

Abstract
Alzheimer disease (AD) is the most common type of dementia and is characterized pathologically by the presence of neurofibrillary tangles (NFTs), senile plaques (SPs), and loss of synapses. The main component of SP is amyloid-beta peptide (Aβ), a 39 to 43 amino acid peptide, generated by the proteolytic cleavage of amyloid precursor protein (APP) by the action of beta- and gamma-secretases. The presenilins (PS) are components of the γ-secretase, which contains the protease active center. Mutations in PS enhance the production of the Aβ42 peptide. To date, more than 160 mutations in PS1 have been identified. Many PS mutations increase the production of the β-secretase-mediated C-terminal (CT) 99 amino acid-long fragment (CT99), which is subsequently cleaved by γ-secretase to yield Aβ peptides. Aβ has been proposed to induce oxidative stress and neurotoxicity. Previous studies from our laboratory and others showed an age-dependent increase in oxidative stress markers, loss of lipid asymmetry, and Aβ production and amyloid deposition in the brain of APP/PS1 mice. In the present study, we used APPNLh/APPNLh x PS-1P246L/PS-1P246L human double mutant knock-in APP/PS-1 mice to identify specific targets of brain protein carbonylation in an age-dependent manner. We found a number of proteins that are oxidatively modified in APP/PS1 mice compared to age-matched controls. The relevance of the identified proteins to the progression and pathogenesis of AD is discussed.
Keywords: Oxidative stress, Amyloid β-peptide, Alzheimer’s disease, Presenilin-1, Redox proteomics, Protein carbonyls
Introduction
Alzheimer disease (AD) is the most common type of dementia that currently affects 5.3 million Americans, a number that is predicted to increase to 16–20 million Americans in just a few decades [1]. AD is characterized pathologically by the presence of neurofibrillary tangles (NFTs), senile plaques (SPs), and loss of synapses [2]. Senile plaques are extracellular and are composed primarily of amyloid-beta (Aβ) peptide, a 39 to 43 amino acid peptide. Aβ is generated by the proteolytic cleavage of a transmembrane protein, amyloid precursor protein (APP), by the action of beta- and gamma-secretases. Aβ has been proposed to induce oxidative stress and neurotoxicity [3-5]. The evidence of Aβ being a key player in AD pathogenesis was proposed principally based on familial Alzheimer disease (FAD) individuals, who have autosomal dominant mutations in amyloid precursor protein (APP) or presenilin (PS) genes 1 and 2 [6, 7], a mutation in PS-1 being more aggressive. In addition, individuals with Down syndrome, an autosomal trisomy of 21 chromosome, a locus of the APP gene, present pathological hallmarks of AD after about age 40 [8]. Studies from our laboratory and others suggested that Aβ is neurotoxic and its neurotoxicity is mediated through its ability to induce free radical oxidative stress, including protein oxidation and lipid peroxidation [3-5, 9, 10]. In AD brain, oxidative stress is indexed by significant increase in the markers of protein oxidation [4], lipid peroxidation [11-13], nucleic acid oxidation, and neuronal dysfunction or death. Protein carbonyls are a general and widely used index to determine the extent of oxidative modification of proteins both under in in vivo and in vitro conditions [14-20]. In AD brain protein carbonyls levels were reported to be elevated [3, 10, 15, 16, 21]. Smith and collaborators [22] reported a strong protein carbonyl signal in neurofibrillary tangles, neuronal cell bodies, and apical dendrites as well as neuronal and glial nuclei in hippocampal sections of AD brains. A recent study showed that the levels of protein carbonyls are significantly increased in both synaptic and non-synaptic mitochondria in the frontal cortex of AD [23]. Our laboratory first applied redox proteomics to identify specifically carbonylated proteins in AD brain [24-26], which led to the identification of a number of proteins involved in various metabolic pathways as altered in AD brain. Further, we also applied redox proteomics approach to identify targets of protein oxidation in different stages of AD, i.e., mild cognitive impairment and early AD, in addition to the in vivo and in vitro models of AD, consistent with the notion that oxidative stress is a key component of the pathogenesis and progress of this dementing disorder.
In the present study we used APPNLh/APPNLh x PS-1P246L/PS-1P246L human double mutant knock-in APP/PS-1 mice to identify carbonylated brain proteins in an age-dependent manner. APPNLh/APPNLh x PS-1P264L/PS-1P24L6 human double knock-in mice were generated using Cre-loc knock-in technology (Cephalon, Inc., Westchester, PA) to humanize the mouse Aβ sequence (NLh) and create a PS-1 proline to leucine mutation in residue 264 (P264L), a mutation identified in human FAD [27, 28]. These double knock-in mice yield proper cleavage of the APP protein to generate Aβ. Previously, we and others demonstrated that APP/PS-1 mice have age-dependent increased oxidative stress markers, loss of lipid asymmetry, Aβ production and amyloid deposition in the brain [29-31]. Anatharaman et al., (2006) showed that the mice deposit Aβ at 9 months of age, with frank deposits at 12 months of age [30]. Further, neuronal cultures from 1 day old APP/PS-1 mice pups showed increased basal protein oxidation and lipid peroxidation and are more vulnerable to oxidation by exogenous oxidants compared with neurons from wild-type mice [32]. The redox proteomics results presented here are discussed with reference to pathogenesis and progression of AD.
Materials and Methods
Materials
All chemicals, proteases, and antibodies used in these studies were purchased from Sigma-Aldrich (St. Louis, MO, USA) with exceptions noted. Criterion precast polyacrylamide gels, TGS and XT MES electrophoresis running buffers, ReadyStrip™ IPG strips, mineral oil, Precision Plus Protein™ All Blue standards, Sypro Ruby® Protein Stain, nitrocellulose membranes, dithiothreitol (DTT), iodoacetamide (IA), Biolytes, and urea were purchased from Bio-RAD (Hercules, CA, USA). 2, 4-Dinitrophenylhydrazine (DNPH) and the primary antibody used for the protein-bound 2,4-dinitrophenylhydrazone (DNP) products were purchased from Chemicon International (Temecula, CA, USA).
Animals
The APP/PS-1 mice used in this study are the APPNLh/APPNLh X PS-1P264L/PS-1P264L double mutant mice were generated by using the Cre-lox knock-in technology to humanize the mouse Aβ sequence and to create a PS-1 mutation identified in human AD by Cephalon (Westchester, PA, USA). All mice used in this study were males. These mice were maintained on a CD-1/129 background. All mice were maintained on a 12-h light: dark cycle in Bioclean units with sterile-filtered air and provided food and water ad libitum. All protocols were implemented in accordance with NIH guidelines and approved by the University of Kentucky Institutional Animal Care and Use Committee. Following euthanasia with CO2, the brain was removed quickly, weighed, and snap-frozen in liquid nitrogen prior to analysis. Six animals per age groups (1, 6, 9, 12, and 15 months old) for both wild-type and APP/PS-1 double knock-in mice were used in this study. The above age groups in this study were chosen based on previous studies conducted in our laboratory (oxidative stress markers and Aβ deposition) [30, 32].
Experimental Procedures
Sample Preparation
Brain tissue homogenates (10%) were prepared in buffer A [0.32 M sucrose, 0.10 mM Tris HCl (pH 8.0), 0.10 mM MgCl2, 0.08 mM EDTA, 10 μg/ml leupeptin, 0.5 μg/ml pepstatin, and 11.5 μg/ml aprotinin; pH 8.0] using a Wheaton glass homogenizer, followed by vortexing and sonication of the samples for 10 s at 20% power with a Fisher 550 Sonic Dismembrator (Pittsburgh, PA, USA). Protein concentrations were determined according to the Pierce BCA method (Rockford, IL, USA).
