

Abstract
Animal models of chronic kidney disease (CKD) are important experimental tools that are used to investigate novel mechanistic pathways and to validate potential new therapeutic interventions prior to pre-clinical testing in humans. Over the past several years, mouse CKD models have been extensively used for these purposes. Despite significant limitations, the model of unilateral ureteral obstruction (UUO) has essentially become the high throughput in vivo model, as it recapitulates the fundamental pathogenetic mechanisms that typify all forms of CKD in a relatively short time span. In addition, several alternative mouse models are available that can be used to validate new mechanistic paradigms and/or novel therapies. Several models are reviewed – both genetic and experimentally induced – that provide investigators with an opportunity to include renal functional study end-points together with quantitative measures of fibrosis severity, something that is not possible with the UUO model.
Keywords: fibrosis, collagen, animal models, extracellular matrix, chronic kidney disease, unilateral ureteral obstruction
Introduction
The high prevalence of chronic kidney disease (CKD) underscores our failure to provide therapies to effectively halt, prevent and/or reverse renal fibrosis – the universal pathway that leads to permanent loss of normal renal parenchyma and the relentless decline in renal function. Basic science investigators are challenged to establish new partnerships that will facilitate the translation of their discoveries into improved healthcare outcomes for people. It is anticipated that recent advances in genetic, cellular, molecular and systems biology will eventually make it possible to custom-design human therapies for CKD based on individual “molecular signatures” that define the renal fibrogenic response for a given patient with CKD.
An essential step in the journey toward human clinical trials is pre-clinical testing in experimental models that often begin in vitro, followed by validation in relevant animal models. Over the past two decades, mice have provided extremely valuable model systems for the experimental nephrology field, given its known genome, the vast repertoire of commercially available experimental tools – such as antibodies and genetic probes – and the powerful tools of genetic engineering that make it possible to manipulate gene expression as a way to investigate biological function. However, there are some significant limitations to the use of mouse CKD models, especially because many strains fail to develop renal fibrosis when exposed to disease triggers that have worked in other species, such as puromycin-induced nephrotic syndrome, streptozotocin-induced diabetic nephropathy, anti-Thy-1 nephritis and 5/6 nephrectomy. The purpose of this review is to discuss current uses and limitations of mouse CKD models.
Renal Fibrosis: The Primary Outcome
Interstitial extracellular matrix expansion is the classical histological hallmark of chronic kidney injury and is usually the best structural correlate of the degree of renal functional loss and a strong predictor of progression risk [1]. Insufficient attention has been paid to the specific molecular composition of the extracellular matrix that constitutes the “scarred” renal interstitium. The primary matrix sub-class is the collagen family – especially the fibrillar collagens I and III, and basement membrane collagen IV – that accumulates within the interstitium as CKD advances. This information indicates that specific procollagen mRNA levels may be useful surrogate measures of fibrosis severity, as transcription is the rate-limiting step in collagen synthesis (Figure 1) [2, 3]. Quantitative analysis of kidney sections stained with specific anti-collagen antibodies is recommended as a confirmatory step, quantified in a blinded fashion, ideally using a computer-assisted image analysis program. Total kidney collagen content is commonly used as an outcome measure, calculated using the hydroxyproline assay (Figure 2). In this assay, collagen content is estimated based on the assumption that it contains approximately 12.7% hydroxyproline based on weight. Results are most commonly expressed as collagen content (μg) per kidney wet weight (mg). An alternative is to express the data as total collagen per kidney. Ideally, each study should also include a semi-quantitative histologic evaluation of fibrosis severity, using collagen chain-specific immunostains or a general collagen stain. For the latter, Sirius Red is preferred over Mason-trichrome, as there is less “noise” in the system – the interstitium is essentially negative in control mice and excessive interstitial collagen is readily visible by polarized light microscopy (Figure 2). Determining the optimal standard method to evaluate fibrosis in human renal biopsies, where sample sizes are even smaller, is another important topic under active discussion [4, 5].
Figure 1. Interstitial collagens I and III are major components of the interstitial scar.

The fibrillar interstitial collagens I and III contribute to the formation of the interstitial matrix scaffold in normal kidneys (upper right photomicrograph); both are greatly expanded and contribute to interstitial scar formation in chronically damaged kidneys (lower right photomicrograph). Kidney mRNA levels (graph) provide a reasonable estimate of synthesis rates, as transcription is considered the rate-limiting step in collagen production. The examples are data from the unilateral ureteral obstruction (UUO) model in C57BL/6 mice (reproduced from [2, 3] reproduced with copyright permission).
Figure 2. Quantitative analysis of kidney collagen content.
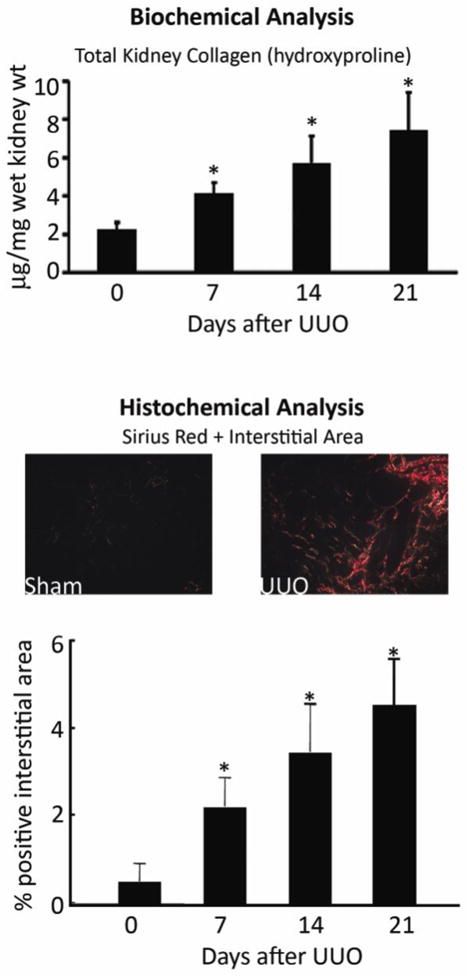
A global assessment of all kidney collagens is frequently performed using a biochemical assay to measure the content of hydroxyproline in kidney tissue, extrapolating using the assumption that 12.7% of collagen is composed of hydroxyproline (upper graph). Alternatively, kidney tissue can be stained with Sirius Red, which identifies mature, cross-linked collagen fibrils. Easily visualized by polarized light microscopy, interstitial staining is minimal in normal kidneys (left photomicrograph), which facilitates detection of abnormal deposits in diseased kidneys (right photomicrograph). Availability of computer software programs facilitates quantification collagen-positive tubulointerstitial areas (lower graph). The graphs are composites of the data from several of our published [2, 14, 97–99] and unpublished studies in C57BL/6 mice. D, days after UUO surgery.
Other points that need to be considered when designing an animal study where fibrosis severity is the primary outcome include:
More than one time-point should be investigated. The complex network of inter-related fibrogenic pathways has many built-in redundancies. It is doubtful that a significant difference observed during the first week of a disease process but absent thereafter is an observation that would engender much enthusiasm for a pre-clinical trial. Too many papers have been published using a single point of observation – for example, day seven after unilateral ureteral obstruction (UUO). There are situations where significant differences at day 7 are no longer present at later time-points, and perhaps more important, significant differences not present on day 7 that become increasingly different at days 14 and 21 [6, 7].
Determine whether the fibrogenic process is focal or diffuse. As in the evaluation of human disease, an important point to address during the study design is the minimum kidney tissue sample size required to provide adequate representation of events in the entire kidney. Even in the highly-predictable model of renal fibrosis induced by UUO we have found a 30% difference in regional collagen concentrations (Figure 3). Thus, for studies in this model, it is important that the same anatomical region of the kidney (upper pole for example) be used for each study animal. When the fibrogenic response is highly variable from one region of the kidney to another, larger sample sizes (potentially the entire kidney) will need to be analyzed. If the disease process differentially affects the cortex and medulla, this variable needs to be taken into consideration.
Remember that the renal capsule and pelvis contain collagen and should be excluded from the analysis.
