell cycle (420 cells). The mean protein mass (whole cell) in these 28 cells is 102.6 pg (σ = 46.4, CV = 0.45), only 7% higher than that of the corresponding G1 peak for the full 420-cell distribution (Fig. 3B). Both protein and nucleic acid mass in these near-mitotic cells also have smaller CV, and hence less variability in whole cell mass compared to non-mitotic cells (see Figure 3 text).
Population analysis according to image information
The potential of UV-derived image information for population analysis is illustrated in Figure 5. A single field of cells at 32× magnification is shown by nucleic-acid mass map and Hoechst channel (Fig. 5 A and B). The table (Fig. 5C) lists nucleic acid mass (whole-cell, nucleus, and nucleolus), protein mass (whole-cell, nucleus, and nucleolus) and integrated Hoechst fluorescence values for the same field. Many of the mass readings in the table cross-correlate with expectations from the cell images at identifiable stages of cell cycle. The first data column in the table also lists the apparent cell “size” (i.e., two-dimensional image area) as it would be observed in a conventional microscope image. This can be compared to the actual mass of the cell (dry mass, i.e., summed columns 2 and 3).
Figure 5. Mass distributions in the whole cell, nucleus and the nucleolus.

(A) nucleic-acid mass map and (B) Hoechst channel for a representative field of CHO–K1 cells imaged at 32× magnification. (C) A table listing nucleic acid mass, protein mass and fluorescence values. Keys 1–18 correspond to labels on the images. Mass values are reported for the whole cell (all compartments), nucleus (with nucleolus), and nucleolus, as determined from the Hoechst image. (D) and (E) show scatter plots that relate cell size (2-dimensional projection) to total protein and total nucleic acid content. Although mass generally increases as a function of size, there can be more than a 2× difference in protein mass for cells of a given size.
We examined this more directly by examining how strongly 2-dimensional size correlated with cell mass across the full population (Fig. 5D–E). While there is a strong relationship (R = 0.82), the dispersion between “size” and actual protein mass increases for larger cells. There can be more than a 2× difference between apparent size and mass even near the median. Averaged over the field of Figure 5 (all stages of cell cycle), the nucleic acid density (fg/μm2) is increased by 20% in the nucleolus relative to the averaged whole nucleus; the protein density (fg/μm2) is increased by 11% in the nucleolus relative to the whole nucleus. The size to mass relationship is weaker for cells near mitosis (Fig. 5E. See line of cells with pixel size below 2500 but with nucleic acid spanning 25 – 50 pg). Much as for image cytometry with a fluorescent stains (13, 42), it is clear that image information from mass distributions in the sub-compartments of the cell may be useful in distinguishing finer metabolic states and stages of replication.
Spatial correlation of nuclear stains with UV-derived mass measurements
The UV mass mapping technique allows the imaging of intracellular structure without bias and to a resolution of approximately 200 nm (see Methods/Discussion). The UV-derived whole-cell total protein and whole-cell total nucleic acid measurements were tested for correlation with the integrated SO and Hoechst fluorescence (Fig. 6, n=260 cells).
Figure 6. Microstructural comparisons reveal differences between mass map and nuclear stains.

