

Abstract
Background
Loss of expression of the desmosomal protein plakophilin-2 (PKP2) leads to decreased gap junction-mediated (GJ) coupling, and alters the amplitude and kinetics of sodium current in cardiac myocytes. Whether these modifications, alone or in combination, are sufficient to act as arrhythmogenic substrates, remains undefined.
Objective
To characterize arrhythmia susceptibility and reentry dynamics consequent to loss of PKP2 expression, and assess the relative contribution of cell uncoupling versus alterations in sodium current in generation of reentry.
Methods
Monolayers of neonatal rat ventricular myocytes were treated with oligonucleotides that either prevented, or failed to prevent PKP2 expression. Numerical simulations modeled experimentally-observed modifications in INa, GJ coupling, or both (models PKP2-Na, PKP2-GJ and PKP2-KD, respectively). Relative roles of sodium current density vs kinetics were further explored.
Results
Loss of PKP2 expression increased incidence of rotors and decreased frequency of rotation. Mathematical simulations revealed that single premature stimuli initiated rotors in models PKP2-Na and PKP2-KD, but not PKP2-GJ. Changes in sodium current kinetics, rather than current density, were key to reentry initiation. Anatomical barriers led to vortex shedding, wavebreaks and rotors when INa kinetics, but not GJ coupling or INa density, were altered.
Conclusions
PKP2-dependent changes in sodium current kinetics lead to slow conduction, increased propensity to functional block, and vortex shedding. Changes in GJ or INa density played only a minor role on reentry susceptibility. Changes in electrical properties of the myocyte caused by loss of expression of PKP2 can set the stage for rotors even if anatomical homogeneity is maintained.
Keywords: plakophilin-2, functional reentry, computer modeling, vortex shedding
Introduction
Plakophilin-2 (PKP2) is as an integral part of the desmosome and, in addition, a scaffolding protein for cell signaling molecules.1,2 Previous studies have shown that mutations in desmosomal proteins, including PKP2, lead to gap junction remodeling,3,4,5 and that loss of PKP2 expression causes a decrease in gap junction mediated coupling between cells.6 More recently we demonstrated that loss of PKP2 expression also leads to a decrease in amplitude and a shift in kinetics of the sodium current in adult ventricular myocytes.7 Mutations associated with changes in pkp2 are identified in over 50% of cases of familial arrhythmogenic right ventricular cardiomyopathy (ARVC),8 an inherited arrhythmogenic disease characterized by ventricular tachyarrhythmias, sudden cardiac death, and replacement of the myocardium with fibroid and fatty tissue.9 The life-threatening arrhythmic episodes in ARVC often occur in its concealed phase, in the absence of extensive structural damage.3 Yet, whether the observed modifications in junctional coupling and sodium current, either alone or in combination, are sufficient to act as an arrhythmogenic substrate, remains to be defined.
Our general hypothesis is that changes in the molecular integrity of the desmosome may disrupt molecular complexes relevant to electrical function sufficiently to initiate and maintain reentrant activity. Here, we utilized a combination of optical mapping experiments and numerical simulations to characterize changes in arrhythmia susceptibility and reentry dynamics consequent to loss of PKP2 expression. Our results support the hypothesis that changes in cellular electrophysiology associated with loss of PKP2 expression can be arrhythmogenic. Our data further indicate that changes in sodium current kinetics (rather than gap junction coupling or sodium current density) play a key role in the initiation of the arrhythmic event. Our studies lead us to speculate that functional reentry can be a primary mechanism for arrhythmias consequent to loss or mutations in the PKP2 protein.
Methods
Detailed experimental methods are provided in the Online Supplement. Briefly, monolayers of NRVMs were either left untreated (UNT), treated with an adenovirus containing a non-silencing construct (φshRNA), or treated with a PKP2 silencing construct (shRNA). Propagation was studied by conventional optical mapping.7
Numerical simulations were performed utilizing models based on ionic currents recorded from both neonatal and adult cardiomyocytes. Propagation was studied in 2D arrays. The behavior of neonatal rat ventricular myocytes (NRVM) was simulated based on the new Deo model.11 The electrical behavior of adult myocytes was based on the Luo-Rudy10 model, modified as follows (see Table 1): i) Changes in INa density and kinetics to match those observed in cardiac myocytes after loss of PKP2 expression (PKP2-Na).7 ii) 60% reduction in cell-cell electrical coupling6 without changes in sodium current properties (PKP2-GJ). iii) 60% reduction in cell-cell electrical coupling and PKP2-dependent INa alterations (PKP2-KD).6,7 Results obtained with the PKP2-Na model were further dissected by separating the effect of 50% reduction in INa density only (kinetics kept as in control; model called “PKP2-Na-amp”) from that resulting from PKP2-related alterations in INa inactivation and recovery kinetics, while sodium current density was maintained as control (PKP2-Na-kntx).
