

Abstract

An efficient and broadly useful two-step ligation protocol is developed. Important mechanistic issues of ligation were probed from diastereomeric competition studies on the formation of the ligation products. We also report an instance of kinetically controlled ligation through the exploitation of selectivity differences between related N-termini. This study potentially provides a valuable approach to facilitate polypeptide synthesis by minimizing protecting group manipulations and intermediate isolations..
Keywords: leucine, native chemical ligation, desulfurization, kinetic control
We have been studying the total synthesis of complex glycopeptide and glycoprotein targets.[1] Pursuant to this goal, we hope to discover broadly useful methods to join large peptide and glycopeptide fragments while minimizing the need for side chain protection.[2] A field-changing contribution to the problem of polypeptide ligation, termed native chemical ligation (NCL), was provided by Kent and co-workers in 1994 (Figure 1).[3] NCL involves the merger of a peptide domain possessing a C-terminal thioester fragment with a second peptide bearing an N-terminal cysteine residue. The key mechanistic features of NCL, (trans-thioesterification and S→N acyl transfer) are adumbrated in Figure 1. Clearly if the primary thio group could be desulfurized, the NCL method can be used to accommodate alanine ligation.[4] Of course, for this to work well, other sulfur moieties within the construct must withstand the desulfurization reaction. A major advance in this regard was accomplished in a metal-free fashion, using classical mechanistic insights in free radical mediated desulfurization.[5]
Figure 1.

NCL and Alanine ligation.
We then set about to apply, more generally, the overall logic of NCL to other proteogenic amino acids. The thought was to synthesize non-proteogenic amino acids, bearing strategically placed thiol groups, to serve as the N-terminal residues in the ligation event. In this way the logic of NCL could, in principle, be broadly extended. In each case, the concluding step would exploit our metal free desulfurization method. Indeed, this was accomplished for valine, lysine, and threonine ligations.[6]
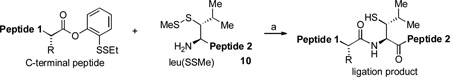
We next turned to the possibility of extending NCL logic to accomplish de facto leucine ligation.[7] We first prepared the diastereomeric β-thio-leucine surrogates, 1 [leu(SSMe)] and 2 [which we term leu(SSMe)*] (Scheme 1a). The synthesis of 1 commenced with commercially available (2S,3S)-3-hydroxy-leucine (3) and passed through 4 and 5 as shown in Scheme 1. Compound 2 was prepared through an analogous sequence from (2S,3R)-3-hydroxyleucine (6).[4c, 6a]
Scheme 1.

Synthesis of peptide substrates. Reagents and conditions: (A) (a) Boc2O, Na2CO3, THF/H2O, RT, 91%; (b) TMSE-OH, DCC, DMAP, CH2Cl2, 0 °C→RT, 99%; (c) MsCl, Et3N, CH2Cl2, 0 °C; (d) AcSK (excess), DMF, RT, 40 °C→60 °C, 82% over two steps; (e) NaOH, MeOH, 0 °C; (f) MMTS, DIEA, CH2Cl2, RT, 79% over two steps; (g) TBAF, THF, RT, 98%. (B) (a) MeOH, DCC, DMAP, CH2Cl2; (b) Piperidine, CH2Cl2; (c) Boc-Leu(SSMe)-OH, HATU, DIEA, DMSO; (d) TFA:H2O:TIS (95:2.5:2.5); (e) EDCI, HOOBt, CHCl3/TFE. MMTS = methane methylthiosulfonate.
