

Abstract
Protein-tyrosine phosphatase receptor type Z (Ptprz) has multiple substrate proteins, including G protein-coupled receptor kinase-interactor 1 (Git1), membrane-associated guanylate kinase, WW and PDZ domain-containing 1 (Magi1), and GTPase-activating protein for Rho GTPase (p190RhoGAP). We have identified a dephosphorylation site at Tyr-1105 of p190RhoGAP; however, the structural determinants employed for substrate recognition of Ptprz have not been fully defined. In the present study, we revealed that Ptprz selectively dephosphorylates Git1 at Tyr-554, and Magi1 at Tyr-373 and Tyr-858 by in vitro and cell-based assays. Of note, the dephosphorylation of the Magi1 Tyr-858 site required PDZ domain-mediated interaction between Magi1 and Ptprz in the cellular context. Alignment of the primary sequences surrounding the target phosphotyrosine residue in these three substrates showed considerable similarity, suggesting a consensus motif for recognition by Ptprz. We then estimated the contribution of surrounding individual amino acid side chains to the catalytic efficiency by using fluorescent peptides based on the Git1 Tyr-554 sequence in vitro. The typical substrate motif for the catalytic domain of Ptprz was deduced to be Glu/Asp-Glu/Asp-Glu/Asp-Xaa-Ile/Val-Tyr(P)-Xaa (Xaa is not an acidic residue). Intriguingly, a G854D substitution of the Magi1 Tyr-858 site matching better to the motif sequence turned this site to be susceptible to dephosphorylation by Ptprz independent of the PDZ domain-mediated interaction in cells. Furthermore, we found by database screening that the substrate motif is present in several proteins, including paxillin at Tyr-118, its major phosphorylation site. Expectedly, we verified that Ptprz efficiently dephosphorylates paxillin at this site in cells. Our study thus provides key insights into the molecular basis for the substrate recognition of Ptprz.
Keywords: Phosphotyrosine, Phosphotyrosine Signaling, Receptors, Signal Transduction, Protein-tyrosine Phosphatase (Tyrosine Phosphatase)
Introduction
Protein-tyrosine phosphorylation is a dynamic process governed by the balanced actions of protein-tyrosine kinases (PTKs),2 and protein-tyrosine phosphatases (PTPs), and critical to the regulation of numerous physiological processes (for review, see Ref. 1). The specificity of the signal transduction depends on the ability of each PTK or PTP to phosphorylate or dephosphorylate precisely particular sites on specific substrates, respectively. Elucidation of the specificity for individual PTPs has been an important subject of investigation; however, even the identification of PTP substrates is still methodologically difficult. To our knowledge, substrate specificity of PTPs has been characterized only for PTP1B (2).
Receptor-like PTPs (RPTPs) are a structurally and functionally diverse family of enzymes comprised of eight subfamilies. PTP receptor type Z (Ptprz, also called PTPζ or RPTPβ) is a RPTP classified in the R5 subfamily together with Ptprg (PTPγ). Three isoforms of Ptprz are generated by alternative splicing from a single Ptprz gene: the two transmembrane isoforms Ptprz-A and Ptprz-B and the secretory isoform Ptprz-S (also known as phosphacan or 6B4 proteoglycan), all of which are expressed as chondroitin sulfate proteoglycans in the brain (3, 4 and references cited therein). The physiological importance of these gene products has been demonstrated through studies with Ptprz-deficient mice (5–8), which display impairments in hippocampal functions in a maturation-dependent manner (6, 7). In peripheral tissues, gastric mucosal cells also express a nonproteoglycan form of Ptprz-B, though at lower levels. Ptprz-deficient mice are resistant to gastric ulceration caused by VacA, a cytotoxin secreted by Helicobacter pylori, indicating that Ptprz functions as the receptor of VacA for gastric mucosal damage (8).