Table 1.
| Model condition | 100% coupling | 40% coupling | INa density reduction | INa inactivation shift | INa recovery from inactivation slowing | |
|---|---|---|---|---|---|---|
| i | Control | + | ||||
| ii | PKP2-Na | + | + | + | + | |
| iii | PKP2-GJ | + | ||||
| iv | PKP2-KD | + | + | + | + | |
| v | PKP2-Na-amp | + | + | |||
| vi | PKP2-Na-kntx | + | + | + | ||
Results
Reentrant activity in cardiac cells after loss of PKP2 expression
Loss of PKP2 led to a significant increase in the incidence of reentrant activity in monolayers of NRVMs. Monolayers were either kept untreated in culture (UNT), or treated with adenoviral particles. Viruses contained oligonucleotides that either did not affect PKP2 expression (φshRNA), or caused a reduction in PKP2 abundance (shRNA). For all experiments, PKP2 expression levels were tested by western blot on the same cells undergoing optical mapping (Supplemental Figure 1). In all cases, shRNA treatment led to a reduction of >80% of PKP2 when compared to either UNT or φshRNA-treated cells. Figure 1A shows sample phase maps demonstrating functional reentry (rotors) in the three groups tested. Rotors were initiated spontaneously, or following pacing to up to 10 Hz.7 These rotors were observed in only 1 out of 9 UNT and 2 out of 12 φshRNA-treated preparations. The number rose to 4 out of 8 in shRNA cells (Panel B, p<0.05). Reentrant activity was sustained for the duration of the recordings for all groups. Dominant frequency (DF) analysis (panel C) indicated that the characteristics of reentrant activity were modified by loss of PKP2 expression; indeed, the DF of reentry decreased from 12.82±0.34 Hz in untreated cells (Mean±SEM; n=6) and 10.89±1.47 Hz in φshRNA-treated cells (n=4) to 6.94±1.4 Hz (n=6) following loss of PKP2 expression (p<0.001 UNT vs shRNA; p<0.001 φshRNA vs shRNA). These data suggest that, in monolayers of NRVMs, loss of PKP2 expression is conducive to reentry initiation and maintenance even in the absence of anatomical obstacles.
Figure 1.

Optical mapping in monolayers of neonatal rat ventricular myocytes. A) Representative phase maps show reentry in monolayers of untreated (UNT), non-silenced (φshRNA), or PKP2-silenced (shRNA) cells. Scale bar: 13.9 mm. B) Incidence of spontaneous or pacing-induced reentry. C) Dominant frequency of rotors. UNT vs shRNA, p<0.001*. φshRNA vs shRNA, p<0.001*. UNT vs φshRNA, not significant (p>0.05).
Modeling the effects of PKP2 knockdown in NRVMs
To better establish an analogy between simulated and experimental data, an NRVM-specific model was implemented (details in online supplement and in Ref 11). Sodium and gap junction current parameters were modified as per patch clamp data obtained in our laboratory (Supplemental Figure 2A, and Ref. 6). Conduction velocity measurements in the NRVM model agreed with those obtained from biological NRVM preparations (Supplemental Figure 2B and Figure 3 of Ref. 7). Finally, simulations presented in Supplemental Figure 2C and Supplemental Figure 3 show a decrease in rotation frequency of reentry, and a prolongation of action potential duration (APD80) during reentry in the PKP2-KD model, similar to that recorded in the experimental preparation (compare with Figure 1 and Supplemental Figure 3A and 3C). Overall, these results show that changes in electrophysiology of NRVM monolayers consequent to loss of PKP2 expression are closely reproduced in the mathematical model when sodium and gap junction currents are modified as per experimental results.