Interstitial fibrosis is not just about collagen. Numerous other extracellular matrix molecules are co-deposited in the interstitium in CKD and this is an aspect of the fibrogenic process that has received little attention [8]. Like the collagens, these additional molecules are not simply building blocks that create an inert interstitial scaffold, but they may also serve more active roles by mediating biological effects through interactions with specific cellular receptors or via interactions with other extracellular molecules. Some of these extracellular matrix molecules are known to promote fibrosis (such as secreted protein acidic and rich in cysteine [9]); some (such as vitronectin) have numerous reported biological functions and accumulate along with collagen, yet genetic deficiency has no net effect on fibrosis [10]; while other matrix proteins attenuate fibrosis (such as decorin and biglycan [11]. It is possible that future studies will determine that quantitative measures of a different molecular component of the renal interstitial extracellular matrix better predicts long-term renal outcomes than collagen. In the meantime, focusing on collagen is the best that we have.
Figure 3. Interstitial fibrosis is heterogeneous.
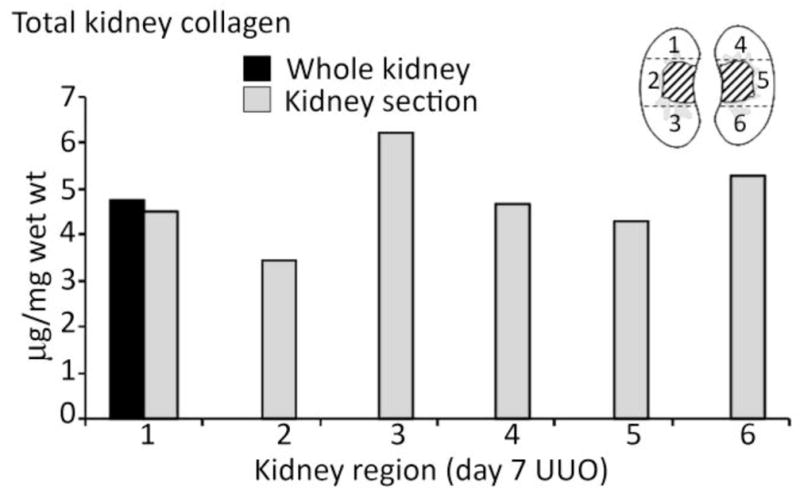
Interstitial fibrosis severity can vary widely from one area of the kidney to another, which underscores the importance of an adequate tissue sample size. Even in the relatively uniform injury induced by UUO of seven days’ duration, total kidney collagen varied by ~30% between different regions within the same kidney, as shown for a kidney that was harvested from a C57BL/6 mouse. The diagram depicts how the kidney was sagittally divided in half and separated into zones that correspond to each numbered bar in the graph (unpublished data).
Renal Parenchymal Loss, Not Fibrosis Severity Per Se, is the Critical Outcome
A small amount of fibrosis is beneficial if it curtails acute injury before more serious kidney damage ensues (i.e., healthy wound repair). The fibrotic response becomes maladaptive when it occurs at the expense of nephron integrity. Thus, optimally designed studies should not only include quantitative measures of the extent of fibrosis but also functional end-point – glomerular filtration rates, high-performance liquid chromatography-determined serum creatinine levels [12], quantitative measures of tubular function or possibly surrogate measures such as proteinuria. Each of these poses technical challenges in the mouse. Furthermore, although the widely-used UUO model causes extensive parenchymal damage, the intact contralateral kidney compensates functionally. Several methods have been used to estimate tubular area and integrity as an alternative approach to evaluate the severity of renal parenchymal loss associated with fibrosis. However, none have undergone rigorous validation against a gold standard measure of renal function. Examples of tubular density surrogate measures include tubular area, levels of expression of tubular proteins such as E-cadherin and reactivity with tubule-specific lectins (Figure 4) [13–16]. Although studies have established significant negative correlations between the degree of interstitial fibrosis and tubular cell density [17], whether there is a tight correlation between the latter two remains to be determined.
Figure 4. Surrogate measures of tubular integrity.

Functional compensation by the normal contralateral kidney and absence or urine flow in the obstructed kidney mean that traditional renal function outcome measures do not reflect the degree of chronic kidney damage in the UUO model. An alternative approach is to estimate the severity of the chronic kidney damage by quantifying the area of intact tubules. Possible approaches include direct measurement of tubular diameters histologically (upper photomicrograph illustrates one approach with the normal kidney on the left, a damaged kidney on the right, interstitial collagen staining in green and measured tubular diameters identified by the red lines); quantification of the area of E-cadherin-positive healthy tubules identified histologically (middle photomicrograph is from a day 14 UUO kidney) or by immunoblotting (middle graph), as shown for C57BL/6 kidneys 3 – 14 days (D) after UUO; or by estimating tubular areas using lectin staining of specific tubular segments (lotus tetragonolobus reacting with the brush border of proximal tubules indicated by the red staining of a normal mouse kidney, shown in the lower left photomicrograph; dolichos biforus identifies the stalk of the ureteric bud/collecting duct in a whole mount of a developing kidney in the lower right photomicrograph). Data in A, B and C right panel are reproduced from [13, 14, 16, 23] respectively, with copyright permission.
Unilateral Ureteral Obstruction (UUO): The High Throughput Screening Model
The UUO model recapitulates all of the key features of the typical fibrogenic response (Figure 5) which include: 1) increased interstitial capillary permeability [18] and the influx of inflammatory mononuclear cells, especially macrophages; 2) up-regulated synthesis of pro-fibrotic molecules including TGF-β and several others [19]; 3) the de novo appearance within the interstitium of a population of matrix-producing myofibroblasts; 4) increased matrix protein synthesis while endogenous matrix turnover pathways fail to keep pace with the increased demand that would prevent matrix accumulation [20]; 5) progressive tubular loss due to apoptosis and perhaps autophagy that is no longer balanced by tubular epithelial cell proliferation [14, 21] (Figure 6); 6) progressive interstitial capillary rarefaction, hypoxia and oxidant stress (Figure 5) [22, 23]. Other advantages of this model include the fact that each of these events is highly predictable and reproducible and leads to significant fibrosis and nephron loss in a relatively short period (7–14 days). Although the tempo may vary depending on the background strain (slower in Balb/c mice for example) [24], disease occurs in all mouse strains that have been reported, including the C57BL/6 strain that is widely used as the background for genetically engineered mice. The primary limitations of this model include the need for survival animal surgery, the aggressive pace of the disease (leading some to question its relevance as a model of human CKD), compensation by the normal contralateral kidney that precludes meaningful renal functional measures and absence of urine from the damaged kidney as a potential fluid for biomarker discovery studies. Another limitation pertains to the fact that renal vascular resistance increases after the surgery, leading to significantly reduced renal blood flow [25]. As a result, experiments based on the use of exogenous therapeutic agents need to take differences in renal delivery rates into consideration. Despite these limitations, the UUO model has become a benchmark that has been intensively investigated. Given the highly-reproducible fibrotic response, it should be possible to generate a catalogue of relative outcomes for the numerous molecular and cellular variations that have already been tested and to establish relative rankings of the fibrogenic potency of candidate molecules of interest. It is recommended that all UUO studies test an early time-point (≈7 days) and a later time-point when parenchymal damage is evident (14 days and possibly later). However, due to the limitations discussed above, investigation of any potentially new pathogenic mechanism should ideally be validated in a second CKD model.
Figure 5. Unilateral ureteral obstruction (UUO): a fast and reproducible model.

Despite some significant limitations, the UUO model reliably induces fibrosis with progressive kidney damage in all mouse strains reported to date, though the specific kinetics may vary between strains. The disease is characterized by the landmark histopathological features of chronic kidney disease that are illustrated above for the C57BL/6 strain and include: interstitial macrophage infiltration (F4/80 marker), interstitial capillary rarefaction (CD31 marker), oxidant stress markers (protein carbonyl and fatty acid-derived hydroxyoctadecadienoic [HODE] acid), interstitial myofibroblasts (αSMA marker), increased total kidney collagen and progressive decline in E-cadherin-positive tubules. These data are previously published and have been reproduced with copyright permission [14, 23] except the CD31 data (from Yamaguchi et al, unpublished). D, days after UUO surgery.
Figure 6. Mechanisms of tubular atrophy.