We previously reported a genetic method to screen for PTP substrates named the “yeast substrate-trapping system” based on the yeast two-hybrid system with two modifications (9, 10): the conditional expression of a PTK to tyrosine-phosphorylate the prey protein, and screening using a substrate-trap PTP mutant as bait. Using this method, we have successfully identified several substrates for Ptprz, including G protein-coupled receptor kinase-interactor 1 (Git1), GTPase-activating protein for Rho GTPase (p190RhoGAP), membrane-associated guanylate kinase, WW and PDZ domain-containing 1 (Magi1), and PDZ and coiled-coil motif containing (Gopc, also called PIST) (9–11). However, only Tyr-1105 in p190RhoGAP has been identified so far as a site of dephosphorylation by Ptprz (7). Thus, our understanding of the molecular basis for the physiological function of Ptprz including its substrate specificity is still limited.
In this study, we revealed Ptprz dephosphorylation sites in Git1 and Magi1, in addition to that in p190RhoGAP. A high degree of similarity in the primary sequence surrounding the target phosphotyrosine residues indicated the presence of a substrate motif in the physiological substrates of Ptprz. We successfully revealed a new Ptprz substrate by using the substrate motif sequence. Our finding thus provides the molecular basis for the substrate recognition of Ptprz.
EXPERIMENTAL PROCEDURES
Antibodies
Rabbit polyclonal antibodies against pY554 on Git1 were prepared as follows. A phosphorylated peptide corresponding to the 547–561 region in rat Git1 (Cys-Glu-Leu-Glu-Asp-Asp-Ala-Ile-Tyr(P)-Ser-Val-His-Val-Pro-Ala-Gly) and a nonphosphorylated form of the peptide were synthesized: The amino (N)-terminal cysteine was added to facilitate linkage to keyhole limpet hemocyanin or activated thiol-Sepharose 4B (GE Healthcare). Rabbits were immunized with the tyrosine-phosphorylated peptide coupled to keyhole limpet hemocyanin and Freund's complete adjuvant. Antibodies were purified from the antisera using phosphorylated peptide-Sepharose. We removed nonspecific antibodies from the purified antibody fraction by passage through a Sepharose column conjugated with the nonphosphorylated peptide and then a column conjugated with a peptide mixture, Cys-Gly-Gly-Gly-Ser-Tyr(P)–Xaa-Ser (Xaa is natural amino acids except cysteine).
Anti-Ptprz-S is a rabbit polyclonal antibody against the extracellular region of Ptprz (4). Rabbit anti-pY118 paxillin was kindly provided by Dr. Hisataka Sabe, Hokkaido University, Hokkaido, Japan (12). We also used commercially available antibodies against phosphotyrosine (PY20; GE Healthcare), the FLAG epitope (mouse monoclonal M2, F3165, and rabbit anti-FLAG, F7425; Sigma), paxillin (rabbit anti-paxillin, sc-5574; Santa Cruz Biotechnology), and the intracellular region of Ptprz (anti-RPTPβ; BD Biosciences).
Mammalian Expression Plasmids
The expression construct for rat Ptprz-B (pZeo-PTPζ) and its substrate trapping mutant (pZeo-PTPζ-DA) were reported previously (9). The phosphatase-inactive mutant of Ptprz-B (pZeo-PTPζ-CS) with Cys-1934 (numbering in rat Ptprz-A) replaced by Ser and the PDZ-binding motif mutant (pZeo-PTPζ-SA) with Ser-2314 replaced by Ala were generated from pZeo-PTPζ using the QuikChange Mutagenesis kit (Stratagene). The construction of FLAG-tagged-Git1 (pFLAG-Git1) and FLAG-tagged Magi1 (pFLAG-Magi1) was described previously (10). The expression constructs for the Git1 and Magi1 mutant proteins were generated from their respective parent plasmids using a QuikChange Multisite-directed Mutagenesis kit (Stratagene). The construct for FLAG-tagged paxillin (pFLAG-paxillin) was generated by inserting the full-length cDNA of mouse paxillin α (GenBank accession no. AF293882) into the pcDNA-FLAG vector (10), which was obtained by RT-PCR from mouse brain total RNA, at the EcoRI and XhoI sites.
Cell Culture and DNA Transfection
HEK293T cells (human embryonic kidney epithelial cells) were grown and maintained on dishes coated with rat tail collagen in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) in a humidified incubator at 37 °C with 5% (v/v) CO2. DNA transfection was performed by the standard calcium phosphate technique as described (13). Transfected cells were replated once on 3.5-cm dishes, cultured for 24 h, and then used for the experiments. v-Src-HEK293T cells with the stable expression of v-Src were obtained by transfecting HEK293T cells with pBaBePurov-src (14) and selection with puromycin (2 μg/ml). In v-src-HEK293T cells, cellular proteins undergo constitutive tyrosine phosphorylation.