In addition to studying the feasibility and quality of the projected leucine ligations, we anticipated that the availability of two epimeric leucine surrogates (compounds 1 and 2) as potential probes, could also provide a basis for studying rather subtle, otherwise hidden issues of native chemical ligation. Accordingly, we prepared the peptides described below (Scheme 1b). In all cases, the acyl donor component of the ligation was presented as a masked thiol ester of a type we had previously described.[2a] The required peptides (vide infra) with C-terminal methyl esters or free carboxylic acids and N-terminal leucine surrogates were prepared using HATU-mediated peptide coupling reactions in the presence of 1 equivalent of synthetic leucine precursor.[1c] The peptides bearing ortho-disulfide phenolic esters at the C-termini were synthesized using EDCI mediation under the non-epimerizing conditions developed by Sakakibara and co-workers. (Scheme 1b)[8]
We began by comparing the quality of ligation of the two leucine epimers by joining both 10 and 11 with peptide 9, presenting a C-terminal phenylalanine residue.[6a] The impact of thio-leucine stereochemistry on the ligation was striking. Thus, under standard conditions, peptide 10 readily underwent coupling with 9 to afford 12. Within 15 minutes, the reaction had achieved ca. 50% conversion by UPLC-MS, and a 75% yield was obtained as shown. In the case of peptide 11, less than 7% conversion was observed after 15 minutes. After 30 h, an 18% yield of 13 could be obtained. The mechanistic implications of this type of finding will be discussed below (vide infra).
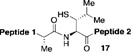
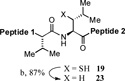
With the preferred leucine amino acid surrogate established (i.e. 1), we next probed the versatility of the leucine ligation protocol. Not surprisingly, the rate and efficiency of ligation was found to be qualitatively dependent on the level of steric hindrance at the C-terminus residue. Thus, under standard conditions, peptide 10 underwent rapid ligation with the C-terminal glycine peptide, 14, to furnish adduct 15 in 95% conversion within 2h (entry 1). Although ligation with the C-terminal alanine peptide, 16, was somewhat less facile, a reasonable conversion of ligation product was obtained (85%, entry 2). As expected, peptides 18 and 20, presenting C-terminal valine and proline residues, were significantly less reactive as acyl donors and the yields suffered accordingly (entries 4 and 5). The ability to efficiently convert the β-thio-leucine surrogates to leucines in the context of their primary ligation products, upon exposure to our standard metal-free desulfurization conditions, is shown in Table 1 (entries 3 and 4).[5]
Table 1.
Substrate scope of leucine ligation and desulfurization.
 | ||||
|---|---|---|---|---|
| entry | C-terminal peptide |
ligation product |
conversion | time |
| 1 | 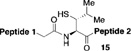 |
95% | 2h | |
| 2 | 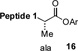 |
 |
85% | 2.5h |
| 3 | 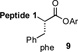 |
 |
83% | 2.5h |
| 4 | 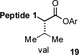 |
 |
50% | 8h |
| 5 | 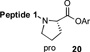 |
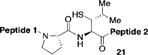 |
21% | 9h |
Reagents and conditions: (a) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5; ratio C-terminal peptide:10 = 1.5:1. (b) TCEP, VA-044, tBuSH, 0.8 h. Peptide 1: GKHLNSAERVE–; Peptide 2: –RKKLQDVHNFVALG–OMe.
The substantial difference in the performance of peptides terminating in surrogate 1 and surrogate 2 as acyl acceptor encouraged us to probe more intensively into some of the mechanistic issues associated with native chemical ligation.[9] The widely held notion is that the trans-thioesterification step is rate determining, and that the acyl transfer of the C-terminal coupling component to nitrogen is rapid (see Figure 1). This accounts for the inability to clearly identify any intermediate thioesters en route to ligation.[9]
To approach this question more precisely, we designed a competition experiment that would allow for rough comparison of the acyl acceptor qualities of the two epimeric N-termini under identical conditions. In the event, peptides 10 and 24, bearing N-terminal leu(SSMe) and leu(SSMe)* residues were treated with the C-terminal glycine peptide 14 under standard ligation conditions. The results are shown in Figure 2. Since the yield of the ligation of the N-terminal leu(SSMe)* epimer is poor (Scheme 2, vide infra), it is not possible to extract hard numbers for the relative rates of acyl acceptor reactivity of epimers leu(SSMe) and leu(SSMe)*. By examining the relative amount of ligation products in the early stages of the experiment, we estimate the ratio to be at least 20:1 in favor of fast reacting epimer leu(SSMe). In the limiting case, assuming the inherent acyl acceptor properties of the thiol groups in leu(SSMe) and leu(SSMe)* were nearly the same, and each intermediate suffered S→N acyl transfer with comparable efficiency, the ratio of effective ligation would be ca. 1:1. The actual comparative yields reported in Scheme 2 strongly suggest that the acyl transfer step is substantially slower in the case of leu(SSMe)* relative to leu(SSMe). This finding can be rationalized, since in the case of leu(SSMe), S→N acyl transfer requires a trans relationship of the substituents on the 5-membered ring, while in the case of the epimeric leu(SSMe)* system, the corresponding isopropyl and peptidic residues are cis. The attenuated rate of acyl transfer in the case of leu(SSMe)* presumably renders its thioester intermediate more vulnerable to competitive adventitious hydrolysis.