Isoelectric Focusing (IEF)
Two hundred micrograms of protein from total brain homogenates were first derivatized by incubation of protein samples with 4x the sample volume of 10 mM DNPH in 2 N HCl for 30 min at room temperature (RT), followed by precipitation of protein with 30% ice-cold TCA. Samples were centrifuged at 23,700 g for 5 min at 4 °C. Pellets were resuspended and rinsed in a Wash Buffer [1:1 (v/v) ethanol:ethyl acetate] a total of four times to remove excess salts. Following the final wash, pellets were dried at RT for ~10 min and rehydrated for 2 h at RT in 200 μl of a rehydration buffer [8 M urea, 2 M thiourea, 50 mM DTT, 2.0% (w/v) CHAPS, 0.2% Biolytes, bromophenol blue], and then sonicated for 10 s at 20% power. Samples (200 μl) were applied to 11 cm pH 3-10 ReadyStrip™ IPG strips (BioRad) and after 1 h, 2 ml of mineral oil was added to prevent sample evaporation. Strips were actively rehydrated at 20°C for 18 h at 50 V, focused at a constant temperature of 20°C beginning at 300 V for 2 h, 500 V for 2 h, 1,000 V for 2 h, 8,000 V for 8 h, and finishing at 8,000 V for 10 h- rapidly. IEF strips were stored at -80 °C until the second dimension of analysis was performed.
2D-PAGE
2D-PAGE was performed to separate proteins on IEF strips based on molecular migration rate. IEF strips were thawed at room temperature and equilibrated for 10 min in equilibration buffer A [50 mM Tris-HCl pH 6.8, 6 M urea, 1% (w/v) SDS, 30% (v/v) glycerol, and 0.5% DTT] and then re-equilibrated for 10 min in equilibration buffer B [50 mM Tris-HCl pH 6.8, 6 M urea, 1% (w/v) SDS, 30% (v/v) glycerol, and 4.5% IA]. All strips were rinsed in a 1x dilution of TGS running buffer before being placed into Criterion precast linear gradient (8-16%) Tris-HCl polyacrylamide gels. Precision Plus Protein™ Standards and samples were run at a constant voltage of 200 V for 65 min in a 1x dilution of TGS running buffer.
SYPRO Ruby® Staining
Following 2D-PAGE, gels were incubated in a fixing solution [7% (v/v) acetic acid, 10% (v/v) methanol] for 60 min at RT. Sypro Ruby® Protein Gel Stain (~50 ml) was added to gels and allowed to stain overnight at RT on a gently rocking platform. Gels were transferred to ~50 ml of deionized water at RT until scanning. Gels were scanned using a UV transilluminator (λex = 470 nm, λem = 618 nm, Molecular Dynamics, Sunnyvale, CA, USA) and stored in deionized water at 4 °C until further use.
2D-PAGE Western Blotting
Following 2D-PAGE, proteins from 2D gels were transferred to a nitrocellulose membrane (0.45 μm) for immunochemical detection of protein carbonyls, using a Trans-Blot Semi-Dry Transfer Cell system at 20 V for 2 h (Bio-RAD, Hercules, CA, USA). Post-transfer, membranes were incubated in a blocking solution of 3% bovine serum albumin (BSA) in Wash Blot [a phosphate-buffered saline (PBS) solution containing 0.04% (v/v) Tween 20 and 0.10 M NaCl] at RT for 2 h. For protein carbonyl detection membranes were incubated with rabbit anti-DNP (1:150) primary antibody, in blocking solution at RT on a rocking platform for 2-3 h. Blots were rinsed three times for 5 min each in Wash Blot, followed by a 1 h incubation with rabbit IgG alkaline phosphatase (1:3000) secondary antibody at RT. Blots were rinsed 3 times for 5 min each in Wash Blot and developed by incubating membrane with bromo-4-chloro-3-indolyl phosphate dipotassium and Nitrotetrazolium Blue chloride (BCIP/NBT) in ALP buffer [0.1 M Tris, 0.1 M NaCl, 5 mM MgCl2 (pH 9.5)]. Blots were allowed to dry overnight at RT prior to scanning into Adobe Photoshop 6.0 with a Canon CanoScan 8800F scanner.
Image Analysis
Carbonylated protein detection
The 2D gels and 2D blots were scanned and saved individually as TIFF files. PD Quest software (Bio-Rad) was used to detect carbonylated proteins. For analysis we have grouped these mice based on age and compared the specific oxidation of a protein obtained from brain of APP/PS1 mice with that from age-matched controls to account for possible effects of chronological aging on protein carbonylation. The average mode of background subtraction was used to normalize intensity values, which represent the amount of protein (total protein on gel and carbonylated protein on the blot) per spot. After completion of spot matching, the normalized intensity of each protein spot from individual gels is divided by the normalized intensity of each protein spot on the blots to obtain specific carbonylation levels. The spots showing significant difference in specific carbonylation levels between APP/PS1 mice and age-matched controls at each age using Student’s t-test statistical analysis (p<0.05) were chosen for identification.
In-Gel Trypsin Digestion
Protein spots identified as statistically significant were excised from 2D-gels with a clean, sterilized blade and transferred to Eppendorf microcentrifuge tubes. In-gel trypsin digestion of selected gel spots was performed as previously described [33]. Briefly, gel plugs were then washed with 0.1 M ammonium bicarbonate (NH4HCO3) at RT for 15 min, followed by incubation with 100% acetonitrile at RT for 15 min. After solvent removal, gel plugs were dried in their respective tubes under a flow hood at RT. Plugs were incubated for 45 min in 20 μl of 20 mM DTT in 0.1 M NH4HCO3 at 56 °C. The DTT/NH4HCO3 solution was then removed and replaced with 20 μl of 55 mM iodoacetamide (IA) in 0.1 M NH4HCO3 and incubated with gentle agitation at RT in the dark for 30 min. Excess IA solution was removed and plugs incubated for 15 min with 200 μl of 50 mM NH4HCO3 at RT. A volume of 200 μL of 100% acetonitrile was added to this solution and incubated for 15 min at RT. Solvent was removed and gel plugs were allowed to dry for 30 min at RT under a flow hood. Plugs were rehydrated with 20 ng/μl of modified trypsin (Promega, Madison, WI, USA) in 50 mM NH4HCO3 in a shaking incubator overnight at 37 °C. Enough trypsin solution was added to completely submerge the gel plugs.
Mass Spectrometry
Salts and contaminants were removed from tryptic peptide solutions using C18 ZipTips (Sigma-Aldrich, St. Louis, MO, USA), reconstituted to a volume of ~15 μL in a 50:50 (water:acetonitrile) solution containing 0.1% formic acid. Desalted samples were analyzed with a NanoMate device from Advion Biosciences (Ithaca, NY, USA) coupled to a LTQ-Orbitrap XL mass spectrometer from Thermo Scientific (Waltham, MA, USA). NanoMate is an automated chip-based device. It was used to generate nanospray from desalted samples and the MS and MS/MS spectra of ionized peptides were acquired with LTQ-Orbitrap XL operated in a data-dependent mode whereby MS spectra were acquired at 60,000 resolution and the 8 most intense precursor ions in MS scan were selected for collision induced dissociation with the following conditions: injection time 50 ms, 35% collision energy, MS/MS spectra were measured in the FT at 7500 resolution, and dynamic exclusion was set for 120 sec. Each sample was acquired for a total of ~2.5 min. MS/MS spectra were searched against the International Protein Index (IPI) Database (downloaded 03/05/09) using SEQUEST with the following specifications: 2 trypsin miscleavages, fixed carbamidomethyl modification, variable Methionine oxidation, parent tolerance 10 ppm, and fragment tolerance of 25 mmu or 0.01 Da. Results were filtered with the following criteria: Xcorr > 1.5, 2.0, 2.5, 3.0 for +1, +2, +3, and +4 charge states, respectively, Delta CN >0.1, and P-value (protein and peptide) <0.01. Accession numbers from IPI were cross-correlated with SwissProt accession numbers for final protein identification.