Immunoprecipitation and Western Blotting
Cells starved in serum-free DMEM for specific periods were lysed with 300 μl (for 3.5-cm culture dishes) of 1% (v/v) Triton X-100 or 1% (v/v) Nonidet P-40 in TBS (10 mm Tris-HCl, pH 7.4, 150 mm NaCl) containing 1 mm vanadate, 10 mm NaF, and protease inhibitors (complete EDTA-free; Roche Applied Science) for 30 min on ice, and the supernatant was collected by centrifugation at 10,000 × g for 15 min. Cell extracts (250 μl) thus obtained were preincubated with 1 μg of anti-FLAG M2 antibody for 1 h. The immunocomplexes were precipitated using 10 μl of protein G-Sepharose 4FF (GE Healthcare), washed with the lysis buffer, and subjected to SDS-PAGE followed by Western blotting with an ECL Western blotting system (GE Healthcare).
In Vitro Dephosphorylation of Immunoprecipitated Proteins
The tyrosine-phosphorylated FLAG-tagged Git1 and FLAG-tagged Magi1 proteins were obtained from pervanadate-treated cells (100 μm for 15 min) by immunoprecipitation and used for in vitro dephosphorylation assays as described (10). Briefly, the beads carrying the immunocomplexes were washed with 25 mm HEPES, pH 6.8, 5 mm EDTA, 50 mm NaCl, 1 mm DTT, and 50 μg/ml bovine serum albumin (BSA). The reaction was then started by adding 1 μg of glutathione S-transferase (GST)-PtprzICR or GST at 37 °C. Subsequently, the tyrosine phosphorylation levels of substrate proteins were analyzed by Western blotting as above. Dephosphorylation by a nonselective phosphatase control was also examined with 1 milliunit of bacterial alkaline phosphatase (Toyobo) in 100 mm Tris-HCl, pH 9.5, 100 mm NaCl, 50 mm MgCl2, and 50 μg/ml BSA.
Recombinant Proteins
GST-fused proteins with the entire intracellular region of Ptprz (GST-PtprzICR) or the second PTP domain and the carboxyl (C)-terminal tail of Ptprz (GST-Ptprz-D2) were expressed in Escherichia coli strain BL21 and purified by glutathione affinity chromatography as described (13). Expression plasmids for Magi1 fragments, Magi1-PDZ1 (amino acid residues 470–640 of mouse Magi1a, GenBank accession no. AB194411), Magi1-PDZ2 (641–838), Magi1-PDZ3 (839–996), Magi1-PDZ4 (997–1137), and Magi1-PDZ45 (1138–1247) were generated by subcloning respective cDNAs prepared from pFLAG-Magi1 (10) into pET28a (Novagen) at the NheI and EcoRI sites to produce the fusion proteins with N-terminal His6 tags. These histidine-tagged proteins were expressed in BL21 and purified using a HisTrap FF column attached to a chromatography apparatus (AKTA prime plus; GE Healthcare).
GST Pulldown Experiments
Pulldown experiments using GST-Ptprz-D2 beads were performed as described (13). Briefly, GST-Ptprz-D2 beads (10 μl of beads, ∼30 pmol of proteins) were incubated with histidine-tagged Magi1 proteins (40 pmol) in 100 μl of 10 mm Tris-HCl, pH 7.4, 150 mm NaCl containing 1% (v/v) Triton X-100 for 30 min. After washing the beads, the bound proteins were eluted by boiling in a SDS-PAGE sample buffer. The proteins were separated by SDS-PAGE and stained with Coomassie Brilliant Blue R-250.