Figure 2.

The competition reaction between leu(SH) and leu(SH)* to form peptide-coupling products. (a) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5. Peptide 1: GKHLNSAERVE–; Peptide 2: RKKLQDVHNFVALG-OMe; Peptide 3: –RKKLQDVHNFVALG–OH.
Scheme 2.

Leucine ligation with two leu(SSMe) diastereomers. Reagents and conditions: (a) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5. Peptide 1: GKHLNSAERVE; Peptide 2: RKKLQDVHNFVALG-OMe.
Furthermore, the rates of the trans-thioesterification steps may also be quite different for the two epimeric surrogates. For intramolecular transfer to occur, the proton on the thiol acyl acceptor group must be transferred to a putative “base” or to the bulk solvent under weakly basic conditions (pH ~ 7.5). A possibility in this regard is that a proton of the neighboring NH3+ group is donated to medium, thus allowing the nitrogen to remove the proton from the sulfur. Once again, in the case of leu(SSMe), such a transfer would require a trans relationship of the large substituents, while in the case of leu(SSMe)*, they are cis (see Figure 2).
We then posed the question as to how the productive epimer, leu(SSMe), might compare with cysteine in a competitive experiment. Required substrates were prepared as described and the experiment crafted as shown in Figure 3.
Figure 3.

The competition reaction between leu(SH) and cys to form peptide-coupling products. (a) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5. Peptide 1: GKHLNSAERVE–; Peptide 3: –RKKLQDVHNFVALG–OH
Because intermolecular trans-thioesterification is supposed to be rate determining, one might anticipate that the primary thiol of the cysteine residue would react more rapidly than the secondary thiol of the leu(SSMe) surrogate.[9] Indeed, as shown in Figure 3, peptide 29, arising from ligation of the cysteine-bearing peptide, 27, was found to be the predominant product (29:28 ~ 4:1). Apropos of the arguments above, explanations can be advanced to rationalize why the margin of difference could well be less than expected on the basis of “A–value type” comparisons of H versus isopropyl.
Putative ring character is clearly involved in the intramolecular S→N acyl transfer step in NCL. As argued above, it could also be involved in the required de-protonation of the thiol group en route to trans thioacylation. In either case such ring formation, in the case of the fast reacting pre-leucine compound, is favored by an argument similar to the classical Thorpe-Ingold effect, since the acyclic array is more substituted.[10] The corresponding pertinent acylic ensemble in the case of the N-terminal cysteine is substantially less substituted. Therefore it might benefit far less from cyclization.
Finally, we were able to demonstrate the complexity-building capacity of this chemistry. The thought was to develop a kinetically controlled reiterative coupling strategy, which would exploit the reactivity differential between the N-terminal-thio-leucine surrogate and cysteine acyl acceptors.[11] In keeping with a major concern of our laboratory, we envisioned, as a goal, the rapid assembly of the human erythropoietin (hEPO) peptide, 34, from three individual fragments through sequential cysteine and thio-leucine ligations.
The experiment was crafted as shown in Scheme 3. We first undertook to connect 32 and 31. The resulting gross product was capped by a second ligation to 30. In the event, a 61% yield of the desired sequential double ligation product 33 was isolated. This product indeed corresponds to the sequence 32+31+30. We were unable to find the single ligation product corresponding to the self-coupling of 31, though it could well have been missed (perhaps due to further oligomerization). Interestingly, we also did not detect a double ligation product arising from a sequence 31+31+30. One product, implicated in ca 5% yield,[12] presumably corresponds to initial cyclization (i.e. thiolactonization) of 31. We also identified a monoligation product arising from the coupling of 32 with 30. In principle, it is the thiolactonization side reaction, which is responsible for “leftover” 32 and 30.
Scheme 3.