(A) Moderate correlation (R=0.69, n = 260 cells) is seen to the integrated Hoechst signal, (B) weaker correlation (R=0.46) to Sytox Orange (SytoxOr) and (C) (as expected) poor correlation (R=0.33) between Hoechst and Sytox Orange channels were observed. (D) Global correlation of mass map to stains. The same cell is imaged at 100× magnification with Sytox Orange (i, 1 μM) and Hoechst 33342 (ii, 16.2 μM). Panel (iii) shows UV-derived nucleic-acid mass and panel (iv) as an equally-weighted linear sum of the normalized images from Hoechst and Sytox Orange. The summed image in (iv) shows higher global correlation to the nucleic acid mass map (also see Fig. 9). Arrows denote the comparisons and the associated Pearson's correlation score (`normxcorr2' function in MatLab) that are made between the panels within (D). Insets show a frequency distribution of pixel values for the nucleic acid and the fluorescence images. SytoxOr staining was performed on formaldehyde fixed cells without permeabilization or addition of RNase. A summed Hoechst+SytoxOr image (E) and nucleic acid mass map (F) of a mitotic cell. Line plots of the same pixels taken from the fluorescence images (G; blue: Hoechst, magenta: SytoxOr) and from the nucleic acid mass map and the equally weighted, summed fluorescence image (H; gray: summed image, black: nucleic acid). Line plots taken from the yellow line in A and B. It appears that the two nuclear stains cannot be summed in any combination so as to reproduce the contours of the nucleic acid mass map.
Hoechst 33342 fluorescence exhibited the strongest linear correlation to whole cell nucleic acid mass (Fig. 6A, R = 0.69). Not surprisingly, this relationship is weaker than when nuclear-isolated nucleic acid is compared to Hoechst (Fig. 4D, R = 0.87). SO and Hoechst staining are not strongly correlated (R = 0.33) to each other when summed over the whole cell (Fig. 6C). This is consistent with SO showing differential staining efficiency to different nucleic acids.
To explore the detailed overlay of nucleic acid stains and the nucleic acid mass map, we identified a series of mitotic cells (Fig. 6D). We then compared the nucleic acid maps to a Hoechst image, a SO image, and image of an evenly weighted linear summation of (normalized) Hoechst and SO (n = 10 cells, all maps normalized to maximum). Neither Hoechst nor SO alone resulted in an image that was entirely similar to the nucleic acid map. The insets contain scatterplots of the pixel intensity between the image pairs (scaled from purple to red to indicate number of coincident pixels, Fig. 6D). The SO inset shows that low fluorescence values co-vary with mass, but at higher mass values the distribution flattens. The opposite is true for the Hoechst scatterplot, which shows agreement for high fluorescence and map values. The summed image (Hoechst + SO, Fig. 6E) creates a better (approaching linear) representation of the total nucleic acid (Fig. 6D and 6F). However the detailed line plots suggest that no linear combination of these two nuclear stains (Fig. 6G) will actually represent the total nucleic acid (Fig. 6H). That is, it is not possible to recover the full nucleic acid map with an arbitrary normalized summation of the Hoechst and SO images. The nucleic acid maps capture detail that is not revealed by either stain. Further examples can be seen in Fig. 2.
DISCUSSION
We have compared population-level UV mass mapping to analogous cytometry data and found good qualitative agreement in the shape of histogram distributions. However the UV-derived measurement provides an absolute calibration to mass. The distributions exhibited a primary G1 peak at a total nucleic acid (DNA and RNA) value of ~26.6 pg for CHO-K1 whole cells, with a little less than half of this mass (11.4 pg) in the nucleus. To isolate the DNA component we measured RNase treated cells, and determined a G1 DNA mass of 7.4 pg. These results are consistent with the published numbers for the genomic DNA and the RNA/DNA ratio measured in similar CHO cells using flow cytometry and Feulgen densitometry (16, 21 – 23, 31, 42 – 43).
In the whole-cell measurements the total nucleic acid by UV did not correlate well with the integrated brightness of the generic nucleic acid dye (SO). We saw a tighter correlation between total nucleic acid and Hoechst staining, particularly for nuclear-isolated nucleic acid. This is consistent with analysis of signal in a flow cytometer, which shows a wider distribution of signal intensities with a general nucleic acid stain, than with a DNA specific stain like Hoechst (11). However a closer examination also shows some marked differences between the UV-derived mass and Hoechst, particularly in staining of the nucleolus and other fine structure. Since these different images of chromatin can be obtained simultaneously, they should be useful to gain a better understanding of nucleic acid structures.