In Vitro Dephosphorylation Assays Using pCAP-Peptides
Phosphocoumaryl-aminopropionic acid (pCAP, a fluorogenic mimic of phosphotyrosine) residue and pCAP-containing peptides were synthesized as described (15). The N-terminal amino group of the synthetic peptides was acetylated, and the C-terminal carboxyl group was amidated. Purification of the peptides was performed by reverse phase high performance liquid chromatography (HPLC) to >90% purity. The pCAP-peptide substrates (50 μm) were preincubated in a three-component buffer of pH 6.5 (0.1 m acetate, 0.05 m Tris, and 0.05 m bis-tris) containing 5 mm DTT and 0.01% (v/v) Briji35 for 10 min at 30 °C, and the reaction was initiated by adding purified GST-PtprzICR (5 nm). The time course of the hydrolysis of pCAP-peptide substrates was recorded continuously by monitoring the increase in fluorescence at 460 nm (excitation at 334 nm) (FI-4500 spectrofluorometer; Hitachi, Tokyo, Japan). The reaction rates were determined from the slope during the initial linear stage (100 s).
RESULTS
Dephosphorylation of Git1 at Tyr-554 by Ptprz
We previously isolated a positive clone encoding the central region of Git1 (see Fig. 1A) in the yeast substrate screening for Ptprz (9). The recombinant protein derived from this clone was phosphorylated by p60c-Src and efficiently dephosphorylated by the whole intracellular catalytic region of Ptprz (PtprzICR) in vitro (9), indicating the presence of a substrate site(s) in the six tyrosine residues. In addition to these residues, four tyrosine residues outside the central region were predicted as potential phosphorylation sites by NetPhos2.0 with scores higher than 0.40. So, we examined 10 tyrosine residues in total as potential phosphorylation sites as follows.
FIGURE 1.

Identification of tyrosine phosphorylation sites in Git1 and their dephosphorylation by Ptprz. A, schematic representation of Git1 and the mutant constructs used in this study. Numbers correspond to amino acid residues of rat Git1. ARF-GAP, ADP-ribosylation factor GAP; Ank, ankyrin repeat; SHD, spa2 homology domain; FAH, FAT-homology domain. The region encoded by the positive clone isolated in the yeast substrate screening (9) is shown by a solid line underneath. B, tyrosine phosphorylation of Git1 mutants induced in cells by pervanadate treatment. Cells transfected with the expression construct for FLAG-tagged Git1 or its mutants shown in A were treated with 100 μm pervanadate for 15 min. Git1 proteins were immunoprecipitated using a mouse anti-FLAG M2 monoclonal antibody from the cell extracts and analyzed by Western blotting with a mouse anti-phosphotyrosine PY20 monoclonal antibody and rabbit anti-FLAG polyclonal antibodies. C, tyrosine phosphorylation of Git1 in cells expressing v-Src. The immunoprecipitation and Western blotting were performed as above. The lane order is the same as in B. D, dephosphorylation of Git1 by PtprzICR in vitro. The immunoprecipitants of the tyrosine-phosphorylated wild-type and mutant Git1 proteins were incubated with GST (as a negative control), GST-PtprzICR (the entire intracellular region of Ptprz fused to GST), or alkaline phosphatase (AP; as a positive control) for 37 °C for 20 min and then analyzed by Western blotting as above.
We first prepared an expression plasmid for Git1Y10F (see Fig. 1A) by replacing all 10 tyrosine residues of Git1 with phenylalanine residues and transfected it into HEK293T cells. When the tyrosine phosphorylation of Git1Y10F was examined by Western blotting following immunoprecipitation, we verified that this replacement resulted in the almost total disappearance of tyrosine phosphorylation in cells in the presence of pervanadate, a membrane-permeable nonspecific PTP inhibitor (compare lane 12 with lane 1 for wild-type Git1 in Fig. 1B).
We next constructed a series of Git1Y9F mutants in which individual phenylalanine residues among the 10 were reconverted to tyrosine (see Fig. 1A). The tyrosine phosphorylation analysis of these mutants revealed that phosphorylation occurs predominantly at Tyr-554, moderately at Tyr-392 and Tyr-607, and faintly at Tyr-519 of Git1 in cells treated with pervanadate (Fig. 1B). When the tyrosine phosphorylation of cellular proteins was enhanced by co-expression of v-Src (a constitutively active PTK), a similar pattern of Git1 phosphorylation was observed, except for further negligible phosphorylation at Tyr-519 (Fig. 1C), indicating that the tyrosine phosphorylation profile of Git1 is almost the same under PTP-inactivated and PTK-activated conditions.