Modeling the effects of PKP2 knockdown in adult myocytes
The adequate correlation between experimental and numerical models led us to explore, using the mathematical model, aspects of cardiac propagation and arrhythmogenesis that could not be assessed in biological preparations due to technical limitations. First, we characterized whether changes in conduction velocity in NRVM monolayers would be reproduced in a system where adult (rather than neonatal) myocytes were modeled. A mathematical description of altered sodium current was created based on results obtained from adult myocytes treated with shRNA for PKP2.7 Figure 2 shows the peak current-voltage relationship, steady-state inactivation and time dependent recovery from inactivation of INa as formulated for the computer model (panel A), or recorded experimentally from adult cardiac myocytes7 (panel B). Blue and red lines in B depict data obtained from control cells, and from cells recorded after loss of PKP2 expression, respectively. Data obtained from numerical simulations either in control (blue) or in a model incorporating PKP2-related changes in sodium and gap junction currents (PKP2-KD) are depicted in A. Experimental data were obtained at room temperature. The function describing the voltage-dependence of steady-state inactivation was therefore adjusted to that predicted for physiological temperatures (37°C) using a temperature compensation factor (Q10).10 The model adequately reproduced the INa characteristics recorded experimentally and was used to study action potential morphology and propagation in a simulated monolayer of cardiac myocytes. Of note, rate-dependent changes in APD80 were similar in the biological and the numerical experiment (Figure 3). In neither case did we observe PKP2-KD-related changes in action potential duration (compare blue and red lines in Figure 3).
Figure 2.

Model of experimentally-observed PKP2-dependent effects on sodium current. A) Voltage clamp data from adult cardiomyocytes (reproduced from Ref. 7; adjusted to 37°C) show approximately 50% reduction in sodium current density, a negative shift in voltage dependence of steady state inactivation and a prolongation of time-dependence of recovery from inactivation in PKP2 knockdown (PKP2-KD) cells. B) Experimentally-observed INa alterations were accurately reproduced by the modified single cell computer model. V1/2: potentials at half inactivation.
Figure 3.

APD80 analysis during pacing in A) experimental monolayers and B) computer models. APDs decreased with increasing frequency in all cases.
Figure 4A shows numerical simulation results aimed at characterizing action potential morphology during 1 Hz pacing in a monolayer of adult myocytes. Upstroke velocity (dV/dt) decreased significantly from 70 mV/ms in control to 34 mV/ms in model PKP2-Na. As a result, maximum AP amplitude (Vmax) occurred approximately 15 ms later in the PKP2-Na model when compared to control (inset). We further dissociated PKP2-Na effects by individually simulating reduction in INa density alone (PKP2-Na-amp) versus alterations in INa kinetics while maintaining current density values as control (PKP2-Na-kntx). As expected, upstroke velocity in PKP2-Na-amp (37 mV/ms) and PKP2-Na-kntx (40 mV/ms) were intermediate between those in control and PKP2-Na models. As a next step, we characterized rate-dependent changes in conduction velocity in the various models under study.
Figure 4.

A) Representative AP traces elicited by 1 Hz pacing stimuli in computer model showing slower AP Upstrokes when models PKP2-Na-amp, PKP2-Na-kntx or PKP2-Na where implemented. Inset: AP upstroke. B) Conduction velocity (CV) obtained in 2D models at varying pacing frequencies. Note that CV was reduced due to PKP2-dependent sodium alterations (PKP2-Na and PKP2-KD) compared to gap junction reduction (PKP2-GJ). Rate-dependent decrease in CV was mostly related to changes in sodium current kinetics, rather than peak current amplitude.
Figure 4B shows CV values in all six conditions at varying pacing frequencies, but with similar stimuli strength. Irrespective of pacing frequencies, CV was reduced significantly in the PKP2-Na model when compared to control (8.5±3.5 cm/s for 1–4 Hz; control: 25.0 cm/s). CV decreased even further (5.8±2.7 cm/s for 1–4 Hz) when electrical coupling was also reduced (PKP2-KD). This condition qualitatively reproduced that observed in biological and numerical experiments on NRVMs (see Ref. 7 and Supplemental Figure 2). Moreover, in model PKP2-Na-kntx, CV was significantly reduced from 22.7 cm/s at 1 Hz to 6 cm/s at 4 Hz. In contrast, a more moderate reduction in CV was observed when only electrical coupling (PKP2-GJ; 17.0 cm/s) or only INa density (PKP2-Na-amp; 19.6 cm/s) was reduced. An increase in pacing rate up to 5 Hz did not cause significant changes in CV in control, PKP2-GJ and PKP2-Na-amp models; however, conditions that included alterations in INa kinetics (PKP2-Na, PKP2-KD and PKP2-Na-kntx) resulted in a frequency-dependent reduction in CV with failure of propagation above 4 Hz (red, orange and pink lines in Figure 4B). These results suggest that changes in sodium current kinetics are primarily responsible for the rate-dependent changes in conduction velocity observed experimentally. Whether this would translate into differences in arrhythmia susceptibility was analyzed in the simulations described below.