Statistical Analysis
All data are presented as mean ± S.D and statistical analyses was performed using a two-tailed Student’s t-test, wherein p<0.05 was considered significant for oxidative modification levels. Protein and peptide identifications obtained with the SEQUEST search algorithm with p<0.01 were considered as statistically significant. To further validate SEQUEST identification, the location of protein spots (i.e., MW, and isoelectric point) on the 2D-gels was manually checked based on expected MW and pI values from SwissProt database information.
Results and Discussion
In this study we identified the brain proteins that showed increased protein carbonyls in an age-dependent manner in human double mutant knock in APP/PS1 mice using a redox proteomics approach [34]. In order to control for the known elevation of protein carbonyls in brain aging [20] we compared a specific age group of APP/PS1 mice with the respective age-matched controls. The selection of age groups in this study was based on earlier studies from our laboratory and others that showed age-dependent increase in amyloid beta-levels and makers of oxidative stress [30, 31, 35]. In a previous study, we showed that APP/PS1 mice have increased markers of oxidative stress even at postnatal day one compared to wild type mice [32]. The APP/PS1 mice show an age-dependent increase in markers of oxidative stress and this increase correlated with the increased levels of amyloid beta-peptide levels [35]. The increase in oxidative stress markers is relatively high at 9 month of age, and based on the amyloid beta-peptide load at this time point, we consider this age as the beginning of the AD stage. In contrast, at 6 months of age, the levels of oxidative stress in brain is somewhat less than at 9 months, and we considered this age to be equivalent conceivably to mild cognitive impairment (MCI), arguably the earliest form of AD [30, 35].
The proteins that were found to be specifically carbonylated in APP/PS1 mice are listed in Table 1, and the mass spectrometric characteristics of these proteins are listed in Table 2. Figures 1, 2, 3, 4, and 5 show the proteins that were found to have increased carbonylation in 1M, 6M, 9M, 12M and 15M-old APP/PS1 mice, respectively, compared to the corresponding aged-matched controls. PDQuest analyses showed that at 1 month of age two protein spots have increased carbonylation, i.e., beta-actin and 14-3-3 zeta/delta/gamma. At 6M, 9M, 12M and 15M-old APP/PS1, we found 2, 3, 5, and 5 proteins, respectively, as specifically carbonylated proteins. The locations of most protein spots agree with reported pI and molecular weight (MW) values; however, in some cases spots differ likely due to protein posttranslational modifications, fragments, or degradation products that directly influence the net charge and MW of the protein.
Table 1.
Proteomic Identification of Oxidatively Modified Brain Proteins at 1M, 6M, 9M, 12M and 15M of age from APP/PS-1 mice compared to aged-matched controls.
| Age of mice | Protein Identified | Specific oxidation (Fold) | P value |
|---|---|---|---|
| 1M | Beta Actin | 1.6 | 0.05 |
| 14-3-3 zeta/delta/gamma | 2.2 | 0.05 | |
| 6M | Alpha enolase | 0.69 | 0.01 |
| Pyruvate dehydrogenase E1 | 0.81 | 0.03 | |
| 9M | Alpha enolase | 0.11/0.015 | 0.006/0.05 |
| 14-3-3 Zeta/gamma/delta | 0.01 | 0.005 | |
| Pin 1 | 2.54 | 0.05 | |
| 12M | Alpha enolase | 5.39 | 0.02 |
| 14-3-3 Zeta/gamma/delta | 2.55 | 0.02 | |
| Pin 1 | 15.21 | 0.01 | |
| Beta Synuclein | 6.09 | 0.03 | |
| ATP Synthase Subunit alpha | 35.28 | 0.05 | |
| 15M | Alpha enolase | 35.45 | 0.003 |
| 14-3-3 Zeta/gamma/delta | 36.54 | 0.04 | |
| Pin 1 | 61.37 | 0.04 | |
| Beta Actin | 301.32 | 0.02 | |
| ATP Synthase Subunit alpha | 9.61 | 0.04 |
P-value is calculated using a Student’s t-test. n=6 is used for the analysis.
Table II.
Mass spectrometric details of identified oxidatively modified brain proteins in APP/PS-1 mice.
| Age of mice | Accession # | Protein Identified | m/z Value | Number of identified peptides | Score | MW (kDa) | pI | Probabilitya |
|---|---|---|---|---|---|---|---|---|
| 1M | P60710 | Beta Actin | 721.4 | 25 | 70.2 | 42 | 5.2 | 3.00e-04 |
| P63101 | 14-3-3 zeta/delta/gamma | 540.2 | 23 | 70.3 | 27.9 | 4.7 | 2.00e-07 | |
| 6M | P17182 | Alpha enolase | 902.4 | 1 | 10 | 47 | 6.3 | 4.00e-05 |
| Q9D051 | Pyruvate dehydrogenase E1 | 774.5 | 8 | 50 | 39 | 6.4 | 2.00e-07 | |
| 9M | P17182 | Alpha enolase | 902.4 | 1 | 10 | 47 | 6.3 | 4.00e-05 |
| P63101 | 14-3-3 Zeta/gamma/delta | 540.2 | 23 | 70.3 | 27.9 | 4.7 | 2.00e-07 | |
| P17742 | Pin 1 | 655 | 20 | 50 | 18 | 7.8 | 1.00e-005 | |
| 12M | P17182 | Alpha enolase | 902.4 | 1 | 10 | 47 | 6.3 | 4.00e-05 |
| P63101 | 14-3-3 Zeta/gamma/delta | 540.2 | 23 | 70.3 | 27.9 | 4.7 | 2.00e-07 | |
| P17742 | Pin 1 | 655 | 20 | 50 | 18 | 7.8/7.9 | 1.00-005 | |
| Q91ZZ3 | Beta Synuclein | 665.7 | 28 | 10 | 14 | 4.2 | 1.00e-009 | |
| Q03265 | ATP Synthase Subunit alpha | 6985.8 | 7 | 30 | 59.72 | 9.53 | 8.00e-07 | |
| 15M | P17182 | Alpha enolase | 902.4 | 1 | 10 | 47 | 6.3 | 4.00e-05 |
| P63101 | 14-3-3 Zeta/gamma/delta | 540.2 | 23 | 70.3 | 27.9 | 4.7 | 2.00e-07 | |
| P17742 | Pin 1 | 655 | 20 | 50 | 18 | 7.8 | 1.00e-005 | |
| P60710 | Beta Actin | 721.4 | 25 | 70.2 | 42 | 5.2 | 3.00e-04 | |
| Q03265 | ATP Synthase Subunit alpha | 6985.8 | 7 | 30 | 59.72 | 9.53 | 8.00e-07 |
The probability associated with a false protein identification using the SEQUEST search algorithm.
Figure 1.