In the high-resolution images, it is interesting to see the large component of protein even in condensed chromatin. Figure 1 shows a mitotic cell with condensed DNA that is strongly visible as high density in the nucleic acid map. This region of condensed nucleic acid structure shows up only with low contrast (~11% OD change) in the 220/200nm images or in the protein maps, which reflects the fact that the total protein in condensed chromosomes during mitosis can be nearly as high as the average value in the cytoplasm. Similarly the nucleic acid maps for the mitotic cells in Fig. 4F look quite different from the Hoechst images of the same cells.
After some experimentation we identified an algorithm consisting of 220/260-nm to be superior for calculating nucleic acid and protein mass at single-cell optical density. With a novel noise analysis method (Fig. 3E–F) and empirically, we found that the classical method of measuring protein in solution (260/280-nm algorithm) is noisy. Furthermore the nucleic-acid contribution to extinction is small at 220-nm, so that there is only a few-percent difference between the UV transmission image and the protein mass maps at this wavelength. This makes the nucleic acid mass maps insensitive to the fine-tuning of the protein calibration (e.g., hypochromic shift) discussed below.
While there is a lower noise component in the 220-nm-derived protein mass map, there is a potential need to make a correction for the hypochromic effect on proteins in physiological conditions (16, 44 – 50). At both 280-nm and 220-nm wavelengths there is some sensitivity of the effective value of ε depending on folding pattern and pH (especially related to solvent access to tryptophan residues and via pH sensitivity of the protonation of carboxyl groups). These shifts can be up to ten times larger at the 220-nm wavelength. Furthermore the literature values for ε-220 nm, even without corrections, are somewhat unsettled. Kuipers and Gruppen (27) have recently revisited the field for solution measurements and have made the case for quantitation of protein, with appropriate controls, at the shorter wavelengths (220-230nm). When averaging protein content across large cell samples, we found agreement between the 220/260-nm and 260/280-nm algorithms (within 17%) without applying a hypochromic shift specific to intracellular protein in CHO-K1. However we have accepted the conclusions of Kuipers and Gruppen (27) that absolute protein determinations should reference the 280-nm protein absorption and, based on our highly averaged results, determined a value for protein in CHO-K1 of ε-220 = 346000 M−1 cm−1. It would be valuable to verify this number with other techniques that retain the folding structure and pH of intracellular mammalian protein.
In principle, the protein determinations by 220-nm absorption provide new information through their generic spectral signature. At 220 nm both peptides and amino acid residues contribute to protein absorption, with peptide absorption accounting for nearly the same fractional absorption as the aromatic contribution. This makes the 220/260-nm algorithm less sensitive to the trytophan/tyrosine/phenylalanine percentage concentration, but more sensitive to solvent exposure, pH and α-helical content (32, 29). The subtraction of protein mass maps generated by the two algorithms (or maps at additional wavelengths) could be used to see differences in aromatic content, pH, or α-helical content in the internal compartments of the cell. In a previous publication (1) we also collected native protein fluorescence images and estimated fluorescence quantum yield as a potential indication of aromatic content of intracellular protein. Therefore comparison of any of these images of intracellular protein may be capable of generating more specific information on intracellular protein distribution. This may be particularly useful in the study of protein aggregation diseases such as Huntington's, Alzheimer's and Parkinson's diseases.
While our primary interest in this study has been absolute measurements in fixed cells, a natural question is whether the technology can be applied to live cells. Deep-UV is well known to be more photoactive than visible light, particularly in the wavelength range of pyrimidine absorption - where the most frequent mutagenic photoproduct is cis-syn cyclobutane pyrimidine dimers (51). However the common DNA stains that are now used in live-cell imaging, (e.g., Hoechst 33342), have a well-documented cytotoxicity (52 – 53), as well as staining biases and uneven distribution from efflux pumping (13 – 17). Our previous study (1), published time-lapse movies of mammalian cells showing normal motile responses and mitosis while cells were undergoing multiple exposures in our UV apparatus even at the most actinic 260-nm and 280-nm wavelengths. Pyrimidine absorption at 220-nm is reduced by a decade from its peak at 260 nm.