Reentry by premature stimulus
In the next phase, we explored the characteristics of reentrant excitation in monolayers of adult model cells. The stimulation protocol was similar to that used to generate reentrant activity in the NRVM model (Supplemental Figure 2). Changes in density and kinetics of sodium current (PKP2-Na) led to a considerable reduction in frequency of rotation, from 10.42 Hz in control to 3.33 Hz (Compare Figure 5A with 5B). In contrast, a reduction in electrical coupling (PKP2-GJ) caused only a minor change in dominant frequency (Figure 5C; DF=9.3 Hz). As such, changes in sodium current were considered the key mechanism for the reduction in DF observed in the PKP2-KD model (2.95 Hz; 5D). Moreover, as illustrated in 5E, the major component responsible for the decrease in DF in the PKP2-Na model was the modification in sodium channel kinetics, rather than current amplitude (compare results with model PKP2-Na-kntx to those in model PKP2-Na-amp).
Figure 5.

Spiral wave reentry initiated by S1–S2 crossfield protocol in 2D computer models for: A) control, B) PKP2-Na, C) PKP2-GJ and D) PKP2-KD. Dominant frequencies for six cases are summarized in panel E. Note that frequency of rotor decreased slightly in PKP2-GJ and PKP2-Na-amp, but significantly in PKP2-Na-kntx, PKP2-Na and PKP2-KD. Scale bars: 100 ms.
Separate simulations assessed reentry initiation by an S1–S2 protocol applied at the same stimulus site and field direction. Here we tested the hypothesis that PKP2-dependent changes in sodium current kinetics can reduce excitability in a rate-dependent manner, leading to arrhythmias. Figure 6 shows the results. In panel A, a control monolayer was paced regularly (S1; 2 Hz), followed by a single premature stimulus (S2) 200 ms after the last S1. Action potential duration of the premature beat was significantly abbreviated, and propagated through the monolayer (see successive frames in 6A). Stimuli applied at shorter intervals failed to induce a propagated response. Changes in sodium current properties drastically altered the dynamics of excitation. In this case, propagation of the premature beat was partially blocked by the tail of the previous excitation wave near the boundaries (panel B at 340 ms). This allowed for propagation of the S2 response back into non-refractory areas, and development of figure-of-eight reentry into newly excitable tissue (see 450 and 550 ms panels). Supplemental Figure 4 shows an APD80 isochrone map following the last S1 wave. Notice the prolonged APD of the S1 beat near the boundary, relative to that in the center, leading to a shorter excitable-gap, and functional conduction block of S2 at that site. The APD prolongation is attributed to both increased curvature of the tail and the no-flux boundary conditions, both reducing the electrotonic repolarizing currents imposed on the tail (see Supplementary Discussion). The tail curvature- and boundary-induced conduction block was not unique to the dish geometry, as evidenced by the blockade of an expanding wave in a square-shaped tissue of PKP2-KD (Supplemental Figure 5). On the other hand, planar and circular waves with uniform curvatures along their fronts and tails (which, we speculate, are unlikely to occur in the heart) did not produce reentry, as they did not lead to heterogeneity in APD and excitable–gap (not shown).
Figure 6.

Reentry induction by a single premature beat. The model was paced at 1 Hz (S1) followed by a premature stimulus (S2) using the same pacing electrode (shown in red). A) Control: The premature beat was either completely blocked (not shown) or propagated throughout. B) PKP2-Na: Conduction of premature beat was blocked at the edges, thus initiating reentry (see frames 340–450 ms). A sustained reentry can be seen in successive frames.