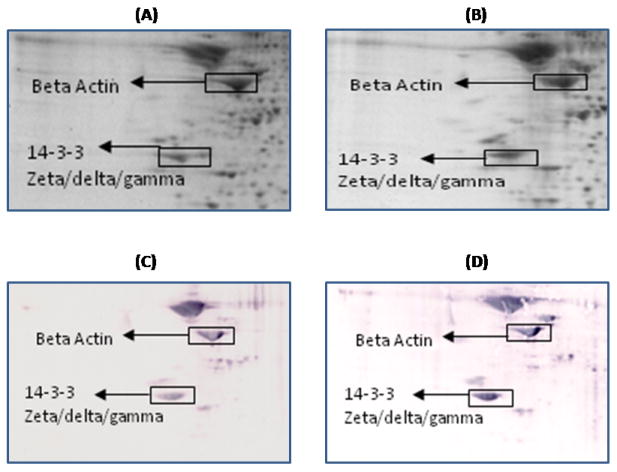
Representative 2D gel images of the brain proteome from 1M-old wild type (A) and 1M-old APP/PS1 mice (B). C and D represent the oxyblots of brain from 1M-old wild type and 1M-old APP/PS1 mice, respectively, showing geographical location of proteins on 2D gel and blot identified by mass spectrometry that showed differences in specific carbonylation. Spots that showed a significant difference in specific carbonylation levels are boxed and labeled with the corresponding protein identity. (n=6 separate WT and APP/PS1 mice).
Figure 2.
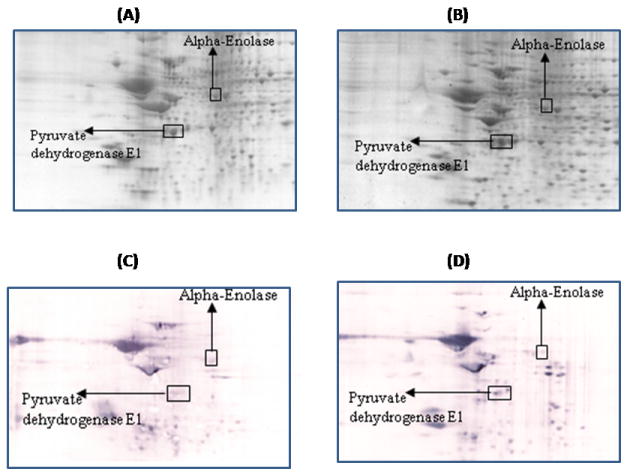
Representative 2D gel images of the brain proteome from 6M-old wild type (A) and 6M-old APP/PS1 mice (B). C and D represent the oxyblots of brain from 6M-old wild type and 6M-old APP/PS1 mice respectively, showing geographical location of proteins on 2D gel and blot identified by mass spectrometry that showed differences in specific carbonylation. Spots that showed a significant difference in specific carbonylation levels are boxed and labeled with the corresponding protein identity. (n=6 separate WT and APP/PS1 mice).
Figure 3.
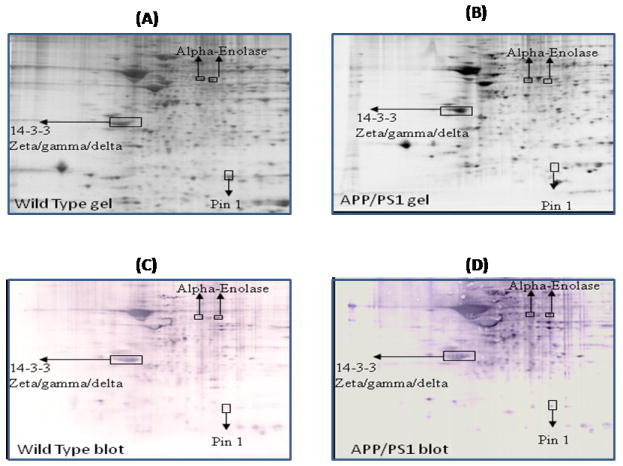
Representative 2D gel images of the brain proteome from 9M-old wild type (A) and 9M-old APP/PS1 mice (B). C and D represent the oxyblots of brain from 9M-old wild type and 9M-old APP/PS1 mice respectively, showing geographical location of proteins on 2D gel and blot identified by mass spectrometry that showed differences in specific carbonylation. Spots that showed a significant difference in specific carbonylation levels are boxed and labeled with the corresponding protein identity. (n=6 separate WT and APP/PS1 mice).
Figure 4.
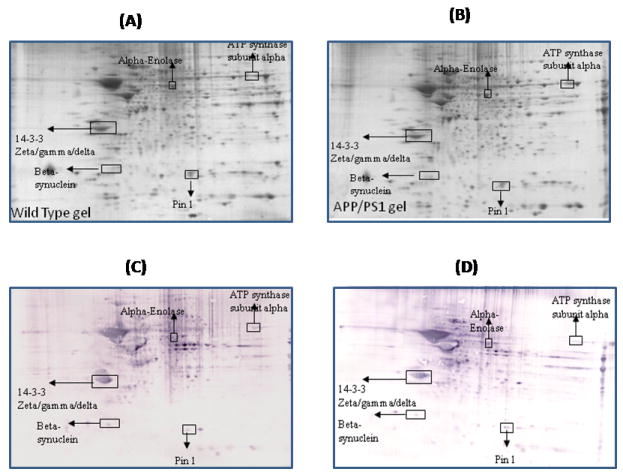
Representative 2D gel images of the brain proteome from 12M-old wild type (A) and 12M old APP/PS1 mice (B). C and D represent the oxyblots of brain from 12M-old wild type and 12M-old APP/PS1 mice respectively, showing geographical location of proteins on 2D gel and blot identified by mass spectrometry that showed differences in specific carbonylation. Spots that showed a significant difference in specific carbonylation levels are boxed and labeled with the corresponding protein identity. (n=6 separate WT and APP/PS1 mice).
Figure 5.
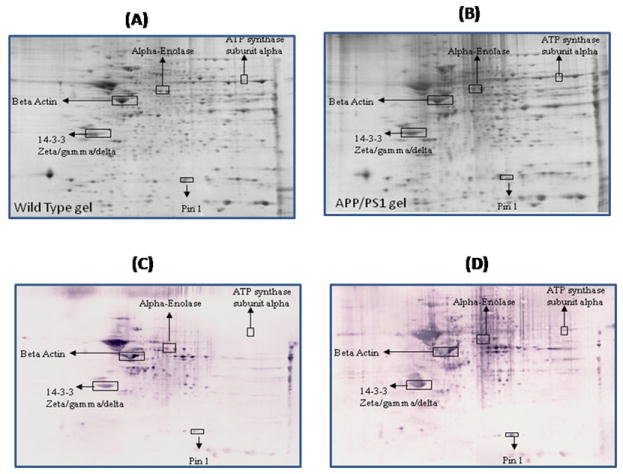
Representative 2D gel images of the brain proteome from 15M-old wild type (A) and 15M-old APP/PS1 mice (B). C and D represent the oxyblots of brain from 15M-old wild type and 15M-old APP/PS1 mice respectively, showing geographical location of proteins on 2D gel and blot identified by mass spectrometry that showed differences in specific carbonylation. Spots that showed a significant difference in specific carbonylation levels are boxed and labeled with the corresponding protein identity. (n=6 separate WT and APP/PS1 mice).