Renal tubules are estimated to occupy 85–90% of the normal kidney volume and irreversible tubular loss is the sine qua non of chronic kidney disease. A key distinction between acute and chronic disease is the process of tubular self-repair by proliferation. Using the UUO model as an example, when proliferating tubular epithelia are labeled by an injection of bromodeoxyuridine (Brdu) that is incorporated into the nucleus of dividing cells that can be detected by immunostaining (left photomicrograph), their proliferative capacity has been shown to progressively decline with chronic injury. During this same time-period the number of tubular cells undergoing apoptotic death, detected by nuclear terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining increase (middle photomicrograph and bar graph) (data are from [14]). It has recently been reported that tubular epithelia also undergo a process of autophagy that can be identified by the expression of (LC3-II), detected by immunostaining (right photomicrograph) or immunoblotting (right graph). It is still not clear if autophagy is a protective or destructive response in the context of chronic kidney disease (data are reproduced from [21, 100]). Copyright permission was obtained for reproduction of these data.
Alternative Mouse CKD Models
Genetic Models
Genetic deletion of the gene encoding the α3 chain of collagen IV (Col4α3 −/−) in mice results in an autosomal recessive form of chronic kidney disease that mimics human hereditary nephritis (Figure 7) [26]. The timing of death due to renal failure is variable depending upon the background strain: around 8 months for the commonly used C57BL/6 strain. Renal fibrosis severity can be accelerated by unilateral nephrectomy (personal observations). Recent studies using Col4α3 −/− mice have provided significant insights into several fibrosis regulatory pathways – such as αvβ6 and α1β1 integrins, metalloproteinases, the collagen receptor DDR1, the bone morphogenic protein antagonist USAG-1 and the renin-angiotensin system [27–32]. Lamb2 knockout mice, a model of human Pierson syndrome, harbor a genetic deletion in the basement membrane protein laminin beta2 but this mutation causes an aggressive form of renal disease that is lethal in the early postnatal period [33].
Figure 7. Col4 α−/ − hereditary nephritis model.

Mouse harboring a genetic mutation in the gene that encodes the glomerular basement membrane protein collagen IV alpha 3 (col4α3−/ −) develop hereditary nephritis with proteinuria and progressive renal failure. The tempo of the kidney disease is mouse strain-dependent. Compared to the asymptomatic heterozygous littermates (col4α3+/−) the mutant mice develop progressive interstitial fibrosis (shown in the Masson trichrome-stained photomicrographs), reduced tubular volume and interstitial volume expansion by matrix proteins. Data are from a study in 10-week old 129Sv/J mice that also harbor an asymptomatic heterozygous mutation in the 6 integrin gene that are reproduced from [27] with copyright permission.
Mouse models of autosomal dominant and autosomal recessive polycystic kidney disease and nephronophthisis are available [34]. However, some develop early renal failure and death which limits their utility for investigating CKD mechanistic pathways. Although mechanisms of cystogenesis and ciliary function are of great interest, it is clear that the accompanying inflammatory and fibrogenic changes in the renal interstitium are critical mechanisms of CKD progression, just as they are in non-cystic forms of CKD [35]. Thus greater use could be made of mouse cystic kidney disease models for validating new mechanistic paradigms.
Studies in mouse models of lupus nephritis have provided many valuable insights into the primary pathogenesis of autoimmune kidney disease. Death due to renal failure has been a valuable end-point for several of these studies. However, a new level of complexity is introduced if these models are used to investigate secondary pathways of CKD progression: it is always possible that the experimental manipulation will modify the severity of the primary immune response that initiates glomerular injury and downstream differences in the severity of the chronic tubulointerstitial disease may be an indirect consequence [36, 37].
As the genetic basis of human kidney disease continues to unfold, new mouse models will undoubtedly become available that could serve as additional experimental CKD models. Current challenges include the need for models with practical time courses – early death (as occurs in the nephrin−/− mice [38]), or disease that develops over long periods of time are not practical for CKD progression studies. Some mouse genetic mutations fail to recapitulate the severe renal phenotype that is observed in humans, such as a mutation in the cystinosin gene that causes human cystinosis, although chronic disease is evident after one year of age when the mutation is expressed on the C57BL/6 background [39, 40]. In this era of explosion in the field of podocyte biology, several interesting mouse models have been created, including many with mutations that are restricted to the podocyte [41], Data on the presence and severity of secondary tubulointerstitial fibrosis and renal insufficiency are not yet available for many of these mice, though it is reasonable to anticipate that useful new CKD models will become available. Integrin-linked kinase and phosphoinositide3-kinase C2alpha knockout mice have been reported to develop chronic renal insufficiency [42, 43]. Other mice with abnormal podocyte function that may prove useful for CKD studies include knockout mice (CD2-associated protein, podocin, α-actinin-4, dicer, VEGF-A, semiphorin A), gain-of-function mice (NFAT, sidekick-1, VEGF-A, Notch-1, angiopoietin-2, angiopoietin-like-4, transient receptor potential cation 6) and podocyte-depleted mice [44].
Spontaneous mutations in inbred strains of mice have occasionally been developed as models of CKD. Two good examples are the kd/kd mice that develop chronic tubulointerstitial disease (reported to have mutation in a mitochondrial gene) [45, 46] and ICGN mice with nephrotic syndrome and progressive renal insufficiency (genetic defect localized to the tensin2 gene) [47]. Spontaneous and genetically engineered models of hydronephrosis and/or renal dysplasia are emerging that may also prove useful for CKD studies [48–51].
A significant limitation of the use of the genetic CKD models is the time and expense of breeding strategies to generate mice that also express a second genetic defect, one that is in a pathway of potential interest to CKD progression mechanisms. These mice may also require extensive back-crossing (10 generations) to create two genetic mutations on a single background strain. This is an especially important pre-requisite in mouse kidney disease models, as background strain may greatly affect disease severity. There is also a large body of experimental and human data documenting gender-specific differences in kidney disease severity, which makes it critical that outcomes in males and females be analyzed separately [52].
Other Mouse CKD Models
1) 5/6 Nephrectomy
The rat remnant kidney model has been one of the most valuable and extensively investigated experimental CKD models. It was especially informative in studies that established the important role of the renin-angiotensin system in proteinuria and kidney disease progression. However, this model poses significant challenges in mice. Mouse background strain exerts a strong influence on disease severity. Although there are some conflicting data, the widely-used C57BL/6 strain appears to be relatively fibrosis-resistant following 5/6 nephrectomy [53]. Susceptible strains include CD-1, 129Sv, and Swiss-Webster [54, 55]. A second challenge is the small amount of kidney tissue that is available after 5/6 nephrectomy, which makes it difficult to do little more than measure fibrosis severity (typical weight of a normal kidney is ~90–120 mg in a 25 g mouse).
2) Adriamycin nephropathy
The mouse adriamycin nephropathy CKD model offers the advantage of two kidneys and the presence of proteinuria (Figure 8) [56]. Its main challenge is that reliable CKD has only been reported in the Balb/c strain (resistance has been mapped to the Prkdc gene [57]); thus, studies in genetically engineered mice require time-consuming back-crossing before these studies begin. We have found that the Balb/c Jackson strains develop acute renal failure that can lead to early death; the Charles River Balb/c is better for longer term studies (6–8 weeks when fibrosis is the primary end-point). The adriamycin model has been widely used for studies focusing on the role of mononuclear cells in the pathogenesis of chronic kidney disease [58].
Figure 8. Adriamycin nephropathy.

Following a single intravenous injection of adriamycin (10 mg/kg), Balb/c mice develop nephrotic proteinuria, interstitial inflammation and interstitial fibrosis (represented as % interstitial volume) and renal failure (indicated as creatinine clearance). These figures are reproduced from Wang et al [56] with copyright permission. The photomicrograph is from taken from a mouse 4 weeks after adriamycin injection (unpublished).
3) Nephrotoxic serum nephritis
One of the classical experimental models of acute glomerulonephritis that has been extensively investigated in rabbits and rats, the nephrotoxic serum nephritis (NTS) model is more challenging to work with in mice. The severity of the initial glomerular disease is highly variable, depending upon the particular antiserum that is used – ranging from intense deposition of the IgG along the glomerular basement membrane but absolutely no evidence of glomerular injury, to severe crescentic glomerulonephritis. Not available commercially, the mouse NTS model requires that investigators either generate, purify and characterize their own antiserum or collaborate with investigators who have already done this. Most published studies have focused on the phase of acute glomerular injury (which may be associated with impaired renal function in association with crescentic disease), while data on long-term follow-up to determine if the mice develop progressive tubulointerstitial fibrosis, tubular atrophy and renal insufficiency are sparse. Some studies have reported interstitial inflammation [59–61] and elevated serum creatinine levels but the latter are likely a consequence of severe glomerular damage [62, 63]. Our preliminary studies with NTS generated by the Shankland laboratory suggest that progressive tubulointerstitial disease requires repeated NTS injections [64].