When individual Git1 mutant proteins immunoprecipitated from pervanadate-treated cells were incubated with PtprzICR in vitro, PtprzICR markedly dephosphorylated wild-type Git1 and Git1Y9F-Y554 (in which only Tyr-554 is potentially phosphorylated), but not Git1Y9F-Y392, or -Y607 (Fig. 1D), indicating that PtprzICR has selectivity for Tyr(P)-554 in Git1. This notion was also supported by the finding that the Git1-Y554F mutant, in which only Tyr-554 is replaced by phenylalanine, was no longer dephosphorylated (Fig. 1D). We verified that all samples were completely dephosphorylated by alkaline phosphatase.
To directly assess the phosphorylation status of wild-type Git1 at Tyr-554 and its dephosphorylation by Ptprz, we generated a specific antibody against Tyr(P)-554 of Git1 (anti-pY554 Git1). When Git1 proteins were expressed in cells together with the wild-type Ptprz-B (Zwt) or PTP-inactive CS mutant of Ptprz-B (Zcs), overall tyrosine phosphorylation patterns of the total cellular proteins were seemingly identical among samples by Western blotting (Fig. 2A). However, immunoblotting with anti-pY554 Git1 revealed the phosphorylation at Tyr-554 to be decreased in cells expressing wild-type Ptprz-B, but not the CS mutant (Fig. 2B). Our previous results with a pan-anti-phosphotyrosine antibody, PY20, showed that the tyrosine phosphorylation of Git1 is markedly suppressed by Ptprz-B (11). Taken together, these results indicate that wild-type Git1 is preferentially phosphorylated at Tyr-554 and it is efficiently dephosphorylated by Ptprz. In contrast, the Git1-Y554F mutant showed no detectable signals with anti-pY554, also demonstrating the specificity of the antibody (Fig. 2B).
FIGURE 2.
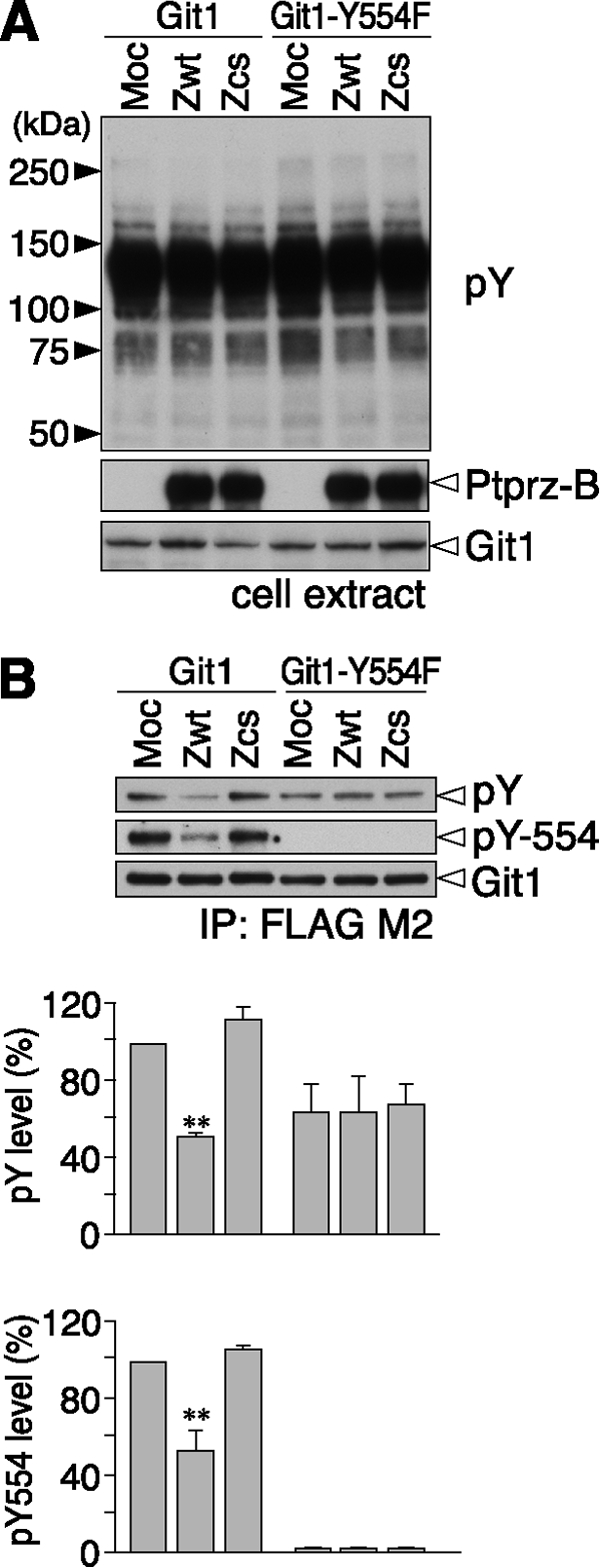
Dephosphorylation of the Git1 Tyr-554 site by Ptprz. A, expression construct for FLAG-tagged wild-type Git1 or its Y554F mutant was transfected into v-Src-HEK293T (a stable HEK293T cell line expressing v-Src) cells, together with the wild-type Ptprz-B (Zwt), PTP-inactive C1934S mutant of Ptprz-B (Zcs), or control vector (Moc). Cells were scraped and lysed after starvation in the serum-free medium for 3 h. The tyrosine phosphorylation of cellular proteins was examined by Western blotting using PY20, and protein expression was verified with rabbit anti-Ptprz-S and anti-FLAG antibodies. B, tyrosine phosphorylation of Git1 and Git1-Y554F was analyzed as above with PY20, rabbit anti-pY554-Git1, and anti-FLAG antibodies. The data are presented as the mean ± S.E. (error bars; n = 3). **, p < 0.01 compared with control Moc cells.