Compared to one month-old wild type mice the levels of proteins carbonyls are found to be significantly increased in beta-actin in APP/PS1 mice (Figure 1). This protein also is found to have significant increased carbonylation at 15M of age in APP/PS1 mice (Figure 5). Beta-actin is a cytoskeletal protein that plays an important role in numerous cellular processes involving membrane dynamics such as cell motility and morphogenesis [36, 37]. This protein has been found in both neurons and glial cells as a core subunit of microfilaments. Presynaptic terminals, dendritic spines and growth cones are neuronal areas rich in actin. For proper learning and memory process synapses play an important role, and they are always undergoing remodeling. Actin is well known to precisely control the development and location of synapses, which are key for proper neuronal network formation and activity and consequently in learning and memory in the brain [38-40]. At synapses, actin also plays an important role in synaptic activities such as organizing the postsynaptic density [41], anchoring postsynaptic receptors [42], facilitating the trafficking of synaptic cargos [43] and localizing translation machinery [44]. Hence, oxidation of actin conceivably could affect both retrograde and anterograde trafficking, neurotransmitter systems, and consequently to loss of neuronal cells. Further, oxidation of actin may reduce the formation and maturation of dendritic spines [45, 46], which are crucial for synaptic plasticity and memory formation [47, 48] and, consequently, important in AD clinical signs and symptoms. Various memory disorders has been shown to involve defects in the regulation of the actin cytoskeleton [49]. Previous studies reported the oxidation of actin in AD brain [26].
Pyruvate dehydrogenase is found to be carbonylated at 6M of age in APP/PS1 mice compared to an age-matched control (Figure 2). This multienzyme complex involves five cofactors and catalyzes the oxidative decarboxylation of pyruvate to acetyl-CoA, the key step in glucose metabolism that allows the complete oxidation of glucose from glycolysis to the TCA cycle resulting in the greater production of ATP. Therefore, based on prior studies of brain proteins from subjects with AD in which oxidative modification led to loss of activity [50-53], oxidation of this protein likely leads to its functional impairment and consequently to decreased glucose metabolism as observed in AD [54, 55]. A previous study reported decreased activity and increased oxidation of pyruvate dehydrogenase in AD brain [56]. Further, increased levels of pyruvate and lactate have been reported in CSF of AD patients [57, 58].
One of the identified brain proteins from APP/PS1 mice which shows an age-dependence increase in oxidation is alpha-enolase (Table 1). This protein showed a significantly decreased oxidation at 6 M and 9M of age, i.e, during the early stage of the disease (Figure 2, and 3), when the brain is more plastic. In contrast, a significantly increased level of protein carbonylation of alpha-enolase at 12M and 15M was observed (Figure 4 and 5). Further, the level of protein carbonylation of enolase increases with age in APP/PS1 mice as does neuronal deposition of Aβ. It is tempting to speculate that the decreased oxidation of enolase and other proteins observed at earlier ages could be due to increased levels of carbonyl reductase, an enzyme that catalyzes reduction of protein carbonyl groups. Alternatively, this finding could be due to increased ability of the 20S proteasome to degrade oxidized proteins early in the age of the mouse. However, the increased level of specific carbonylation of enolase at 12 and 15M of age of APP/PS1 mice possibly could be explained based on the literature, which clearly documents impaired activity and function of the proteasome and also carbonyl reductase in AD brain [51, 59-61]. The observation of initial decreased oxidation of enolase suggests that models of the initial stages of the disease, 6M and 9M age of APP/PS1 mice, may reflect the plasticity and robust defense system of the brain, trying to prevent neuronal functional impairment by elevation of the levels of cellular defense systems. However, with age, we propose that an excess load of Aβ exceeds the cell’s ability to fight oxidative stress, consequently leading to increased oxidation of proteins and loss of ability to function with subsequent loss of neurons and increased AD pathology. In the case of enolase, this enzyme is well known for its involvement in glycolysis, in which it catalyzes the conversion of 2-phosphoglycerate to phosphoenolpyruvate, the second of the two-energy intermediates that generates ATP in glycolysis. However, a number of studies showed that enolase is a multifunctional protein, and is also involved in the regulation of proteolytic clearance of Aβ and induction of pro-survival pathways (see [62] for a review). Previous studies from our laboratory and others have shown that oxidative modification of α-enolase in MCI, EAD and AD results in a loss of enzyme function and, hence, in reduced amounts of ATP [24-26, 52, 62-66]. Further, increased oxidation of alpha-enolase also occurs in other models of AD [57, 67]. Several pathologies are linked to enolase-dependent pathways [68, 69].
Another protein that is involved in energy metabolism, i.e., ATP synthase α-chain, a component of Complex V of the mitochondrial electron transport chain, is also found to be oxidatively modified in these knock in mice. The oxidation of this protein is found to be significant at 12M and 15M (Figures 4 and 5). As noted above, this is the same time period when the load of amyloid beta-peptide is significantly higher in brains of these mice suggesting that this neurotoxic peptide contributes to the oxidation of these proteins [35]. Previous proteomics studies reported increased nitration of this protein in AD brain [52], and ATP synthase α-chain has previously been shown to be associated with NFT in AD [70]. Moreover, the levels of complex V have been shown to be low in isolated mitochondria from AD [71]. Both the oxidation and decreased levels of ATP synthase alpha may lead to altered interactions between the different subunits of complex V, thereby contributing to the reduced activity of ATP synthase and compromised ATP synthesis. Further, oxidation of this protein also could lead to increased generation of ROS due to an altered proton gradient and interruption created in electron flow in the electron transport chain and, if severe, could lead to neuronal death. Moreover, dysfunction of mitochondria is reported to alter APP metabolism, enhancing the intraneuronal accumulation of Aβ-peptide and enhancing neuronal vulnerability [72]. Recent studies showed the presence of oligomeric Aβ -peptide in mitochondria, and previous studies reported increased Aβ levels in brains of these mice [73-76]. Though these studies did not measure Aβ levels in subcellular compartments, we speculate that the levels of Aβ are elevated in mitochondria in brain of APP/PS1 mice, especially from 9-15 months of age, which would, consequently, trigger mitochondrial functional impairment and cell death. Our laboratory proposed the Aβ42-induced oxidative stress hypothesis of AD [3], and we propose that after the formation of Aβ42 oligomers, these oligomers are inserted into the lipid bilayer, which then initiates the process of lipid peroxidation [4]. One of the products of lipid peroxidation is 4-hydroxy 2-trans nonenal (HNE), which is highly reactive with biomolecules. In the case of mitochondria, lipid peroxidation initiated by amyloid beta-peptide might affect the structure and function of proteins by HNE modification to form Michael adducts [9]. Further, aldehydic HNE bound to proteins can introduce further carbonylation of the proteins. Therefore, it is tempting to speculate that the Aβ42 present in the mitochondrial lipid bilayer might lead to indirect oxidation of ATP synthase alpha subunit, via HNE modification, leading to elevated protein carbonyls on this protein and consequent decreased ATP production. Such alterations in the mitochondrial electron transport system and altered mitochondrial functions could lead to increased ROS [74, 76]. These alterations, in turn, can lead to alterations in membrane fluidity, leakage of apoptosis-inducing molecules such as cytochrome C and apoptosis-inducing factor from mitochondria, all of which may contribute to decreased cellular energetics, neuronal apoptosis, and generation of ROS in AD. Studies from our laboratory and others also showed decreased activities of respiratory chain enzymes and oxidation of mitochondria isolated from lymphocytes of AD patients [71, 77], consistent with the results observed in brain of APP/PS1 mice reported here.
Oxidation and subsequent loss of function of pyruvate dehydrogenase, enolase and ATP synthase alpha could result in decreased ATP production, a finding consistent with altered glucose utilization and metabolism in AD patients [78, 79]. Decreased levels of ATP clearly would affect normal cellular activities. For examples, stores of ATP at nerve terminals are key to proper neural communication. Hence, the decrease in ATP as a consequence of carbonylation of both enolase and ATP synthase alpha may contribute to loss of synapses and memory impairment and cognitive decline [80] observed in amnestic MCI, EAD and AD. In addition to synapse loss, reduced ATP levels would impair ion-motive ATPase activity with subsequent altered cell potential, loss of membrane lipid asymmetry, intra- and intercellular communication, and elevated intracellular Ca2+ [54, 81].