4) Diabetic nephropathy
Generating reliable mouse models of chronic and progressive diabetic nephropathy has been particularly challenging, and a limitation in the field given the high prevalence of CKD due to diabetic nephropathy in humans. The pancreatic B cell toxin streptozocin reliably causes hyperglycemia in most mouse strains, yet the degree of renal fibrosis is modest [65]. This has led to studies in a vast array of mutant mice (spontaneous mutations as well as those that are genetically engineered) [66]. It has recently been reported that CD1 and eNOS−/− C57BL/6 mice develop more severe nephropathy after STZ treatment, though studies often need to be continued for several months before significant renal fibrosis can be detected [67, 68]. BTBR mice with the ob/ob leptin-deficiency mutation develop type 2 diabetes and more aggressive nephropathy than most mouse models (focal interstitial fibrosis by 22 weeks) [69].
5) Other proteinuria models
The model of overload proteinuria, that was modified from the rat model of repeated intraperitoneal injections to induce interstitial fibrosis, is not as aggressive in mice [70]. Due to high mouse acute mortality rates with less purified forms of bovine serum albumin, we have limited our studies to highly-purified, endotoxin-free preparation and use several days to reach the final daily dose of 1g/100gm body weight. Uninephrectomy typically accentuates fibrosis, but even after 6–8 weeks, interstitial fibrosis is focal and does not appear to progress significantly with an additional month of injections (unpublished observations). The influence of mouse strain on disease susceptibility is unknown. In contrast, it appears that virtually all strains of mice fail to develop significant proteinuria and interstitial fibrosis with puromycin aminonucleoside – a nephropathy model that has been especially valuable in rat studies. It has also been difficult to achieve significant nephropathy in mice using anti-Thy-1 antiserum (a rat model of mesangial proliferative glomerulonephritis) and anti-Fx1A antiserum (a rat model of membranous nephropathy): repeated injections of these antisera have been reported to produce interstitial inflammation and fibrosis in rats. Mouse models of renal vasculitis have recently been developed, based on the induction of immune responses to myeloperoxidase [71–73], and glomerular thrombotic microangiopathy using complement factor H-deficient mice [74, 75]. Whether these mice develop significant CKD is not yet clear.
6) Chronic Nephrotoxicity
Given the great interest in the drugs, environmental toxins, heavy metals and alternative medicines as potential causes of CKD in humans, a limited number of studies have attempted to induce nephrotoxic renal disease in mice. Studies have primarily focused on the acute injury phase and the extent of chronic tubulointerstitial disease has not been carefully evaluated. Two of the most promising as CKD models are folic acid nephropathy [76–79] (Figure 9) and aristolochic acid (the active ingredient implicated in Chinese herb nephropathy). Both cause acute kidney injury that may progress to interstitial fibrosis [80]. Cisplatin has been a popular acute kidney injury mouse model that should be evaluated for long-term sequelae [81].
Figure 9. Chronic folic acid-induced nephropathy.

C57BL/6 mice given a single intraperitoneal injection of folic acid (250 mg/kg) develop acute kidney injury (shown by the serum BUN levels) but recovery is not complete. On day 14 BUN levels are still significantly elevated (64 ± 9 versus 28 ± 1 mg/dl at baseline). The kidneys show interstitial fibrosis based on computer-assisted analysis of the Masson trichrome (MT) stained interstitial area (middle graph and upper photomicrographs), accompanied by significant numbers of αSMA+ interstitial myofibroblasts (lower photomicrographs). Data are reproduced from [76] with copyright permission.
7) CKD as a Sequel to Acute Kidney Injury
A topic of great current interest is the relative risk of CKD after a severe episode of ischemic acute kidney injury. Recent human studies indicate the recovery is often not complete and that long-term interstitial fibrosis and progressive renal insufficiency may ensue [82–85]. The ischemia-reperfusion model of acute kidney injury has been extensively investigated in rats and, more recently, in mice; renal fibrosis has been demonstrated as an outcome [86, 87] (Figure 10). Given the clinical significance of this issue, it is likely that future studies will refine methods to achieve reliable fibrotic outcomes in mouse models of acute kidney injury and to determine if such lesions progress over time.
Figure 10. Interstitial fibrosis flowing severe acute kidney injury (AKI).
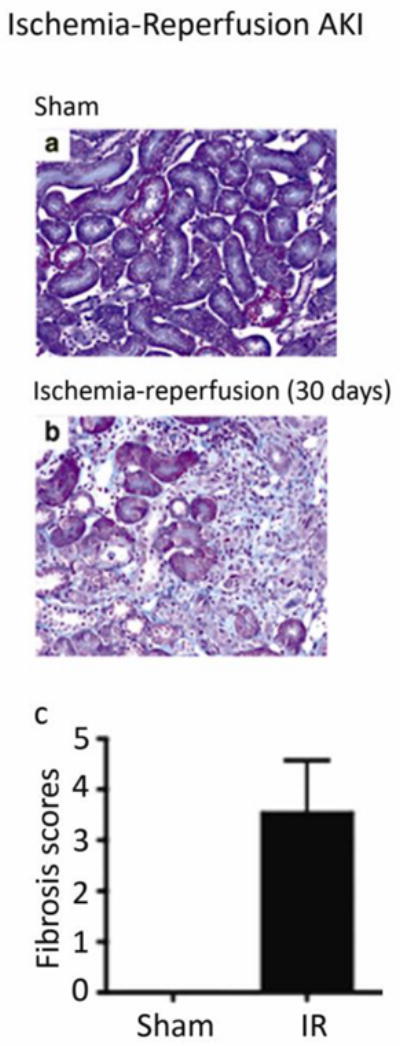
Acute renal ischemia (induced in C57BL/6 mice by cross-clamping the renal artery for 30 minutes), followed by reperfusion, has been extensively investigated as a model of acute kidney injury. Similar to humans, it is increasingly appreciated that renal recovery may not be complete; it may lead to chronic tubulointerstitial damage. In the study that is shown, (reproduced from [87] with copyright permission), the contralateral kidney was removed at the time of ischemia induction and fibrosis was detected by Masson trichrome staining 30 days later.
Reversible Models of Renal Fibrosis
Several studies in animal models have documented that regions within the kidney affected by abnormal extracellular matrix expansion can undergo a remodeling process such that normal renal architecture is restored. Perhaps the most compelling examples are the rat model of anti-Thy-1 nephritis [88] and early human diabetic nephropathy treated successfully by pancreatic transplantation [89]. In both of these examples, intraglomerular regions of expanded mesangial matrix re-model with time. Similarly, experimental models of acute tubulointerstitial injury induced by reversible nephrotic syndrome, reversible obstruction or reversible ischemia have been characterized by areas of “interstitial fibrosis” during the acute phase of the disease that are less prevalent several weeks after recovery [90]. Such data provide convincing evidence that renal extracellular matrix undergoes active remodeling. Further evidence is based on measures of significant kidney matrix protein synthesis rates (mRNA levels, in vivo collagen labeling studies with heavy water) in normal kidneys that do not manifest pathological fibrosis. These observations have significant clinical implications, highlighting the importance of diagnosis and treatment during the early stages when fibrosis is potentially reversible. It also underscores how little is currently known about the differential molecular composition and the potential for reversal of the processes of glomerular matrix expansion, acute interstitial “edema”, glomerulosclerosis and interstitial fibrosis. By analogy to chronic liver disease, the point at which chronic kidney damage and fibrosis transition to irreversible “cirrhosis phase”, is a critical question to be answered.
Since the specific proteolytic pathways responsible for matrix remodeling in the kidney are unknown (many “candidate” matrix metalloproteinases and serine proteases have been ruled out [20, 91–93]), the availability of reliable mouse models of interstitial fibrosis progression and regression are needed but not yet well-characterized. Long-term follow-up studies using the models of acute reversible glomerulonephropathy and tubular injury models should prove useful. The potential to reverse surgically-induced UUO appears promising but remains technically challenging with a high failure rate and thus the need to ensure that the degree of reversal is similar in all of the study animals [94, 95]. Promising methods include the use of removable vascular clamps (but the tenuous blood supply to the ureter limits the duration that occlusion that is tolerated) and surgical ureteral re-implantation. Background strain may influence the degree of recovery, as shown for C57BL/6 mice that develop worse fibrosis and less recovery after UUO reversal compared to Balb/c mice [24].