The speed and resolution that can be achieved in deep-UV mass mapping is notable. It is possible to generate population-level data with a few exposures of tens of milliseconds duration. In our system a further optimization of resolution to near 110 nm should be possible even before super-resolution techniques are considered. Some of the most important target applications may be; (i) direct generation of biological standards including gemome size, (ii) multiparameteric analysis of cell-cycle and metabolic subpopulations at the level of the internal compartments of the cell and, (iii) high-resolution studies of chromatin. It would be interesting to expand comparative imaging between the deep-UV mass maps and nucleic acid stains, and to include more specific DNA reporters.
ACKNOWLEDGMENT
This work was supported, in part, by the National Institutes of Health under grants RR-028147 and HG-001389. We thank William Holber, Jason Silterra, and Steven Horne of Energetic Inc. for stimulating discussions and for contributing the laser-plasma source.
Research Support: This work was supported, in part, by the National Institutes of Health under grants RR-028147 and HG-001389.
REFERENCES
- 1.Zeskind BJ, Jordan CD, Timp W, Trapani L, Waller G, Horodincu V, Ehrlich DJ, Matsudaira P. Nucleic acid and protein mass mapping by live-cell deep-ultraviolet microscopy. Nat Methods. 2007;4:567–569. doi: 10.1038/nmeth1053. [DOI] [PubMed] [Google Scholar]
- 2.Gallagher SR. Quantitation of DNA and RNA with absorption and fluorescence spectroscopy. In: Ausubel FM, editor. Current Protocols in Molecular Biology. John Wiley & Sons Inc.; New York: 2011. pp. A3D.1–A3D.14. [DOI] [PubMed] [Google Scholar]
- 3.Alupoaci CE, Garcia-Rubio LH. Growth behavior of microorganisms using UV-VIS spectroscopy: escherichia coli. Biotechnol Bioeng. 2004;86:163–167. doi: 10.1002/bit.20001. [DOI] [PubMed] [Google Scholar]
- 4.Lang-Pauluzzi I. The behaviour of the plasma membrane during plasmolysis: a study by UV microscopy. J Microsc. 2000;198:188–198. doi: 10.1046/j.1365-2818.2000.00677.x. [DOI] [PubMed] [Google Scholar]
- 5.Hatfield JM, Schulze L, Ernst D. Measurement of the ultraviolet absorption in specific parts of both living and fixed mammalian cells, using a specially designed microspectrophotometer. Exp Cell Res. 1970;59:484–486. doi: 10.1016/0014-4827(70)90659-2. [DOI] [PubMed] [Google Scholar]
- 6.Davies HG. Ultra-violet microspectrography of living tissue culture cells. Part I. Radiation measurements. Discuss Faraday Soc. 1950;9:442–449. [Google Scholar]
- 7.Doležel J, Greilhuber J. Nuclear genome size: Are we getting closer? Cytometry A. 2010;77A:635–642. doi: 10.1002/cyto.a.20915. [DOI] [PubMed] [Google Scholar]
- 8.Tiersch TR, Chandler RW, Wachtel SS, Elias S. Reference standards for flow cytometry and application in comparative studies of nuclear DNA content. Cytometry. 1989;10:706–710. doi: 10.1002/cyto.990100606. [DOI] [PubMed] [Google Scholar]
- 9.Rasch EM. Feulgen-DNA cytophotometry for estimating C values. Meth Mol Biol. 2004;247:163–201. doi: 10.1385/1-59259-665-7:163. [DOI] [PubMed] [Google Scholar]