Synthesis of EPO(95–120) peptide. Reagents and conditions: (a) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5, 0.5 h. (b) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5, 0.5 h; MESNa, H2O:MeCN (1:1), 1 min, 61% over two steps. (c) TCEP, VA-044, tBuSH, 1h, 82%.
It is well to note that every self-coupling of 31+31 serves to deplete two equivalents of the “competing substrates.” Hence, in principle, even a 4:1 acyl acceptor reactivity ratio of 32:31 would be leveraged to provide an ~ 8:1 factor “against” the hypothetical (unobserved) product of a single ligation of 31+30.[13] Obviously, any further level of oligomerization of 31 would provide greater leveraging of the selectivity for forming 33 rather than the unobserved products (31 + 31 + 30 or 31+30).
In summary, the chemistry described above started with the central concept of native chemical ligation. To extend this core idea, we have devised an effective route to generating the required C-terminal thioester acyl donor via an intramolecular O →S transfer in the same step where the cysteine (or surrogate) thiol is exposed. Sterochemically defined leucine ligation surrogates were synthesized and used to probe important mechanistic issues involved in native chemical ligation. Selectivities in acyl accepter efficacies in NCL were explored from several considerations. First, evidence has been brought to bear that the rate of the S→N intramolecular acyl transfer (Figure 1, step b) can be undermined by imposing a steric hindrance restraint at the stage where a formation of mechanistically required ring is required. Similar issues may also impact on the first step of the process, i.e. trans thioesterafication (Figure 1, step a). We postulate that, perhaps, even in this opening step, quasi ring formation may be a central element in orchestrating a deprotonation of the acyl acceptor thiol group (see Figure 2, structure 24). Finally, we further suggest that increased substitution in the acyclic system can perhaps be a factor in the transthioesterification step by favoring ring formation in the transfer of the thiol proton by intramolecular means (see Figure 3).[10]
A combination of these considerations served to establish a selectivity margin between N-terminal cysteine, itself, and the N-terminal thiol containing precursor of leucine. Combining the principles, we realized a rather promising sequential ligation, wherein peptide A (cf. 31) couples with peptide B (cf. 32) to give peptide C (not purified), which in turn couples with peptide D (cf. 30) to give rise to peptide F (cf. 33). The implications of these findings for the synthesis of critical, biologically relevant peptides and glycopeptides are being pursued.[14]
Supplementary Material
Footnotes
Support for this research was provide by the National Institute of Health (CA28824 to SJD).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Wu B, Tan Z, Chen G, Chen J, Hua Z, Wan Q, Ranganathan K, Danishefsky SJ. Tetrahedron Lett. 2006;47:8009. doi: 10.1016/j.tetlet.2006.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen J, Chen G, Wu B, Wan Q, Tan Z, Hua Z, Danishefsky SJ. Tetrahedron Lett. 2006;47:8013. doi: 10.1016/j.tetlet.2006.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tan Z, Shang S, Halkina T, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5424. doi: 10.1021/ja808704m. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yuan Y, Chen J, Wan Q, Tan ZP, Chen G, Kan C, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5432. doi: 10.1021/ja808705v. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kan C, Trzupek JD, Wu B, Chen G, Tan ZP, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5438. doi: 10.1021/ja808707w. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Nagorny P, Fasching B, Li X, Chen G, Aussedat B, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5792. doi: 10.1021/ja809554x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Warren JD, Miller JS, Keding SJ, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:6576. doi: 10.1021/ja0491836. [DOI] [PubMed] [Google Scholar]; b) Wu B, Chen J, Warren JD, Chen G, Hua Z, Danishefsky SJ. Angew. Chem. 2006;118:4222. doi: 10.1002/anie.200600538. Angew. Chem. Int. Ed.2006, 45, 4116. [DOI] [PubMed] [Google Scholar]; c) Wu B, Warren JD, Chen J, Chen G, Hua Z, Danishefsky SJ. Tetrahedron Lett. 2006;47:5219. doi: 10.1016/j.tetlet.2006.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chen G, Wan Q, Tan Z, Kan C, Hua Z, Ranganathan K, Danishefsky SJ. Angew Chem. 2007;119:7527. doi: 10.1002/anie.200702865. Angew. Chem. Int. Ed.2007, 46, 7383. [DOI] [PubMed] [Google Scholar]; e) Wan Q, Chen J, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:15814. doi: 10.1021/ja804993y. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Li X, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:5446. doi: 10.1021/ja800612r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]; b) Tam JP, Lu YA, Liu CF, Shao J. Proc. Natl. Acad. Sci. USA. 1995;92:12485. doi: 10.1073/pnas.92.26.12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Yan LZ, Dawson PE. J. Am. Chem. Soc. 2001;123:526. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]; b) Pentelute BL, Kent SB. Org. Lett. 2007;9:687. doi: 10.1021/ol0630144. [DOI] [PubMed] [Google Scholar]; c) Crich D, Banerjee A. J. Am. Chem. Soc. 2007;129:10064. doi: 10.1021/ja072804l. [DOI] [PubMed] [Google Scholar]
- 5. Wan Q, Danishefsky SJ. Angew. Chem. 2007;119:9408. doi: 10.1002/anie.200704195. Angew. Chem. Int. Ed.2007, 46, 9248. For the seleno analog, see: Metanis N, Keinan E, Dawson PE. Angew. Chem. Int. Ed. 2010 doi: 10.1002/anie.201001900. Early View.