It remains unclear whether it is the quality (especially post-translational modifications), specific molecular composition (that is, which specific matrix proteins are present) and/or overall quantity of extracellular matrix in the renal “scars” that determines their susceptibility or resistance to proteolytic remodeling. This is a question that can be addressed using creative mouse models. For example, Thornhill et al [96] reported significant nephron recovery after the relief of partial UUO in neonatal mice. It is critical to appreciate that renal fibrosis regression will only have clinical benefit if the surrounding renal parenchyma regenerate to leave nephrons intact and/or further loss is prevented as a consequence of fibrosis regression. These are the necessary outcomes to prevent ongoing renal functional decline.
Additional Considerations in Mouse CKD Models
In addition to the call for futures studies to do a better job of including measures of nephron structural integrity and renal function together with quantitative assessment of kidney fibrosis severity, studies based on mouse models should measure blood pressure (which can be performed reliably and non-invasively using automated tail cuffs) and protein excretion rates (best reported as urinary protein/creatinine ratios due to the challenge of collecting accurately-timed urine collections and fasting the animals during the collection to avoid contamination from dietary proteins). Given the widely-recognized importance of proteinuria and hypertension as accelerators of human CKD, the opportunity to collect these data in the mouse studies should not be overlooked.
Summary
As powerful tools to decipher the genetic basis of human kidney disease are available and growing at an incredible pace, it will be ever more important to determine the biological function of the encoded proteins. The complexities of in vivo biological systems underscore the need for good animal models of human diseases to address these important questions. CKD studies in mice offer many advantages despite the limitations discussed in this review. We would like to advocate for establishing standard metrics to quantify fibrosis severity and renal functional loss to enable outcomes to be compared between studies performed at different institutions, analogous to the standards that we strive to achieve for human clinical trails. The UUO model offers many advantages and can be considered the in vivo high throughput screening tool; important findings should be validated in a second CKD model, analogous to the standards that are expected for human genetics studies.
Acknowledgments
The authors would like to acknowledge the research grants that have supported some of their own work that was cited: National Institutes of Health RO1DK54500 and P50DK44757 (AAE), K08DK073497 and RO3DK083648 (DO), K08DK080926 (IY), R03DK58925 (J L-G), the National Kidney Foundation Young Investigator Award (DO), the American Society of Nephrology Young Investigator Award (IY), Seattle Children’s Research Institute and the Robert O. Hickman Endowed Chair in Pediatric Nephrology. Several people contributed to our own studies that were cited in this review, including two talented research technicians (Xiaohe Cia and Sarah Collins) and several research fellows/visiting scientists (Drs. Takashi Oda, Heungsoo Kim, Guoqiang Zhang, Shunya Matsuo, and Masateru Yamada). We would like to acknowledge Dr. Shelia Violette and Dr. Kent Doi who provided high resolution digital photomicrographs.
References
- 1.Risdon RA, Sloper JC, de Vardener HE. Relationship between renal function and histologic changes found in renal-biopsy specimens from patients with persistent glomerulonephritis. Lancet. 1968;2:363–366. doi: 10.1016/s0140-6736(68)90589-8. [DOI] [PubMed] [Google Scholar]
- 2.Oda T, Jung YO, Kim H, Cai x, Lopez-Guisa J, Ikeda Y, Eddy AA. PAI-1 deficiency attenuates the fibrogenic response to ureteral obstruction. Kidney Int. 2001;60:587–596. doi: 10.1046/j.1523-1755.2001.030002587.x. [DOI] [PubMed] [Google Scholar]
- 3.Jones CL, Buch S, Post M, McCulloch L, Liu E, Eddy AA. The pathogenesis of interstitial fibrosis in chronic purine aminonucleoside nephrosis. Kidney Int. 1991;40:1020–1031. doi: 10.1038/ki.1991.310. [DOI] [PubMed] [Google Scholar]
- 4.Farris AB, Adams CD, Brousaides N, Della Pelle PA, Collins AB, Moradi E, Smith RN, Grimm PC, Colvin RB. Morphometric and visual evaluation of fibrosis in renal biopsies. J Am Soc Nephrol. 2011;22:176–186. doi: 10.1681/ASN.2009091005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fogo AB, Alpers CE. Navigating the challenges of fibrosis assessment: land in sight? J Am Soc Nephrol. 2011;22:11–13. doi: 10.1681/ASN.2010111132. [DOI] [PubMed] [Google Scholar]
- 6.Okamura DM, Pasichnyk K, Lopez-Guisa JM, Collins S, Hsu DK, Liu FT, Eddy AA. Galectin-3 preserves renal tubules and modulates extracellular matrix remodeling in progressive fibrosis. Am J Physiol Renal Physiol. 2011;300:F245–253. doi: 10.1152/ajprenal.00326.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.López-Guisa JM, Bugge TH, Isacke CH, Collins S, Cai S, Eddy AA. Endo180/uPAR-associated protein is an important regular of renal fibrogenesis. J Am Soc Nephrol. 2008;19:32A. [Google Scholar]
- 8.Eddy AA. Progression in chronic kidney disease. Adv Chronic Kidney Dis. 2005;12:353–365. doi: 10.1053/j.ackd.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 9.Socha MJ, Manhiani M, Said N, Imig JD, Motamed K. Secreted protein acidic and rich in cysteine deficiency ameliorates renal inflammation and fibrosis in angiotensin hypertension. Am J Pathol. 2007;171:1104–1112. doi: 10.2353/ajpath.2007.061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez-Guisa JM, Rassa AC, Cai X, Collins SJ, Eddy AA. Vitronectin accumulates in the interstitium but minimally impacts fibrogenesis in experimental chronic kidney disease. Am J Physiol Renal Physiol. 2011 Jan 26; doi: 10.1152/ajprenal.00701.2010. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaefer L, Mihalik D, Babelova A, Krzyzankova M, Grone HJ, Iozzo RV, Young MF, Seidler DG, Lin G, Reinhardt DP, Schaefer RM. Regulation of fibrillin-1 by biglycan and decorin is important for tissue preservation in the kidney during pressure-induced injury. Am J Pathol. 2004;165:383–396. doi: 10.1016/S0002-9440(10)63305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Breyer MD, Bottinger E, Brosius FC, 3rd, Coffman TM, Harris RC, Heilig CW, Sharma K. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 13.Morrissey J, Hruska K, Guo G, Wang S, Chen Q, Klahr S. Bone morphogenetic protein-7 improves renal fibrosis and accelerates the return of renal function. J Am Soc Nephrol. 2002;13(Suppl 1):S14–21. [PubMed] [Google Scholar]
- 14.Matsuo S, Lopez-Guisa JM, Cai X, Okamura DM, Alpers CE, Bumgarner RE, Peters MA, Zhang G, Eddy AA. Multifunctionality of PAI-1 in fibrogenesis: evidence from obstructive nephropathy in PAI-1-overexpressing mice. Kidney Int. 2005;67:2221–2238. doi: 10.1111/j.1523-1755.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- 15.Holthofer H. Lectin binding sites in kidney. A comparative study of 14 animal species. J Histochem Cytochem. 1983;31:531–537. doi: 10.1177/31.4.6827083. [DOI] [PubMed] [Google Scholar]