- 10.Holden MJ, Haynes RJ, Rabb SA, Satija N, Yang K, Blasic JR. Factors affecting quantitation of total DNA by UV spectroscopy and PicoGreen fluorescence. J Agric Food Chem. 2009;57:7221–7226. doi: 10.1021/jf901165h. [DOI] [PubMed] [Google Scholar]
- 11.Steck K, El-Naggar A. Simultaneous DNA/RNA analysis of solid and hematoreticular malignancies. Meth Mol Biol. 1998;91:167–179. doi: 10.1385/0-89603-354-6:167. [DOI] [PubMed] [Google Scholar]
- 12.Crissman HA, Steinkamp JA. Rapid, simultaneous measurement of DNA, protein, and cell volume in single cells from large mammalian cell populations. J Cell Biol. 1973;59:766–771. doi: 10.1083/jcb.59.3.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santistenban MS, Montmasson MP, Giroud F, Ronot X, Brugal G. Fluorescence image cytometry of nuclear DNA content versus chromatin pattern: a comparative study of ten fluorochromes. J Histochem Cytochem. 1992;40:1789–1797. doi: 10.1177/40.11.1431064. [DOI] [PubMed] [Google Scholar]
- 14.Krishan A. Effect of drug efflux blockers on vital staining of cellular DNA with Hoechst 33342. Cytometry. 1987;8:642–645. doi: 10.1002/cyto.990080618. [DOI] [PubMed] [Google Scholar]
- 15.Darzynkiewicz Z, Traganos F, Kapuscinski J, Staiano-Coico L, Melamed MR. Accessibility of DNA in situ to various fluorochromes: relationship to chromatin changes during erythroid differentiation of friend leukemia cells. Cytometry. 1984;5:355–363. doi: 10.1002/cyto.990050411. [DOI] [PubMed] [Google Scholar]
- 16.Martin RM, Leonhardt H, Cardoso MC. DNA labeling in living cells. Cytometry A. 2005;67A:45–52. doi: 10.1002/cyto.a.20172. [DOI] [PubMed] [Google Scholar]
- 17.Mascetti G, Carrara S, Vergani L. Relationship between chromatin compactness and dye uptake for in situ chromatin stained with DAPI. Cytometry. 2001;44:113–119. doi: 10.1002/1097-0320(20010601)44:2<113::aid-cyto1089>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 18.Loureiro J, Rodriguez E, Doležel J, Santos C. Comparison of four nuclear isolation buffers for plant DNA flow cytometry. Ann Bot London. 2006;98:3679–3689. doi: 10.1093/aob/mcl141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bainard JK, Fazekas AJ, Newmaster SG. Methodology significantly affects genome size estimates: quantitative evidence using bryophytes. Cytometry A. 2010;77A:725–732. doi: 10.1002/cyto.a.20902. [DOI] [PubMed] [Google Scholar]
- 20.Klein E, Klein G. Nucleic acid content of tumour cells. Nature. 1950;166:832–833. doi: 10.1038/166832a0. [DOI] [PubMed] [Google Scholar]
- 21.Duck-Chong C, Pollak JK, North RJ. The relation between intracellular ribonucleic acid distribution and amino acid incorporation in the liver of the developing chick embryo. J Cell Biol. 1964;20:25–35. doi: 10.1083/jcb.20.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauer KD, Dethlefsen LA. Total cellular RNA content: correlation between flow cytometry and ultraviolet spectroscopy. J Histochem Cytochem. 1980;28:493–498. doi: 10.1177/28.6.6156196. [DOI] [PubMed] [Google Scholar]
- 23.DiSanto ME, Stein R, Chang S, Hypolite JA, Zheng Y, Zderic S, Wein AJ, Chacko S. Alteration in expression of myosin isoforms in detrusor smooth muscle following bladder outlet obstruction. Am J Physiol Cell Physiol. 2003;285:1397–1410. doi: 10.1152/ajpcell.00513.2002. [DOI] [PubMed] [Google Scholar]