Vulnerability to reentry
The protocol described in Figure 6 was used to define susceptibility to either propagation (P), conduction block (CB) or reentry (R) at various S1–S2 intervals (Figure 7). Uncoupling alone only shifted the S1–S2 interval at which action potentials failed to propagate, but did not create a window of vulnerability for reentry. In contrast, changes in sodium current properties greatly increased reentry susceptibility, and the range of S1–S2 intervals at which reentry occurred increased only slightly when changes in electrical coupling were further incorporated into the model. Dissociation of PKP2-Na alterations revealed that reduction in INa amplitude alone (PKP2-Na-amp) did not produce a vulnerability window. However, a window was observed for S1–S2 intervals between 180 and 200 ms when changes in sodium channel kinetics were incorporated into the model (see Figure 7). Overall, these results suggest that a) changes in ionic current properties elicited by loss of PKP2 expression are arrhythmogenic and b) disruption of sodium current kinetics represents the key contributor to arrhythmia generation in this model.
Figure 7.

Window of vulnerability in barrier-free 2D models. Pacing protocol as in Figure 6. Control, PKP2-GJ and PKP2-Na-amp monolayers showed conduction block (CB) or uninterrupted propagation (P) of premature beat for all S1–S2 intervals, with no reentry formation. In PKP2-Na and PKP2-Na-kntx, reentry (R) was formed for a range of S1–S2 intervals. Window of intervals leading to reentry (R) was wider in PKP2-KD.
Fibrosis, vortex shedding and PKP2 down-regulation
Non-myocyte infiltrates are common in hearts with PKP2 mutations.1,9 Here, we assessed the possible relevance of myofibroblast infiltrates on arrhythmogenesis. A 10% and 25% of the area of a monolayer was assigned to non-excitable, poorly coupled cells (“myofibroblasts”; see Ref. 24). Addition of non-myocyte cells reduced CV in both control and PKP2-KD models. The reduction in CV was higher in PKP2-KD (41%–67% reduction with 10%–25% myofibroblast density, respectively) than in Control (35%–58% with 10%–25% myofibroblast density, respectively). As seen in Supplemental Figure 6, the presence of simulated myofibroblasts led to fragmentation of the propagating wavefronts; wavebreaks or reentry were observed for model PKP2-KD, but not for control. The latter emphasizes the importance of modifications on ionic currents on the generation of arrhythmias in PKP2-deficient cell systems.
PKP2 deficiency leads to replacement of myocardial mass with fibrous and adipose tissue.9 Additional simulations investigated the effect of changes in sodium and/or gap junction currents on action potential propagation around anatomical obstacles. In control (Figure 8A), a wavefront reached the barrier, propagated along its margins, curved around its sharp edge and continued on the side opposite to the site of initiation. However, in PKP2-Na, the wavefront failed to wrap around the obstacle given the sink-source mismatch provoked by the high curvature at the tip of the wavefront (Figure 8B). As a result, the wavefront detached from the edge and moved forward and away from the obstacle (i.e., vortex shedding). Figure 8C shows another example where an excitation wavefront was interrupted by an obstacle placed amidst excitable cells. In this case, two separate waves were generated as a result of detachment. The fronts curved and faced back toward the area of origin, though did not complete a reentry circuit due to the limited availability of excitable tissue (panel 850 ms). However, placing the area of block on a different position, provided enough substrate area for rotation of the wavefront and sustainment of reentry (see Figure 8D). Interestingly, reduction in sodium current density alone (PKP2-Na-amp) did not produce the vortex shedding effect despite reduction in excitatory current. On the other hand, modifications in INa kinetics (PKP2-Na-kntx) reproduced the wave-detachment phenomena due to vortex shedding. Thus, reduced excitability consequent to alterations in INa kinetics (PKP2-Na and PKP2-Na-kntx model) formed an arrhythmogenic substrate and initiated sustained reentry when confronted with conduction block. Additional changes in electrical coupling did not modify this propagation pattern (PKP2-KD), suggesting that, as in the case of reentry initiated by an S1–S2 protocol (Figures 6 and 7), modification of INa kinetics is the major player in arrhythmogenesis under our experimental conditions.
Figure 8.