- 6.a) Chen J, Wan Q, Yuan Y, Zhu J, Danishefsky SJ. Angew. Chem. 2008;120:8649. doi: 10.1002/anie.200803523. Angew. Chem. Int. Ed.2008, 47, 8521. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Haase C, Rohde H, Seitz O. Angew. Chem. 2008;120:6912. doi: 10.1002/anie.200801590. Angew. Chem. Int. Ed.2008, 47, 6807. [DOI] [PubMed] [Google Scholar]; c) Chen J, Wang P, Zhu JL, Wan Q, Danishefsky SJ. Tetrahedron. 2010;66:2277. doi: 10.1016/j.tet.2010.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yang R, Pasunooti KK, Li F, Liu XW, Liu CF. J. Am. Chem. Soc. 2009;131:13592. doi: 10.1021/ja905491p. [DOI] [PubMed] [Google Scholar]
- 7.During the preparation of this manuscript, Brik and co-workers reported a native chemical ligation at leucine. However, these authors did not investigate the relative rates of the two diastereomeric leucine surrogates as the key selectivity versus reported herein. Harpaz Z, Siman P, Kumar KS, Brik A. Chembiochem. 2010;11:1232. doi: 10.1002/cbic.201000168.
- 8.Sakakibara S. Biopolymers. 1995;37:17. doi: 10.1002/bip.360370105. [DOI] [PubMed] [Google Scholar]
- 9.a) Kent SB. Chem. Soc. Rev. 2009;38:338. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]; b) Johnson EC, Kent SB. J. Am. Chem. Soc. 2006;128:6640. doi: 10.1021/ja058344i. [DOI] [PubMed] [Google Scholar]; c) Muir TW. Annu. Rev. Biochem. 2003;72:249. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]; d) Hackeng TM, Griffin JH, Dawson PE. Proc. Natl. Acad. Sci. USA. 1999;96:10068. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Canne LE, Bark SJ, Kent SBH. J. Am. Chem. Soc. 1996;118:5891. [Google Scholar]
- 10. Beesley RM, Ingold CK, Thorpe JF. J. Chem. Soc.,Trans. 1915;107:1080. Of course, in its pure form, the classical Thorpe-Ingold effect refers to the inclusion of a fully substituted (quaternary) carbon in the cyclizing chain. However, as a general matter, heavier substitution (in the absence of steric hindrance in the resulting ring) tends to favor cyclization for reasons of entropy.
- 11.Bang D, Pentelute BL, Kent SB. Angew. Chem. 2006;118:4089. doi: 10.1002/anie.200600702. Angew. Chem. Int. Ed.2006, 45, 3985. [DOI] [PubMed] [Google Scholar]
- 12.This product was not isolated, per se, but inferred as shown in the Supporting Information.
- 13.Of course, the 4:1 ratio established for the benchmark case in Figure 3 may not be applicable to the competition of 32 and 31.
- 14.a) Wilson RM, Danishefsky SJ. Pure Appl. Chem. 2007;79:2189. [Google Scholar]; b) Gamblin DP, Scanlan EM, Davis BG. Chem. Rev. 2009;109:131. doi: 10.1021/cr078291i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.