- 16.Michael L, Sweeney DE, Davies JA. The lectin Dolichos biflorus agglutinin is a sensitive indicator of branching morphogenetic activity in the developing mouse metanephric collecting duct system. J Anat. 2007;210:89–97. doi: 10.1111/j.1469-7580.2006.00670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mackensen-Haen S, Bohle A, Christensen J, Wehrmann M, Kendziorra H, Kokot F. The consequences for renal function of widening of the interstitium and changes in the tubular epithelium of the renal cortex and outer medulla in various renal diseases. Clin Nephrol. 1992;37:70–77. [PubMed] [Google Scholar]
- 18.Tchao BN, Burger ML, Eddy AA, Yamaguchi I. Vascular endothelial cadherin in the progression of renal interstitial fibrosis. 2010. E-PAS2010:1562.96 (abstract) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eddy AA, Neilson EG. Chronic kidney disease progression. J Am Soc Nephrol. 2006;17:2964–2966. doi: 10.1681/ASN.2006070704. [DOI] [PubMed] [Google Scholar]
- 20.Zhang G, Eddy AA. Urokinase and its receptors in chronic kidney disease. Front Biosci. 2008;13:5462–5478. doi: 10.2741/3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Zepeda-Orozco D, Black R, Lin F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am J Pathol. 2010;176:1767–1778. doi: 10.2353/ajpath.2010.090345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mimura I, Nangaku M. The suffocating kidney: tubulointerstitial hypoxia in end-stage renal disease. Nat Rev Nephrol. 2010;6:667–678. doi: 10.1038/nrneph.2010.124. [DOI] [PubMed] [Google Scholar]
- 23.Okamura DM, Pennathur S, Pasichnyk K, Lopez-Guisa JM, Collins S, Febbraio M, Heinecke J, Eddy AA. CD36 regulates oxidative stress and inflammation in hypercholesterolemic CKD. J Am Soc Nephrol. 2009;20:495–505. doi: 10.1681/ASN.2008010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puri TS, Shakaib MI, Chang A, Mathew L, Olayinka O, Minto AW, Sarav M, Hack BK, Quigg RJ. Chronic kidney disease induced in mice by reversible unilateral ureteral obstruction is dependent on genetic background. Am J Physiol Renal Physiol. 2010;298:F1024–1032. doi: 10.1152/ajprenal.00384.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim W, Moon SO, Lee SY, Jang KY, Cho CH, Koh GY, Choi KS, Yoon KH, Sung MJ, Kim DH, Lee S, Kang KP, Park SK. COMP-angiopoietin-1 ameliorates renal fibrosis in a unilateral ureteral obstruction model. J Am Soc Nephrol. 2006;17:2474–2483. doi: 10.1681/ASN.2006020109. [DOI] [PubMed] [Google Scholar]
- 26.Miner JH, Sanes JR. Molecular and functional defects in kidneys of mice lacking collagen alpha 3(IV): implications for Alport syndrome. J Cell Biol. 1996;135:1403–1413. doi: 10.1083/jcb.135.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hahm K, Lukashev ME, Luo Y, Yang WJ, Dolinski BM, Weinreb PH, Simon KJ, Chun Wang L, Leone DR, Lobb RR, McCrann DJ, Allaire NE, Horan GS, Fogo A, Kalluri R, Shield CF, 3rd, Sheppard D, Gardner HA, Violette SM. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol. 2007;170:110–125. doi: 10.2353/ajpath.2007.060158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cosgrove D, Meehan DT, Delimont D, Pozzi A, Chen X, Rodgers KD, Tempero RM, Zallocchi M, Rao VH. Integrin alpha1beta1 regulates matrix metalloproteinases via P38 mitogen-activated protein kinase in mesangial cells: implications for Alport syndrome. Am J Pathol. 2008;172:761–773. doi: 10.2353/ajpath.2008.070473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeisberg M, Khurana M, Rao VH, Cosgrove D, Rougier JP, Werner MC, Shield CF, 3rd, Werb Z, Kalluri R. Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med. 2006;3:e100. doi: 10.1371/journal.pmed.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gross O, Girgert R, Beirowski B, Kretzler M, Kang HG, Kruegel J, Miosge N, Busse AC, Segerer S, Vogel WF, Muller GA, Weber M. Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. 2010;29:346–356. doi: 10.1016/j.matbio.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka M, Asada M, Higashi AY, Nakamura J, Oguchi A, Tomita M, Yamada S, Asada N, Takase M, Okuda T, Kawachi H, Economides AN, Robertson E, Takahashi S, Sakurai T, Goldschmeding R, Muso E, Fukatsu A, Kita T, Yanagita M. Loss of the BMP antagonist USAG-1 ameliorates disease in a mouse model of the progressive hereditary kidney disease Alport syndrome. J Clin Invest. 2010;120:768–777. doi: 10.1172/JCI39569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gross O, Beirowski B, Koepke ML, Kuck J, Reiner M, Addicks K, Smyth N, Schulze-Lohoff E, Weber M. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003;63:438–446. doi: 10.1046/j.1523-1755.2003.00779.x. [DOI] [PubMed] [Google Scholar]
- 33.Jarad G, Cunningham J, Shaw AS, Miner JH. Proteinuria precedes podocyte abnormalities inLamb2−/− mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Invest. 2006;116:2272–2279. doi: 10.1172/JCI28414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson PD. Mouse models of polycystic kidney disease. Curr Top Dev Biol. 2008;84:311–350. doi: 10.1016/S0070-2153(08)00606-6. [DOI] [PubMed] [Google Scholar]
- 35.Okada H, Ban S, Nagao S, Takahashi H, Suzuki H, Neilson EG. Progressive renal fibrosis in murine polycystic kidney disease: An immunohistochemical observation. Kidney Int. 2000;58:587–597. doi: 10.1046/j.1523-1755.2000.00205.x. [DOI] [PubMed] [Google Scholar]
- 36.Kulkarni O, Anders HJ. Chemokines in lupus nephritis. Front Biosci. 2008;13:3312–3320. doi: 10.2741/2927. [DOI] [PubMed] [Google Scholar]
- 37.Bao L, Zhou J, Holers VM, Quigg RJ. Excessive matrix accumulation in the kidneys of MRL/lpr lupus mice is dependent on complement activation. J Am Soc Nephrol. 2003;14:2516–2525. doi: 10.1097/01.asn.0000089831.96794.0b. [DOI] [PubMed] [Google Scholar]
- 38.Donoviel DB, Freed DD, Vogel H, Potter DG, Hawkins E, Barrish JP, Mathur BN, Turner CA, Geske R, Montgomery CA, Starbuck M, Brandt M, Gupta A, Ramirez-Solis R, Zambrowicz BP, Powell DR. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol. 2001;21:4829–4836. doi: 10.1128/MCB.21.14.4829-4836.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cherqui S, Sevin C, Hamard G, Kalatzis V, Sich M, Pequignot MO, Gogat K, Abitbol M, Broyer M, Gubler MC, Antignac C. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol Cell Biol. 2002;22:7622–7632. doi: 10.1128/MCB.22.21.7622-7632.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nevo N, Chol M, Bailleux A, Kalatzis V, Morisset L, Devuyst O, Gubler MC, Antignac C. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant. 2010;25:1059–1066. doi: 10.1093/ndt/gfp553. [DOI] [PubMed] [Google Scholar]
- 41.D’Agati VD. Podocyte injury in focal segmental glomerulosclerosis: Lessons from animal models (a play in five acts) Kidney Int. 2008;73:399–406. doi: 10.1038/sj.ki.5002655. [DOI] [PubMed] [Google Scholar]
- 42.El-Aouni C, Herbach N, Blattner SM, Henger A, Rastaldi MP, Jarad G, Miner JH, Moeller MJ, St-Arnaud R, Dedhar S, Holzman LB, Wanke R, Kretzler M. Podocyte-specific deletion of integrin-linked kinase results in severe glomerular basement membrane alterations and progressive glomerulosclerosis. J Am Soc Nephrol. 2006;17:1334–1344. doi: 10.1681/ASN.2005090921. [DOI] [PubMed] [Google Scholar]
- 43.Harris DP, Vogel P, Wims M, Moberg K, Humphries J, Jhaver KG, DaCosta CM, Shadoan MK, Xu N, Hansen GM, Balakrishnan S, Domin J, Powell DR, Oravecz T. Requirement for class II phosphoinositide 3-kinase C2alpha in maintenance of glomerular structure and function. Mol Cell Biol. 2011;31:63–80. doi: 10.1128/MCB.00468-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ichikawa I, Ma J, Motojima M, Matsusaka T. Podocyte damage damages podocytes: autonomous vicious cycle that drives local spread of glomerular sclerosis. Curr Opin Nephrol Hypertens. 2005;14:205–210. doi: 10.1097/01.mnh.0000165884.85803.e1. [DOI] [PubMed] [Google Scholar]