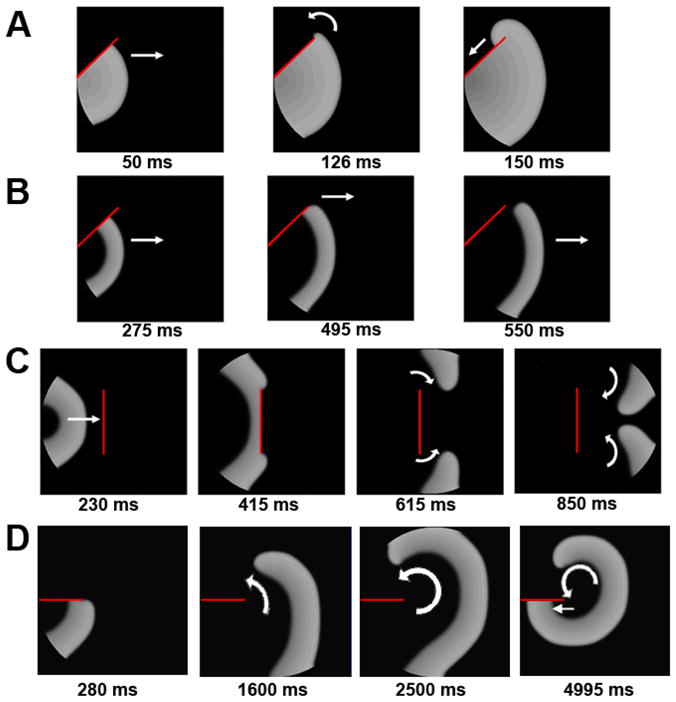
Vortex shedding at the edge of a conduction barrier in 2D models. A) Control: wavefront wraps around the barrier (red line) and continues to propagate. B) PKP2-Na: wavefront detaches at the edge of barrier (vortex shedding). C) Wave-detachment when confronted with a perpendicular barrier in PKP2-Na. D) Sequential beats propagate beyond the barrier in PKP2-Na, eventually leading to reentry.
Discussion
In previous studies, we demonstrated that loss of expression of the desmosomal protein PKP2 leads to changes in gap junction-mediated coupling, and in sodium current properties.6,7 Experiments in monolayers of NRVMs presented here show that loss of PKP2 expression resulted in increased incidence of reentry and a decrease in DF of activation. Numerical simulations further showed that PKP2 knockdown-related changes in sodium current caused a significant reduction in AP upstroke velocity and a decrease in conduction velocity; these changes were only moderately affected if combined with changes in electrical coupling between cells. Moreover, mathematical models showed that PKP2-KD-related changes in sodium current kinetics, and not density, facilitated reentry initiation by a conduction delay and/or vortex shedding, both of which can lead to wavebreaks and rotors. Overall, we present, for the first time, evidence that modifications in ion currents resulting from loss of PKP2 expression constitute a sufficient condition for increased arrhythmogenesis, primarily due to reentrant activity, in a two-dimensional preparation of cardiac cells.
Clinical perspective
Previous studies on ventricular tachycardia associated with ARVC have suggested that the arrhythmia results from anatomical reentrant circuits around damaged tissue.12,13 Here we show that PKP2-dependent alterations in sodium current can provide an arrhythmogenic substrate capable of initiating and maintaining functional reentry in the absence of any structural heterogeneity. Whether this is relevant to the origin of arrhythmias in the concealed phase of ARVC3 is an issue that deserves further investigation. Our results do support the notion of a link between desmosomal integrity and reentry caused by modifications in electrical properties of individual myocytes. Although modifications in sodium current amplitude and kinetics were based on experimental observations obtained following loss of PKP2 expression, loss of sodium channel function is found in various other inherited and acquired diseases.14–18 Whether results presented here can be extended to cases such as Brugada syndrome, idiopathic VF, or other conditions, remains to be determined.
Mechanisms of propagation and reentry initiation
Previous experiments7 have suggested a cross-talk between three components of the intercalated disc: desmosomes, gap junctions and the voltage-gated sodium channel complex. Our simulations show that uniform reduction in electrical coupling alone was not arrhythmogenic; however, alterations in sodium current kinetics form a reentry-prone substrate due to a severe frequency-dependent reduction in excitability characteristics leading to wave blocks at sites with high curvature near boundaries and barriers (see Supplementary Discussion). Our data are in agreement with previous studies showing that CV in cardiac tissue does not change appreciably when connexin43 content is decreased by 50%,19 whereas disorders of the sodium channel can be severely arrhythmogenic.20
Wave-detachments at a non-excitable barrier were not observed in model PKP2-GJ. This may be consequent, at least in part, to the paradoxical improvement in margin of safety for propagation that results from a mild decrease in electrical coupling, as originally proposed by Spach et al.21 In contrast to what happens when electrical coupling is modified, the safety factor for propagation decreases monotonically as sodium channel availability is reduced.22 Our simulations were carried out in homogenous preparations. In that condition, reducing INa amplitude by 50% (PKP2-Na-amp) did not achieve the extent of functional heterogeneity in refractoriness necessary to initiate reentry that was present, on the other hand, when INa kinetics were altered.