Dephosphorylation of Magi1 at Tyr-373 and Tyr-858 by Ptprz
As was the case for Git1, the first isolated clone of Magi1 (encoding amino acid residues 824–1247, see Fig. 3A) showed an increase in its binding ability to a Ptprz substrate-trapping mutant by tyrosine phosphorylation (10), indicating that the encoded region contains a dephosphorylation site(s) of Ptprz. To narrow the candidate substrate region, we first performed in vitro dephosphorylation assays using C-terminal deletion mutants of Magi1. Wild-type Magi1 and two C-terminal deletion derivatives (ΔC1, Δ1193–1247; and ΔC2, Δ984–1247), were efficiently dephosphorylated by PtprzICR, whereas a further C-terminal deletion mutant (ΔC3, Δ789–1247) showed partial resistance to the catalysis (Fig. 3B), suggesting that one of the potential substrate sites is present within the 789–983 region. In this region, only one phosphorylation site at Tyr-858 has been registered in public databases, such as PhosphoSitePlus and PHOSPHONET. So, we focused on Tyr-858 as a potential substrate site by using a mutant in which this tyrosine is replaced with phenylalanine (Magi1-Y858F). As was the case for Magi1-ΔC3, the Magi1-Y858F mutant was less dephosphorylated than wild-type Magi1 (Fig. 3C), indicating that the Tyr-858 site of Magi1 is most likely dephosphorylated by Ptprz. However, dephosphorylation of Magi1-Y858F was certainly detected with PtprzICR at a higher enzyme dosage, and this was completely inhibited with vanadate, suggesting the presence of another substrate site for Ptprz outside the C-terminal region.
FIGURE 3.

Dephosphorylation of Magi1 by Ptprz. A, schematic representation of wild-type Magi1 and its mutant constructs. GK, guanylate kinase; WW, a protein module with two highly conserved tryptophans. The region encoded by the positive clone isolated in the yeast substrate screening (10) is shown by the solid line underneath. B and C, dephosphorylation of Magi1 by PtprzICR. The immunoprecipitants of the tyrosine-phosphorylated Magi1 proteins shown in A were incubated in vitro with GST-PtprzICR for 20 min at 37 °C and analyzed by Western blotting as described in the Fig. 1 legend. The enzymatic reaction by PTP was verified by complete inhibition with vanadate (rightmost lanes in C).