14-3-3 proteins were identified as oxidatively modified at 1M, 9M, 12M and 15M of age of APP/PS1 mice compared to their respective age-matched controls (Figure 1, 3,4, and 5) (Table 1). 14-3-3 Proteins play an important role in regulating various cellular functions such as signal transduction, protein trafficking and metabolism [82]. Interactions of 14-3-3 with the target proteins are important to regulate the function of these target proteins, including acting as a bridge between two target proteins, altering the intrinsic catalytic activity of the target protein, or protection of the target protein from proteolysis and dephosphorylation [83]. 14-3-3 Proteins have been shown to regulate the activity of GSK3-beta, involved in phosphorylation of tau protein. Previous studies showed that the levels of 14-3-3 proteins are increased in AD brain [84], and AD CSF [85]. Further, 14-3-3 is reported to be associated with NFT and to increase the protein kinase A (PKA)-catalyzed phosphorylation of tau protein, suggesting that 14-3-3ζ regulates microtubule assembly dynamics in the brain [83, 86].
Another brain protein identified as exclusively carbonylated at 12M of age in APP/PS1 mice compared to age matched controls is β-Synuclein (Figure 4). It is not clear why this protein is excessively carbonylated at only 12M. β-Synuclein belongs to a family of presynaptic proteins and is found in the neocortex, hippocampus, striatum, thalamus, and cerebellum regions [87, 88]. β-synuclein is associated with cholinergic components particularly in the basal forebrain [89]. The exact function of β-Synuclein is not known; however, several lines of evidence suggest that this protein acts as a chaperone-like protein in membrane-associated processes at the presynaptic terminal, and thereby is possibly involved in vesicular docking at the presynaptic terminal. The levels of β-synuclein are reportedly decreased in AD brain, and this protein is linked to Aβ in AD with Lewy body lesions [90]. Further, studies from our laboratory found β-synuclein as oxidatively modified in an Aβ-synaptosomal model of AD [91]. We also found this protein with altered glycosylation in both AD and MCI brain [92, 93]. As mentioned, if β-synuclein is involved in regulating synaptic vesicle function, oxidation of this would likely alter neuotransmission systems leading to impaired learning and memory, one of the clinical manifestations of AD.
Peptidylprolyl cis-trans isomerase-1 (Pin-1) was identified to be excessively carbonylated at age 9M, 12M and 15M in APP/PS1 mice compared to age-matched wild type (Figure 3,4, and 5). Pin1 catalyzes cis to trans isomerization and vice versa of proline on the carboxyl side of phosphorylated serine or threonine residues of target proteins, thereby playing an important role in regulating the functions of these target proteins [52, 94]. A number of studies showed that Pin1 plays important roles in tau phosphorylation/dephosphorylation, APP regulation and processing, and synapse loss [52, 94-96]. Further, in AD brain the levels of Pin-1 showed an inverse correlation with neuronal vulnerability and degeneration [97], and Pin1 also is oxidatively modified in AD hippocampus [52]. Hence, oxidative dysfunction of Pin1 conceivably could be attributed to the development of pathologies of AD such as SP, NFT, and synapse loss. Further studies by Lu et al., showed that overexpression of Pin1 leads to decreased phosphorylation of Tau protein [95], and suggests that Pin1 might be a promising therapeutic target to delay or combat this devastating disorder.
In conclusion, the present study documents that elevated Aβ is associated with selective oxidative damage of brain proteins. It is interesting to note that despite different levels of brain amyloid reported in these mice at different ages, there are some common targets of oxidation at different ages, suggesting that some proteins are easily prone to oxidation even at low Aβ levels and may be important in progression of AD. Oxidation of some proteins occurs before Aβ deposition, consistent with the notion that small Aβ oligomers are highly toxic and can induce oxidative stress [98-100]. Further, assuming that the APP/PS1 human double mutant knock-in mouse is a good model of AD, our redox proteomics results suggest that oxidation of enolase, 14-3-3 protein and Pin1 occurs early in the progression of this disease and could be important targets to prevent or delay AD this dementing disorder.
Acknowledgments
This research was supported in part by a NIH grant [AG-05119] to D.A.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mebane-Sims I. 2009 Alzheimer’s disease facts and figures. Alzheimers Dement. 2009;5:234–270. doi: 10.1016/j.jalz.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Katzman R, Saitoh T. Advances in Alzheimer’s disease. FASEB J. 1991;5:278–286. [PubMed] [Google Scholar]
- 3.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 4.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 5.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 6.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 7.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lott IT, Head E. Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol Aging. 2005;26:383–389. doi: 10.1016/j.neurobiolaging.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Butterfield DA, Bader Lange ML, Sultana R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim Biophys Acta. 2010;1801:924–929. doi: 10.1016/j.bbalip.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM, et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65:2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 11.Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 12.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- 14.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 15.Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends Mol Med. 2003;9:169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 16.Smith CD, Carney JM, Starkereed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess Brain Protein Oxidation and Enzyme Dysfunction in Normal Aging and in Alzheimer-Disease. P Natl Acad Sci USA. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winterbourn CC, Buss IH. Protein carbonyl measurement by enzyme-linked immunosorbent assay. Methods Enzymol. 1999;300:106–111. doi: 10.1016/s0076-6879(99)00118-4. [DOI] [PubMed] [Google Scholar]
- 18.Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta. 2003;329:23–38. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 19.Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003;25:207–218. doi: 10.1007/s00726-003-0011-2. [DOI] [PubMed] [Google Scholar]
- 20.Butterfield DA, Stadtman ER. Protein Oxidation processes in aging brain. Adv Cell Aging Gerontol. 1997;2:161–191. [Google Scholar]
- 21.Picklo MJ, Montine TJ, Amarnath V, Neely MD. Carbonyl toxicology and Alzheimer’s disease. Toxicol Appl Pharmacol. 2002;184:187–197. doi: 10.1006/taap.2002.9506. [DOI] [PubMed] [Google Scholar]
- 22.Smith MA, Sayre LM, Anderson VE, Harris PL, Beal MF, Kowall N, Perry G. Cytochemical demonstration of oxidative damage in Alzheimer disease by immunochemical enhancement of the carbonyl reaction with 2,4-dinitrophenylhydrazine. J Histochem Cytochem. 1998;46:731–735. doi: 10.1177/002215549804600605. [DOI] [PubMed] [Google Scholar]