Our data show that when sodium current is impaired (PKP2-Na and PKP2-KD), excitation waves can detach from sharp boundaries, leading to wavebreaks (see Figure 8). Detached wavefronts may or may not initiate spiral waves depending upon availability of re-excitable tissue. Notably, reducing gap junctions alone (PKP2-GJ) or sodium current density alone (PKP2-Na-amp) did not produce vortex shedding, consistent with previous studies in tetrodotoxin-treated sheep epicardium.23
Limitations
Our experimental models depart significantly from conditions present in ARVC-afflicted hearts. Moreover, complete loss of PKP2 expression is not a clinical finding, and we do not know whether reduction in PKP2 gene dose (as may be the case in loss of function mutations) or expression of mutant PKP2 proteins, would exert the same effects as those described here. Further studies are necessary to determine whether loss of PKP2 expression is associated with changes in ionic currents other than INa. Occurrence of structural heterogeneities such as fibrosis would further worsen the arrhythmogenic substrate, an event that needs to be investigated more systematically. Additionally, use of a continuum-based computer model properly simulated the condition present in a two-dimensional preparation of myocytes lacking directionality of coupling and displaying isotropic propagation. Translation of these results to a model simulating the regular distribution of intercalated discs, with realistic wave initiation and anisotropic propagation in an adult heart tissue, remains a future next step in our study. These limitations notwithstanding, our data allow us to speculate that modifications in cellular electrophysiology due to loss of interactions between desmosomal and ion channel properties can create an arrhythmogenic substrate, even in the absence of anatomical obstacles.
Conclusions
We have investigated the individual and combined effects of altered sodium current amplitude and kinetics, and reduced gap junction coupling, on arrhythmogenesis caused by PKP2 knockdown. Within the confines of our experimental and computer models, we have shown that a) loss of PKP2 expression resulted in increased propensity for functional reentry, b) reduction in electrical coupling alone, or sodium current amplitude alone was not sufficient to initiate arrhythmias, but PKP2-dependent alterations in sodium current kinetics were key to the formation of the arrhythmogenic substrate and c) increased arrhythmogenicity was primarily consequent to impaired excitability, leading to functional propagation block, vortex shedding and wavebreaks. Overall, our data support the hypothesis that changes in the properties of individual myocytes, consequent to loss PKP2 expression, can create a substrate for functional reentry. Our results warrant further investigation on the role of sodium currents on the initiation of arrhythmias in patients afflicted with PKP2-related heart disease.
Supplementary Material
Acknowledgments
Sources of financial support: Supported by NIH/NHLBI grants P01-HL039707 and P01-HL087226, a Leducq Foundation Transatlantic Network, and the American Heart Association.