- 24.Zubkov MV, Fuchs BM, Eilers H, Burkill PH, Amann R. Determination of total protein content of bacterial cells by SYPRO staining and flow cytometry. Appl Environ Microbiol. 1999;65:3251–3257. doi: 10.1128/aem.65.7.3251-3257.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aitken A, Learmonth M. Part I. Protein determination by UV absorption. In: Walker J, editor. The Protein Protocols Handbook. Humana Press Inc.; Totowa, NJ: 1996. pp. 3–6. [Google Scholar]
- 26.Platt JR, Klevens HB. Absolute absorption intensities of alkylbenzenes in the 2250 – 1700 Å region. Chem Rev. 1947;41:301–10. doi: 10.1021/cr60129a009. [DOI] [PubMed] [Google Scholar]
- 27.Kuipers BJ, Gruppen H. Prediction of molar extinction coefficients of proteins and peptides using UV absorption of the constituent amino acids at 214 nm to enable quantitative reverse phase high-performance liquid chromatography-mass spectrometry analysis. J Agric Food Chem. 2007;55:5445–5451. doi: 10.1021/jf070337l. [DOI] [PubMed] [Google Scholar]
- 28.Saidel J, Goldfarb AR, Waldman S. The absorption spectra of amino acids in the region two hundred to two hundred and thirty millimicrons. J Biol Chem. 1952;197:285–291. [PubMed] [Google Scholar]
- 29.Rosenheck K, Doty P. The far ultraviolet absorption spectra of polypeptide and protein solutions and their dependence on conformation. Proc Nat Acad Sci. 1961;47:1775–1785. doi: 10.1073/pnas.47.11.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tourasse NJ, Li WH. Selective constraints, amino acid composition, and the rate of protein evolution. Mol Biol Evol. 2000;17:656–664. doi: 10.1093/oxfordjournals.molbev.a026344. [DOI] [PubMed] [Google Scholar]
- 31.Traganos F, Darzynkiewicz Z, Melamed MR. The ratio of RNA to total nucleic acid content as a quantitative measure of unbalanced cell growth. Cytometry. 1982;2:212–218. doi: 10.1002/cyto.990020403. [DOI] [PubMed] [Google Scholar]
- 32.Li HJ, Isenberg I, Johnson WC. Absorption and circular dichroism studies on nucleohistone IV. Biochemistry. 1971;10:2587–2593. doi: 10.1021/bi00789a027. [DOI] [PubMed] [Google Scholar]
- 33.Tataurov AV, You Y, Owczarzy R. Predicting ultraviolet spectrum of single stranded and double stranded deoxyribonucleic acids. Biophys Chem. 2008;133:66–70. doi: 10.1016/j.bpc.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 34.Volpe P, Eremenko-Volpe T. Quantitative studies on cell proteins in suspension cultures. Eur. J. Biochem. 1970;12:195–200. doi: 10.1111/j.1432-1033.1970.tb00837.x. [DOI] [PubMed] [Google Scholar]
- 35.Crissman HA, Darzynkiewicz Z, Tobey RA, Steinkamp JA. Correlated measurements of DNA, RNA, and protein in individual cells by flow cytometry. Science. 1985;228:1321–1324. doi: 10.1126/science.2408339. [DOI] [PubMed] [Google Scholar]
- 36.Shapiro HM. Flow Cytometric Estimation of DNA and RNA in Intact Cells Stained with Hoechst 33342 and Pyronin Y. Cytometry. 1981;2:143–149. doi: 10.1002/cyto.990020302. [DOI] [PubMed] [Google Scholar]
- 37.Krishan A, Dandekar P, Nathan N, Hamelik R, Miller C, Shaw J. DNA index, genome size, and electronic nuclear volume of vertebrates from the Miami Metro Zoo. Cytometry A. 2005;65A:26–34. doi: 10.1002/cyto.a.20130. [DOI] [PubMed] [Google Scholar]
- 38.Animal genome size database. http://www.genomesize.com.