- 23.Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69:155–167. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 25.Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 26.Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J Neurochem. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- 27.Reaume AG, Howland DS, Trusko SP, Savage MJ, Lang DM, Greenberg BD, Siman R, Scott RW. Enhanced amyloidogenic processing of the beta-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer’s disease mutations and a “humanized” Abeta sequence. J Biol Chem. 1996;271:23380–23388. doi: 10.1074/jbc.271.38.23380. [DOI] [PubMed] [Google Scholar]
- 28.Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG. Presenilin-1 P264L knock-in mutation: differential effects on abeta production, amyloid deposition, and neuronal vulnerability. J Neurosci. 2000;20:8717–8726. doi: 10.1523/JNEUROSCI.20-23-08717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 30.Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, St Clair DK. Beta-amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am J Pathol. 2006;168:1608–1618. doi: 10.2353/ajpath.2006.051223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bader Lange ML, St Clair D, Markesbery WR, Studzinski CM, Murphy MP, Butterfield DA. Age-related loss of phospholipid asymmetry in APP(NLh)/APP(NLh) x PS-1(P264L)/PS-1(P264L) human double mutant knock-in mice: relevance to Alzheimer disease. Neurobiol Dis. 2010;38:104–115. doi: 10.1016/j.nbd.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohmmad Abdul H, Sultana R, Keller JN, St Clair DK, Markesbery WR, Butterfield DA. Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid beta-peptide (1-42), HO and kainic acid: implications for Alzheimer’s disease. J Neurochem. 2006;96:1322–1335. doi: 10.1111/j.1471-4159.2005.03647.x. [DOI] [PubMed] [Google Scholar]
- 33.Thongboonkerd V, Luengpailin J, Cao J, Pierce WM, Cai J, Klein JB, Doyle RJ. Fluoride exposure attenuates expression of Streptococcus pyogenes virulence factors. J Biol Chem. 2002;277:16599–16605. doi: 10.1074/jbc.M200746200. [DOI] [PubMed] [Google Scholar]
- 34.Dalle-Donne I, Scaloni A, Butterfield DA. Redox Proteomics: From protein modifications to cellular dysfunction and diseases. John Wiley and Sons; Hoboken, NJ: 2006. [DOI] [PubMed] [Google Scholar]
- 35.Abdul HM, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med. 2008;45:1420–1425. doi: 10.1016/j.freeradbiomed.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 37.Carlier MF, Pantaloni D. Control of actin assembly dynamics in cell motility. J Biol Chem. 2007;282:23005–23009. doi: 10.1074/jbc.R700020200. [DOI] [PubMed] [Google Scholar]
- 38.Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010;33:121–129. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 39.Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- 40.Schubert V, Dotti CG. Transmitting on actin: synaptic control of dendritic architecture. J Cell Sci. 2007;120:205–212. doi: 10.1242/jcs.03337. [DOI] [PubMed] [Google Scholar]
- 41.Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- 42.Renner M, Specht CG, Triller A. Molecular dynamics of postsynaptic receptors and scaffold proteins. Curr Opin Neurobiol. 2008;18:532–540. doi: 10.1016/j.conb.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 43.Schlager MA, Hoogenraad CC. Basic mechanisms for recognition and transport of synaptic cargos. Mol Brain. 2009;2:25. doi: 10.1186/1756-6606-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bramham CR. Local protein synthesis, actin dynamics, and LTP consolidation. Curr Opin Neurobiol. 2008;18:524–531. doi: 10.1016/j.conb.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 45.Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23:11759–11769. doi: 10.1523/JNEUROSCI.23-37-11759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ivanov A, Esclapez M, Pellegrino C, Shirao T, Ferhat L. Drebrin A regulates dendritic spine plasticity and synaptic function in mature cultured hippocampal neurons. J Cell Sci. 2009;122:524–534. doi: 10.1242/jcs.033464. [DOI] [PubMed] [Google Scholar]
- 47.van Woerden GM, Hoebeek FE, Gao Z, Nagaraja RY, Hoogenraad CC, Kushner SA, Hansel C, De Zeeuw CI, Elgersma Y. betaCaMKII controls the direction of plasticity at parallel fiber-Purkinje cell synapses. Nat Neurosci. 2009;12:823–825. doi: 10.1038/nn.2329. [DOI] [PubMed] [Google Scholar]
- 48.Wu LJ, Ren M, Wang H, Kim SS, Cao X, Zhuo M. Neurabin contributes to hippocampal long-term potentiation and contextual fear memory. PLoS One. 2008;3:e1407. doi: 10.1371/journal.pone.0001407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newey SE, Velamoor V, Govek EE, Van Aelst L. Rho GTPases, dendritic structure, and mental retardation. J Neurobiol. 2005;64:58–74. doi: 10.1002/neu.20153. [DOI] [PubMed] [Google Scholar]
- 50.Perluigi M, Sultana R, Cenini G, Di Domenico F, Memo M, Pierce WM, Coccia R, Butterfield DA. Redox Proteomics Identification of HNE-Modified Brain Proteins in Alzheimer’s Disease: Role of Lipid Peroxidation in Alzheimer’s Disease Pathogenesis. Prot Clin Appli. 2009;l 3:682–693. doi: 10.1002/prca.200800161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol Dis. 2008;30:107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 52.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006;27:1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 53.Sultana R, Butterfield DA. Oxidatively modified GST and MRP1 in Alzheimer’s disease brain: implications for accumulation of reactive lipid peroxidation products. Neurochem Res. 2004;29:2215–2220. doi: 10.1007/s11064-004-7028-0. [DOI] [PubMed] [Google Scholar]
- 54.Planel E, Miyasaka T, Launey T, Chui DH, Tanemura K, Sato S, Murayama O, Ishiguro K, Tatebayashi Y, Takashima A. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease. J Neurosci. 2004;24:2401–2411. doi: 10.1523/JNEUROSCI.5561-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, Pirraglia E, De Santi S, Reisberg B, Wisniewski T, de Leon MJ. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2009;36:811–822. doi: 10.1007/s00259-008-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, Perrino P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch Neurol. 1988;45:836–840. doi: 10.1001/archneur.1988.00520320022009. [DOI] [PubMed] [Google Scholar]
- 57.Parnetti L, Gaiti A, Polidori MC, Brunetti M, Palumbo B, Chionne F, Cadini D, Cecchetti R, Senin U. Increased cerebrospinal fluid pyruvate levels in Alzheimer’s disease. Neurosci Lett. 1995;199:231–233. doi: 10.1016/0304-3940(95)12058-c. [DOI] [PubMed] [Google Scholar]
- 58.Parnetti L, Reboldi GP, Gallai V. Cerebrospinal fluid pyruvate levels in Alzheimer’s disease and vascular dementia. Neurology. 2000;54:735–737. doi: 10.1212/wnl.54.3.735. [DOI] [PubMed] [Google Scholar]
- 59.Cecarini V, Ding Q, Keller JN. Oxidative inactivation of the proteasome in Alzheimer’s disease. Free Radic Res. 2007;41:673–680. doi: 10.1080/10715760701286159. [DOI] [PubMed] [Google Scholar]
- 60.Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer’s disease. J Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- 61.Balcz B, Kirchner L, Cairns N, Fountoulakis M, Lubec G. Increased brain protein levels of carbonyl reductase and alcohol dehydrogenase in Down syndrome and Alzheimer’s disease. J Neural Trans. 2001:193–201. doi: 10.1007/978-3-7091-6262-0_15. [DOI] [PubMed] [Google Scholar]