Abbreviations
- PKP2
Plakophilin-2
- NRVM
Neonatal rat ventricular myocytes
- UNT
Untreated monolayers of NRVMs
- shRNA
Monolayers of NRVMs treated with an oligonucleotide that prevents PKP2 expression
- φshRNA
Monolayers of NRVMs treated with an oligonucleotide that does not prevent PKP2 expression
- PKP2-Na
Model incorporating PKP2-dependent alterations in sodium current
- PKP2-GJ
Model incorporating 60% reduction in cell-to-cell coupling
- PKP2-KD
Model incorporating PKP2-dependent alterations in sodium current as well as 60% reduction in cell-to-cell coupling
- PKP2-Na-amp
Model incorporating PKP2-dependent reduction in sodium current density alone
- PKP2-Na-kntx
Model incorporating PKP2-dependent alterations in sodium current inactivation and recovery kinetics without reducing the current density
- DF
Dominant frequency
Footnotes
Conflicts of interest:
None
References
- 1.Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107:700–14. doi: 10.1161/CIRCRESAHA.110.223412. [DOI] [PubMed] [Google Scholar]
- 2.Bass-Zubek AE, Hobbs RP, Amargo EV, et al. Plakophilin 2: a critical scaffold for PKC alpha that regulates intercellular junction assembly. J Cell Biol. 2008;181:605–13. doi: 10.1083/jcb.200712133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplan SR, Gard JJ, Protonotarios N, et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease) Heart Rhythm. 2004;1:3–11. doi: 10.1016/j.hrthm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan SR, Gard JJ, Carvajal-Huerta L, et al. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol. 2004;13:26–32. doi: 10.1016/S1054-8807(03)00107-8. [DOI] [PubMed] [Google Scholar]
- 5.Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 6.Oxford EM, Musa H, Maass K, et al. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ Res. 2007;101:703–11. doi: 10.1161/CIRCRESAHA.107.154252. [DOI] [PubMed] [Google Scholar]
- 7.Sato PY, Musa H, Coombs W, et al. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105:523–6. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Tintelen JP, Entius MM, Bhuiyan ZA, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–1658. doi: 10.1161/CIRCULATIONAHA.105.609719. [DOI] [PubMed] [Google Scholar]
- 9.Sen-Chowdhry S, Morgan RD, Chambers JC, McKeena WJ. Arrhythmogenic cardiomyopathy: Etiology, diagnosis and treatment. Annu Rev Med. 2010;61:233–253. doi: 10.1146/annurev.med.052208.130419. [DOI] [PubMed] [Google Scholar]
- 10.Viswanathan PC, Shaw RM, et al. 1999 Effects of IKr and IKs heterogeneity on action potential duration and its rate dependence: a simulation study. Circulation. 1999;99:2466–2474. doi: 10.1161/01.cir.99.18.2466. [DOI] [PubMed] [Google Scholar]
- 11.Hou L*, Deo M*, Furspan P, et al. A major role for HERG in determining frequency of reentry in neonatal rat ventricular myocyte monolayer. Circ Res. 2010 Dec 10;107(12):1503–11. doi: 10.1161/CIRCRESAHA.110.232470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verma A, Kilicaslan F, Schweikert RA, et al. Short- and long-term success of substrate-based mapping and ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia. Circulation. 2005;111:3209–3216. doi: 10.1161/CIRCULATIONAHA.104.510503. [DOI] [PubMed] [Google Scholar]
- 13.Kusano KF, Emori T, Morita H, et al. Ablation of ventricular tachycardia by isolating the critical site in a patient with arrhythmogenic right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2000;11:102–105. doi: 10.1111/j.1540-8167.2000.tb00745.x. [DOI] [PubMed] [Google Scholar]
- 14.Benson DW, Wang DW, Dyment M, et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A) J Clin Invest. 2003;112:1019–1028. doi: 10.1172/JCI18062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 16.Tan HL, Bink-Boelkens MT, Bezzina CR, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001;409:1043–1047. doi: 10.1038/35059090. [DOI] [PubMed] [Google Scholar]
- 17.Wang DW, Viswanathan PC, Balser JR, et al. Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction block. Circulation. 2002;105:341–346. doi: 10.1161/hc0302.102592. [DOI] [PubMed] [Google Scholar]
- 18.Rook MB, Bezzina AC, Groenewegen WA, et al. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc Res. 1999;44:507–517. doi: 10.1016/s0008-6363(99)00350-8. [DOI] [PubMed] [Google Scholar]
- 19.Morley GE, Vaidya D, Samie FH, et al. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol. 1999;10:1361–1375. doi: 10.1111/j.1540-8167.1999.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 20.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol. 2009;6:337–348. doi: 10.1038/nrcardio.2009.44. [DOI] [PubMed] [Google Scholar]
- 21.Spach MS, Dolber PC, Heidlage JF, et al. Propagating depolarization in anisotropic human and canine cardiac muscle: apparent directional differences in membrane capacitance. Circ Res. 1987;60:206–219. doi: 10.1161/01.res.60.2.206. [DOI] [PubMed] [Google Scholar]
- 22.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 23.Cabo C, Pertsov AM, Davidenko JM, et al. Vortex shedding as a precursor of turbulent electrical activity in cardiac muscle. Biophys J. 1996;70:1105–1111. doi: 10.1016/S0006-3495(96)79691-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zlochiver S, Muñoz V, Vikstrom KL, Taffet SM, Berenfeld O, Jalife J. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys J. 2008 Nov 1;95(9):4469–80. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.