In the cellular context, tyrosine phosphorylation of Magi1 was decreased when Ptprz-B was co-expressed (compare first lane with third lane in Fig. 4A) as described previously (11). Because Ptprz associates with PDZ domains of Magi1 through its C-terminal PDZ-binding motif (10), we further evaluated the effect of this binding by using a C-terminal PDZ-binding motif mutant of Ptprz (Zwt-SA). Notably, the dephosphorylation of wild-type Magi1 was partially suppressed by mutation of the PDZ-binding motif (compare second and third lanes in Fig. 4A). Magi1-Y858F was dephosphorylated by Ptprz-B in cells (compare first and third lanes in Fig. 4B), but the decrease in the tyrosine phosphorylation was reduced compared with wild-type Magi1, consistent with the results of the in vitro assay (see Fig. 3C). Intriguingly, unlike Magi1 WT, the level of dephosphorylation of Magi1 Y858F by the Ptprz-B SA mutant was almost equal to that by wild-type Ptprz-B (compare second and third lanes in Fig. 4B), suggesting that the dephosphorylation of Magi1 at Tyr-858 by Ptprz requires PDZ domain-mediated binding between Magi1 and Ptprz. Here, pulldown experiments using the C-terminal Ptprz fragment indicated that the second PDZ domain of Magi1 is responsible for the PDZ domain-mediated binding with Ptprz, along with a weak interaction with the fourth/fifth PDZ domains (Fig. 5).
FIGURE 4.

Dephosphorylation of the Magi1 Tyr-373 and Tyr-858 site by Ptprz. The expression construct for FLAG-tagged wild-type Magi1 (A), or its mutant Y858F (B), Y373F (C), or Y373:858F (D) was transfected into v-Src-HEK293T cells, together with wild-type Ptprz-B (Zwt), a Ptprz-B mutant in its C-terminal PDZ-binding motif (Zwt-SA), or control vector (Moc). Cell extracts were prepared after starvation in the serum-free medium for 3 h. The tyrosine phosphorylation of cellular proteins and protein expression were analyzed with anti-phosphotyrosine, anti-RPTPβ, and anti-FLAG antibodies. The tyrosine phosphorylation of Magi1 proteins was analyzed as above with PY20 and anti-FLAG antibodies, and the data are presented as the mean ± S.E. (error bars; n = 4). *, p < 0.05; **, p < 0.01 compared with control Moc cells.
FIGURE 5.
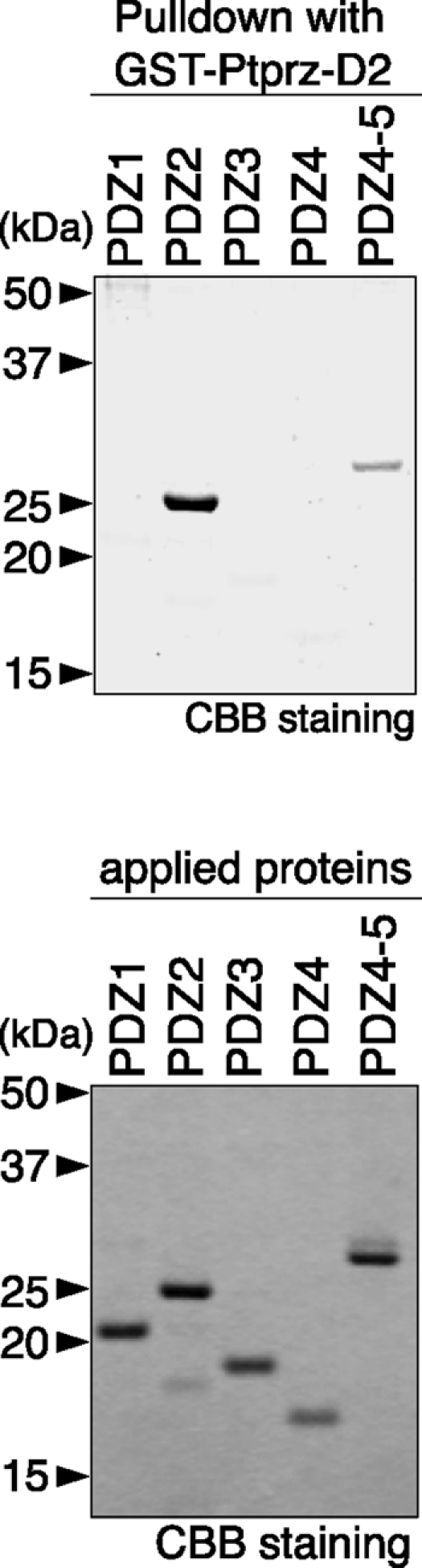
Binding between the C terminus of Ptprz and PDZ domains of Magi1. Purified His-tagged Magi1-PDZ domains shown in Fig. 3A were incubated with GST-Ptprz-D2 (the second PTP domain and the C-terminal tail of Ptprz)-immobilized beads. Bound proteins were analyzed by SDS-PAGE, followed by Coomassie Brilliant Blue R-250 (CBB) staining (upper panel). The lower panel shows the result of CBB staining of the applied Magi1 proteins.