- 39.Greilhuber J, Volleth M, Loidl J. Genome size of man and animals relative to the plant Allium cepa. Can J Genet Cytol. 1983;25:554–560. doi: 10.1139/g83-084. [DOI] [PubMed] [Google Scholar]
- 40.Kato H, Harada M, Tsuchiya K, Moriwaki K. Absence of correlation between DNA repair in ultraviolet irradiated mammalian cells and life span of the donor species. Jpn J Genet. 1980;5:99–108. [Google Scholar]
- 41.Bachmann K. Genome size in mammals. Chromosoma. 1972;35:85–93. doi: 10.1007/BF00329560. [DOI] [PubMed] [Google Scholar]
- 42.Darzynkiewicz Z, Halicka HD, Zhao H. Analysis of cellular DNA content by flow and laser scanning cytometry. Adv Exp Med Biol. 2010;676:137–147. doi: 10.1007/978-1-4419-6199-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker PMB, Davies HG. Ultra-violet microspectrography of living tissue culture cells. Part II. Microspectrographic studies of living cells and ultraviolet-irradiated chick fibroblasts. Discuss Faraday Soc. 1950;9:461–470. [Google Scholar]
- 44.Liu P-F, Avramova LV, Park C. Revisiting absorbance at 230 nm as a protein unfolding probe. Anal Biochem. 2009;389:165–170. doi: 10.1016/j.ab.2009.03.028. [DOI] [PubMed] [Google Scholar]
- 45.Glazer AN, Smith EL. Studies on the ultraviolet difference spectra of proteins and polypeptides. J Biol Chem. 1961;236:2942–2947. [PubMed] [Google Scholar]
- 46.Glazer AN, Smith EL. Effect of denaturation on the ultraviolet absorption spectra of proteins. J Biol Chem. 1960;235:PC43–PC44. [Google Scholar]
- 47.Martin CJ, Bhatnagar GM. Unfolding reactions of proteins. I. Kinetic and equilibrium measurements of diisopropylphosphorylchymotrypsin and chymotrypsinogen in urea. Biochem. 1966;5:1230–1241. doi: 10.1021/bi00868a017. [DOI] [PubMed] [Google Scholar]
- 48.Eisenberg DS, Edsall JT. Spectrophotometric titrations of human serum albumin and reduced carboxylmethylated albumin. Science. 1963;142:50–51. doi: 10.1126/science.142.3588.50. [DOI] [PubMed] [Google Scholar]
- 49.Mora S, Elodi P. Investigation of the near and far ultraviolet denaturation difference spectra of dehydrogenases. Eur J Biochem. 1968;5:574–582. doi: 10.1111/j.1432-1033.1968.tb00408.x. [DOI] [PubMed] [Google Scholar]
- 50.Donovan JW. Changes in ultraviolet absorption produced by alteration of protein conformation. J Biol Chem. 1969;244:1961–1967. [PubMed] [Google Scholar]
- 51.Borgdorff V, Pauw B, van Hees-Stuivenberg S, de Wind N. DNA mismatch repair mediates protection from mutagenesis induced by short-wave ultraviolet light. DNA Repair. 2006;5:1364–1372. doi: 10.1016/j.dnarep.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 52.Durand RE, Olive PL. Cytotoxicity, mutagenicity, and DNA damage by Hoechst 33342. J Histochem Cytochem. 1982;30:111–116. doi: 10.1177/30.2.7061816. [DOI] [PubMed] [Google Scholar]
- 53.Darzynkiewicz Z, Traganos F, Kapuscinski J, Melamed MR. Denaturation and condensation of DNA in situ induced by acridine orange in relation to chromatin changes during growth and differentiation of friend erythoroleukemia cells. Cytometry. 1985;6:195–207. doi: 10.1002/cyto.990060305. [DOI] [PubMed] [Google Scholar]