- 62.Butterfield DA, Lange ML. Multifunctional roles of enolase in Alzheimer’s disease brain: beyond altered glucose metabolism. J Neurochem. 2009;111:915–933. doi: 10.1111/j.1471-4159.2009.06397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choi J, Forster MJ, McDonald SR, Weintraub ST, Carroll CA, Gracy RW. Proteomic identification of specific oxidized proteins in ApoE-knockout mice: relevance to Alzheimer’s disease. Free Radic Biol Med. 2004;36:1155–1162. doi: 10.1016/j.freeradbiomed.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 64.Korolainen MA, Goldsteins G, Nyman TA, Alafuzoff I, Koistinaho J, Pirttila T. Oxidative modification of proteins in the frontal cortex of Alzheimer’s disease brain. Neurobiol Aging. 2006;27:42–53. doi: 10.1016/j.neurobiolaging.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 65.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 66.Sultana R, Reed T, Perluigi M, Coccia R, Pierce WM, Butterfield DA. Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: a regional study. J Cell Mol Med. 2007;11:839–851. doi: 10.1111/j.1582-4934.2007.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, Klein JB, Butterfield DA. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1-42) into rat brain: implications for Alzheimer’s disease. Neurosci. 2005;132:313–324. doi: 10.1016/j.neuroscience.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 68.Opii WO, Nukala VN, Sultana R, Pandya JD, Day KM, Merchant ML, Klein JB, Sullivan PG, Butterfield DA. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. J Neurotrauma. 2007;24:772–789. doi: 10.1089/neu.2006.0229. [DOI] [PubMed] [Google Scholar]
- 69.Ko AC, Hernandez J, Brinton JP, Faidley EA, Mugge SA, Mets MB, Kardon RH, Folk JC, Mullins RF, Stone EM. Anti-gamma-enolase autoimmune retinopathy manifesting in early childhood. Arch Ophthalmol. 2010;128:1590–1595. doi: 10.1001/archophthalmol.2010.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sergeant N, Wattez A, Galvan-valencia M, Ghestem A, David JP, Lemoine J, Sautiere PE, Dachary J, Mazat JP, Michalski JC, Velours J, Mena-Lopez R, Delacourte A. Association of ATP synthase alpha-chain with neurofibrillary degeneration in Alzheimer’s disease. Neurosci. 2003;117:293–303. doi: 10.1016/s0306-4522(02)00747-9. [DOI] [PubMed] [Google Scholar]
- 71.Molina JA, de Bustos F, Jimenez-Jimenez FJ, Benito-Leon J, Gasalla T, Orti-Pareja M, Vela L, Bermejo F, Martin MA, Campos Y, Arenas J. Respiratory chain enzyme activities in isolated mitochondria of lymphocytes from patients with Alzheimer’s disease. Neurology. 1997;48:636–638. doi: 10.1212/wnl.48.3.636. [DOI] [PubMed] [Google Scholar]
- 72.Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, Yankner BA. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron. 2002;33:677–688. doi: 10.1016/s0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- 73.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 74.Reddy PH. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp Neurol. 2009;218:286–292. doi: 10.1016/j.expneurol.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ankarcrona M, Mangialasche F, Winblad B. Rethinking Alzheimer’s disease therapy: are mitochondria the key? J Alzheimers Dis. 2010;20(Suppl 2):S579–590. doi: 10.3233/JAD-2010-100327. [DOI] [PubMed] [Google Scholar]
- 76.Sultana R, Butterfield DA. Oxidatively modified, mitochondria-relevant brain proteins in subjects with Alzheimer disease and mild cognitive impairment. J Bioenerg Biomembr. 2009;41:441–446. doi: 10.1007/s10863-009-9241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sultana R, Mecocci P, Mangialasche F, Cecchetti R, Baglioni M, Butterfield DA. Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer disease: Insights into the role of oxidative stress in ad and initial investigations into a potential biomarker for this dementing disorder. J Alz Dis. 2011 doi: 10.3233/JAD-2011-101425. In Press. [DOI] [PubMed] [Google Scholar]
- 78.Messier C, Gagnon M. Glucose regulation and cognitive functions: relation to Alzheimer’s disease and diabetes. Behav Brain Res. 1996;75:1–11. doi: 10.1016/0166-4328(95)00153-0. [DOI] [PubMed] [Google Scholar]
- 79.Vanhanen M, Soininen H. Glucose intolerance, cognitive impairment and Alzheimer’s disease. Curr Opin Neurol. 1998;11:673–677. doi: 10.1097/00019052-199812000-00011. [DOI] [PubMed] [Google Scholar]
- 80.Hoyer S. Memory function and brain glucose metabolism. Pharmacopsychiatry. 2003;36(Suppl 1):S62–67. doi: 10.1055/s-2003-40452. [DOI] [PubMed] [Google Scholar]
- 81.Chen Y, Jungsuwadee P, Vore M, Butterfield DA, St Clair DK. Collateral damage in cancer chemotherapy: oxidative stress in nontargeted tissues. Mol Interv. 2007;7:147–156. doi: 10.1124/mi.7.3.6. [DOI] [PubMed] [Google Scholar]
- 82.Dougherty MK, Morrison DK. Unlocking the code of 14-3-3. J Cell Sci. 2004;117:1875–1884. doi: 10.1242/jcs.01171. [DOI] [PubMed] [Google Scholar]
- 83.Takahashi Y. The 14-3-3 proteins: gene, gene expression, and function. Neurochem Res. 2003;28:1265–1273. doi: 10.1023/a:1024296932670. [DOI] [PubMed] [Google Scholar]
- 84.Fountoulakis M, Cairns N, Lubec G. Increased levels of 14-3-3 gamma and epsilon proteins in brain of patients with Alzheimer’s disease and Down syndrome. J Neural Trans. 1999;57:323–335. doi: 10.1007/978-3-7091-6380-1_23. [DOI] [PubMed] [Google Scholar]
- 85.Burkhard PR, Sanchez JC, Landis T, Hochstrasser DF. CSF detection of the 14-3-3 protein in unselected patients with dementia. Neurology. 2001;56:1528–1533. doi: 10.1212/wnl.56.11.1528. [DOI] [PubMed] [Google Scholar]
- 86.Fu H, Subramanian RR, Masters SC. 14-3-3 proteins: structure, function, and regulation. Annu Rev Pharmacol Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- 87.Clayton DF, George JM. Synucleins in synaptic plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:120–129. [PubMed] [Google Scholar]
- 88.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li JY, Henning Jensen P, Dahlstrom A. Differential localization of alpha-, beta- and gamma-synucleins in the rat CNS. Neurosci. 2002;113:463–478. doi: 10.1016/s0306-4522(02)00143-4. [DOI] [PubMed] [Google Scholar]
- 90.Rockenstein E, Hansen LA, Mallory M, Trojanowski JQ, Galasko D, Masliah E. Altered expression of the synuclein family mRNA in Lewy body and Alzheimer’s disease. Brain Res. 2001;914:48–56. doi: 10.1016/s0006-8993(01)02772-x. [DOI] [PubMed] [Google Scholar]
- 91.Boyd-Kimball D, Sultana R, Poon HF, Mohmmad-Abdul H, Lynn BC, Klein JB, Butterfield DA. Gamma-glutamylcysteine ethyl ester protection of proteins from Abeta(1-42)-mediated oxidative stress in neuronal cell culture: a proteomics approach. J Neurosci Res. 2005;79:707–713. doi: 10.1002/jnr.20393. [DOI] [PubMed] [Google Scholar]
- 92.Owen JB, Di Domenico F, Sultana R, Perluigi M, Cini C, Pierce WM, Butterfield DA. Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer’s disease and mild cognitive impairment: implications for progression of AD. J Proteome Res. 2009;8:471–482. doi: 10.1021/pr800667a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Di Domenico F, Owen JB, Sultana R, Sowell RA, Perluigi M, Cini C, Cai J, Pierce WM, Butterfield DA. The wheat germ agglutinin-fractionated proteome of subjects with Alzheimer’s disease and mild cognitive impairment hippocampus and inferior parietal lobule: Implications for disease pathogenesis and progression. J Neurosci Res. 2010;88:3566–3577. doi: 10.1002/jnr.22528. [DOI] [PubMed] [Google Scholar]
- 94.Butterfield DA, Abdul HM, Opii W, Newman SF, Joshi G, Ansari MA, Sultana R. Pin1 in Alzheimer’s disease. J Neurochem. 2006;98:1697–1706. doi: 10.1111/j.1471-4159.2006.03995.x. [DOI] [PubMed] [Google Scholar]
- 95.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 96.Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 97.Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, Hunter T, Lu KP. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- 98.Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- 99.Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- 100.Klein WL. Synaptic targeting by A beta oligomers (ADDLS) as a basis for memory loss in early Alzheimer’s disease. Alzheimers Dement. 2006;2:43–55. doi: 10.1016/j.jalz.2005.11.003. [DOI] [PubMed] [Google Scholar]