- 45.Madaio MP, Ahima RS, Meade R, Rader DJ, Mendoza A, Peng M, Tomaszewski JE, Hancock WW, Gasser DL. Glomerular and tubular epithelial defects in kd/kd mice lead to progressive renal failure. Am J Nephrol. 2005;25:604–610. doi: 10.1159/000089709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng M, Jarett L, Meade R, Madaio MP, Hancock WW, George AL, Jr, Neilson EG, Gasser DL. Mutant prenyltransferase-like mitochondrial protein (PLMP) and mitochondrial abnormalities in kd/kd mice. Kidney Int. 2004;66:20–28. doi: 10.1111/j.1523-1755.2004.00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haruna Y, Kashihara N, Satoh M, Tomita N, Namikoshi T, Sasaki T, Fujimori T, Xie P, Kanwar YS. Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proc Natl Acad Sci USA. 2007;104:2331–2336. doi: 10.1073/pnas.0611079104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taniguchi K, Sugiyama F, Kakinuma Y, Uehara S, Nishijho N, Tanimoto K, Murakami K, Fukamizu A, Yagami KI. Pathologic characterization of hypotensive C57BL/6J-agt: angiotensinogen-deficient C57BL/6J mice. Int J Mol Med. 1998;1:583–587. doi: 10.3892/ijmm.1.3.583. [DOI] [PubMed] [Google Scholar]
- 49.Lye CM, Fasano L, Woolf AS. Ureter myogenesis: putting Teashirt into context. J Am Soc Nephrol. 2010;21:24–30. doi: 10.1681/ASN.2008111206. [DOI] [PubMed] [Google Scholar]
- 50.Ingraham SE, Saha M, Carpenter AR, Robinson M, Ismail I, Singh S, Hains D, Robinson ML, Hirselj DA, Koff SA, Bates CM, McHugh KM. Pathogenesis of Renal Injury in the Megabladder Mouse: A Genetic Model of Congenital Obstructive Nephropathy. Pediatr Res. 2010 Aug;:42. doi: 10.1203/PDR.0b013e3181f82f15. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cain JE, Rosenblum ND. Control of mammalian kidney development by the Hedgehog signaling pathway. Pediatr Nephrol. 2010 Dec 15; doi: 10.1007/s00467-010-1704-x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 52.Si H, Banga RS, Kapitsinou P, Ramaiah M, Lawrence J, Kambhampati G, Gruenwald A, Bottinger E, Glicklich D, Tellis V, Greenstein S, Thomas DB, Pullman J, Fazzari M, Susztak K. Human and murine kidneys show gender- and species-specific gene expression differences in response to injury. PLoS One. 2009;4:e4802. doi: 10.1371/journal.pone.0004802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kren S, Hostetter TH. The course of the remnant kidney model in mice. Kidney Int. 1999;56:333–337. doi: 10.1046/j.1523-1755.1999.00527.x. [DOI] [PubMed] [Google Scholar]
- 54.Ma LJ, Fogo AB. Model of robust induction of glomerulosclerosis in mice: importance of genetic background. Kidney Int. 2003;64:350–355. doi: 10.1046/j.1523-1755.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- 55.Leelahavanichkul A, Yan Q, Hu X, Eisner C, Huang Y, Chen R, Mizel D, Zhou H, Wright EC, Kopp JB, Schnermann J, Yuen PS, Star RA. Angiotensin II overcomes strain-dependent resistance of rapid CKD progression in a new remnant kidney mouse model. Kidney Int. 2010;78:1136–1153. doi: 10.1038/ki.2010.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Wang YP, Tay YC, Harris DC. Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int. 2000;58:1797–1804. doi: 10.1046/j.1523-1755.2000.00342.x. [DOI] [PubMed] [Google Scholar]
- 57.Papeta N, Zheng Z, Schon EA, Brosel S, Altintas MM, Nasr SH, Reiser J, D’Agati VD, Gharavi AG. Prkdc participates in mitochondrial genome maintenance and prevents Adriamycin-induced nephropathy in mice. J Clin Invest. 2010;120:4055–4064. doi: 10.1172/JCI43721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cao Q, Wang Y, Zheng D, Sun Y, Wang Y, Lee VW, Zheng G, Tan TK, Ince J, Alexander SI, Harris DC. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J Am Soc Nephrol. 2010;21:933–942. doi: 10.1681/ASN.2009060592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klein J, Gonzalez J, Decramer S, Bandin F, Neau E, Salant DJ, Heeringa P, Pesquero JB, Schanstra JP, Bascands JL. Blockade of the kinin B1 receptor ameloriates glomerulonephritis. J Am Soc Nephrol. 2010;21:1157–1164. doi: 10.1681/ASN.2009090887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Menke J, Lucas JA, Zeller GC, Keir ME, Huang XR, Tsuboi N, Mayadas TN, Lan HY, Sharpe AH, Kelley VR. Programmed death 1 ligand (PD-L) 1 and PD-L2 limit autoimmune kidney disease: distinct roles. J Immunol. 2007;179:7466–7477. doi: 10.4049/jimmunol.179.11.7466. [DOI] [PubMed] [Google Scholar]
- 61.Xie C, Liu K, Fu Y, Qin X, Jonnala G, Wang T, Wang HW, Maldonado M, Zhou XJ, Mohan C. RANTES Deficiency Attenuates Autoantibody-Induced Glomerulonephritis. J Clin Immunol. 2010 Oct 1; doi: 10.1007/s10875-010-9470-x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 62.Giorgini A, Brown HJ, Sacks SH, Robson MG. Toll-like receptor 4 stimulation triggers crescentic glomerulonephritis by multiple mechanisms including a direct effect on renal cells. Am J Pathol. 2010;177:644–653. doi: 10.2353/ajpath.2010.091279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Turner JE, Paust HJ, Steinmetz OM, Peters A, Meyer-Schwesinger C, Heymann F, Helmchen U, Fehr S, Horuk R, Wenzel U, Kurts C, Mittrucker HW, Stahl RA, Panzer U. CCR5 deficiency aggravates crescentic glomerulonephritis in mice. J Immunol. 2008;181:6546–6556. doi: 10.4049/jimmunol.181.9.6546. [DOI] [PubMed] [Google Scholar]
- 64.Ohse T, Vaughan MR, Kopp JB, Krofft RD, Marshall CB, Chang AM, Hudkins KL, Alpers CE, Pippin JW, Shankland SJ. De novo expression of podocyte proteins in parietal epithelial cells during experimental glomerular disease. Am J Physiol Renal Physiol. 2010;298:F702–711. doi: 10.1152/ajprenal.00428.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Collins SJ, Alexander SL, Lopez-Guisa JM, Cai X, Maruvada R, Chua SC, Zhang G, Okamura DM, Matsuo S, Eddy AA. Plasminogen activator inhibitor-1 deficiency has renal benefits but some adverse systemic consequences in diabetic mice. Nephron. 2006;104:e23–34. doi: 10.1159/000093673. [DOI] [PubMed] [Google Scholar]
- 66.Brosius FC, 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sugimoto H, Grahovac G, Zeisberg M, Kalluri R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes. 2007;56:1825–1833. doi: 10.2337/db06-1226. [DOI] [PubMed] [Google Scholar]
- 68.Kosugi T, Heinig M, Nakayama T, Matsuo S, Nakagawa T. eNOS knockout mice with advanced diabetic nephropathy have less benefit from renin-angiotensin blockade than from aldosterone receptor antagonists. Am J Pathol. 2010;176:619–629. doi: 10.2353/ajpath.2010.090578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hudkins KL, Pichaiwong W, Wietecha T, Kowalewska J, Banas MC, Spencer MW, Muhlfeld A, Koelling M, Pippin JW, Shankland SJ, Askari B, Rabaglia ME, Keller MP, Attie AD, Alpers CE. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J Am Soc Nephrol. 2010;21:1533–1542. doi: 10.1681/ASN.2009121290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eddy AA, Kim H, Lopez-Guisa J, Oda T, Soloway PD. Interstitial fibrosis in mice with overload proteinuria: deficiency of TIMP-1 is not protective. Kidney Int. 2000;58:618–628. doi: 10.1046/j.1523-1755.2000.00208.x. [DOI] [PubMed] [Google Scholar]
- 71.Gan PY, Steinmetz OM, Tan DS, O’Sullivan KM, Ooi JD, Iwakura Y, Kitching AR, Holdsworth SR. Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J Am Soc Nephrol. 2010;21:925–931. doi: 10.1681/ASN.2009070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20:289–298. doi: 10.1681/ASN.2008050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, Maeda N, Falk RJ, Jennette JC. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955–963. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pickering MC, de Jorge EG, Martinez-Barricarte R, Recalde S, Garcia-Layana A, Rose KL, Moss J, Walport MJ, Cook HT, de Cordoba SR, Botto M. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med. 2007;204:1249–1256. doi: 10.1084/jem.20070301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.de Jorge EG, Macor P, Paixao-Cavalcante D, Rose KL, Tedesco F, Cook HT, Botto M, Pickering MC. The development of atypical hemolytic uremic syndrome depends on complement C5. J Am Soc Nephrol. 2011;22:137–145. doi: 10.1681/ASN.2010050451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Doi K, Okamoto K, Negishi K, Suzuki Y, Nakao A, Fujita T, Toda A, Yokomizo T, Kita Y, Kihara Y, Ishii S, Shimizu T, Noiri E. Attenuation of folic acid-induced renal inflammatory injury in platelet-activating factor receptor-deficient mice. Am J Pathol. 2006;168:1413–1424. doi: 10.2353/ajpath.2006.050634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koziolek MJ, Muller GA, Zapf A, Patschan D, Schmid H, Cohen CD, Koschnick S, Vasko R, Bramlage C, Strutz F. Role of CX3C-chemokine CX3C-L/fractalkine expression in a model of slowly progressive renal failure. Nephrol Dial Transplant. 2010;25:684–698. doi: 10.1093/ndt/gfp602. [DOI] [PubMed] [Google Scholar]
- 78.Yokoyama T, Kamijo-Ikemori A, Sugaya T, Hoshino S, Yasuda T, Kimura K. Urinary excretion of liver type fatty acid binding protein accurately reflects the degree of tubulointerstitial damage. Am J Pathol. 2009;174:2096–2106. doi: 10.2353/ajpath.2009.080780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yuan HT, Li XZ, Pitera JE, Long DA, Woolf AS. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 alpha. Am J Pathol. 2003;163:2289–2301. doi: 10.1016/s0002-9440(10)63586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou L, Fu P, Huang XR, Liu F, Chung AC, Lai KN, Lan HY. Mechanism of chronic aristolochic acid nephropathy: role of Smad3. Am J Physiol Renal Physiol. 2010;298:F1006–1017. doi: 10.1152/ajprenal.00675.2009. [DOI] [PubMed] [Google Scholar]
- 81.Linkermann A, Himmerkus N, Rolver L, Keyser KA, Steen P, Brasen JH, Bleich M, Kunzendorf U, Krautwald S. Renal tubular Fas ligand mediates fratricide in cisplatin-induced acute kidney failure. Kidney Int. 2011;79:169–178. doi: 10.1038/ki.2010.317. [DOI] [PubMed] [Google Scholar]
- 82.Triverio PA, Martin PY, Romand J, Pugin J, Perneger T, Saudan P. Long-term prognosis after acute kidney injury requiring renal replacement therapy. Nephrol Dial Transplant. 2009;24:2186–2189. doi: 10.1093/ndt/gfp072. [DOI] [PubMed] [Google Scholar]
- 83.Lo LJ, Go AS, Chertow GM, McCulloch CE, Fan D, Ordonez JD, Hsu CY. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int. 2009;76:893–899. doi: 10.1038/ki.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lafrance JP, Djurdjev O, Levin A. Incidence and outcomes of acute kidney injury in a referred chronic kidney disease cohort. Nephrol Dial Transplant. 2010;25:2203–2209. doi: 10.1093/ndt/gfq011. [DOI] [PubMed] [Google Scholar]
- 85.Liu KD. Acute kidney injury: is acute kidney injury a risk factor for long-term mortality? Nat Rev Nephrol. 2010;6:389–391. doi: 10.1038/nrneph.2010.39. [DOI] [PubMed] [Google Scholar]
- 86.Kim J, Seok YM, Jung KJ, Park KM. Reactive oxygen species/oxidative stress contributes to progression of kidney fibrosis following transient ischemic injury in mice. Am J Physiol Renal Physiol. 2009;297:F461–470. doi: 10.1152/ajprenal.90735.2008. [DOI] [PubMed] [Google Scholar]
- 87.Hotta K, Sho M, Yamato I, Shimada K, Harada H, Akahori T, Nakamura S, Konishi N, Yagita H, Nonomura K, Nakajima Y. Direct targeting of fibroblast growth factor-inducible 14 protein protects against renal ischemia reperfusion injury. Kidney Int. 2011;79:179–188. doi: 10.1038/ki.2010.379. [DOI] [PubMed] [Google Scholar]
- 88.Border WA, Noble NA, Yamamoto T, Harper JR, Yamaguchi Y, Pierschbacher MD, Ruoslahti E. Natural inhibitor of transforming growth factor-β protects against scarring in experimental kidney disease. Nature. 1992;360:361–364. doi: 10.1038/360361a0. [DOI] [PubMed] [Google Scholar]
- 89.Fioretto P, Steffes MW, Sutherland DE, Goetz FC, Mauer M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med. 1998;339:69–75. doi: 10.1056/NEJM199807093390202. [DOI] [PubMed] [Google Scholar]
- 90.Eddy AA. Can renal fibrosis be reversed? Pediatr Nephrol. 2005;20:1369–1375. doi: 10.1007/s00467-005-1995-5. [DOI] [PubMed] [Google Scholar]
- 91.Wang X, Zhou Y, Tan R, Xiong M, He W, Fang L, Wen P, Jiang L, Yang J. Mice lacking the matrix metalloproteinase-9 gene reduce renal interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol. 2010;299:F973–982. doi: 10.1152/ajprenal.00216.2010. [DOI] [PubMed] [Google Scholar]
- 92.Surendran K, Simon TC, Liapis H, McGuire JK. Matrilysin (MMP-7) expression in renal tubular damage: association with Wnt4. Kidney Int. 2004;65:2212–2222. doi: 10.1111/j.1523-1755.2004.00641.x. [DOI] [PubMed] [Google Scholar]
- 93.Sakamaki Y, Sasamura H, Hayashi K, Ishiguro K, Takaishi H, Okada Y, D’Armiento JM, Saruta T, Itoh H. Absence of gelatinase (MMP-9) or collagenase (MMP-13) attenuates adriamycin-induced albuminuria and glomerulosclerosis. Nephron. 2010;115:e22–32. doi: 10.1159/000312883. [DOI] [PubMed] [Google Scholar]
- 94.Cochrane AL, Kett MM, Samuel CS, Campanale NV, Anderson WP, Hume DA, Little MH, Bertram JF, Ricardo SD. Renal structural and functional repair in a mouse model of reversal of ureteral obstruction. J Am Soc Nephrol. 2005;16:3623–3630. doi: 10.1681/ASN.2004090771. [DOI] [PubMed] [Google Scholar]
- 95.Tapmeier TT, Brown KL, Tang Z, Sacks SH, Sheerin NS, Wong W. Reimplantation of the ureter after unilateral ureteral obstruction provides a model that allows functional evaluation. Kidney Int. 2008;73:885–889. doi: 10.1038/sj.ki.5002797. [DOI] [PubMed] [Google Scholar]
- 96.Thornhill BA, Forbes MS, Marcinko ES, Chevalier RL. Glomerulotubular disconnection in neonatal mice after relief of partial ureteral obstruction. Kidney Int. 2007;72:1103–1112. doi: 10.1038/sj.ki.5002512. [DOI] [PubMed] [Google Scholar]
- 97.Kim H, Oda T, Lopez-Guisa J, Wing D, Edwards DR, Soloway PD, Eddy AA. TIMP-1 deficiency does not attenuate interstitial fibrosis in obstructive nephropathy. J Am Soc Nephrol. 2001;12:736–748. doi: 10.1681/ASN.V124736. [DOI] [PubMed] [Google Scholar]
- 98.Zhang G, Kim H, Cai X, Lopez-Guisa J, Alpers C, Liu Y, Carmeliet P, Eddy A. Urokinase receptor deficiency accelerates fibrosis in obstructive nephropathy. J Am Soc Nephrol. 2003;14:1254–1271. doi: 10.1097/01.asn.0000064292.37793.fb. [DOI] [PubMed] [Google Scholar]
- 99.Yamaguchi I, Lopez-Guisa JM, Cai X, Collins SJ, Okamura DM, Eddy AA. Endogenous urokinase lacks antifibrotic activity during progressive renal injury. Am J Physiol Renal Physiol. 2007;293:F12–19. doi: 10.1152/ajprenal.00380.2006. [DOI] [PubMed] [Google Scholar]
- 100.Koesters R, Kaissling B, Lehir M, Picard N, Theilig F, Gebhardt R, Glick AB, Hahnel B, Hosser H, Grone HJ, Kriz W. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 2010;177:632–643. doi: 10.2353/ajpath.2010.091012. [DOI] [PMC free article] [PubMed] [Google Scholar]