Our preliminary mass spectrometric studies detected that two tyrosine residues at positions 373 and 858 in FLAG-tagged Magi1 were phosphorylated in cells when v-Src was co-expressed (data not shown). So, we asked whether Tyr-373 is another potential substrate site of Magi1. We constructed a single point mutant Y373F and a double mutant Y373:858F (see Fig. 3A): Unfortunately, significant tyrosine phosphorylation of Y373:858F was still observed in cells stably expressing v-Src, indicating the existence of other phosphorylation sites in this cellular context (Fig. 4D). We verified that the tyrosine phosphorylation of Magi1-Y373F was decreased by Ptprz-B (compare first and third lanes in Fig. 4C), as well as Magi1-Y858F (Fig. 4B). However, that of the double mutant was no longer decreased (compare first and third lanes in Fig. 4D), indicating that the Tyr-373 and Tyr-858 residues in Magi1 are primary substrate sites for Ptprz. Although Ptprz-B dephosphorylated both Magi1-Y373F and -Y858F, the dephosphorylation activity for the Y373F mutant was selectively abolished by mutation of its PDZ-binding motif (compare first and second lanes in Fig. 4C). The dephosphorylation at Tyr-858 by Ptprz thus occurs dependent on the PDZ binding, in contrast to that for the Tyr-373 site.
Identification of the Substrate Site Motif for Ptprz
Alignment of the primary sequences surrounding the target Tyr(P) residues in Git1 and Magi1 identified here, and p190RhoGAP previously (7), demonstrated a marked similarity (Fig. 6A). To understand the substrate specificity of Ptprz in more detail, we performed an in vitro assay using fluorogenic peptide-based PTP substrates. Peptides containing pCAP are highly sensitive fluorogenic probes for PTPase assays, and the PTPase-catalyzed hydrolysis of pCAP-peptide substrates can be monitored by the increase in fluorescence (15).
FIGURE 6.

Identification of the substrate motif for Ptprz. A, primary sequences surrounding the target phosphotyrosine residues located in Git1 (Tyr-554), p190RhoGAP (Tyr-1105), Magi1 (Tyr-373 and Tyr-858), and paxillin (Tyr-118) aligned, together with four substrate candidates, AFAP1L2 (Tyr-56), KIAA1486 (Tyr-277), UBR3 (Tyr-1284), and TJP2/ZO-2 (Tyr-486). Boxed residues are conserved in at least three substrate sequences. B, kinetic analysis for the hydrolysis of Git1-derived peptide substrates by PtprzICR in vitro. The amino acid sequences of the substrate peptides (P1–10) used in this study are shown on the left of the figure. P1, P8, and P9 peptides correspond to Git1550–556, Git1549–556, and Git1548–556, respectively, except that the Tyr residue at 554 is replaced with a phosphotyrosine mimic, pCAP. P1–P7 and P10 peptides are Git1 peptide derivatives, in which the substituted amino acid residues are shown in bold. The assay was performed as described under “Experimental Procedures.” C, proposed consensus substrate-site motif for the PTP catalytic domain of Ptprz.
We first selected the Git1550–556 sequence as the template (P1 in Fig. 6B), and examined the contribution of individual amino acid side chains (from position −4 to +1) to catalytic affinity by replacing each amino acid residue with other residues. The largest effect was observed when the acidic Asp at −4 or −3 was replaced with the nonpolar Ala: the hydrolysis was drastically reduced by 60∼70% compared with that of the wild-type Git1550–556 peptide (P2 and P3). Substitution of the hydrophobic Ile at −1 with hydrophilic Asp had a moderate inhibitory effect (P5). Because phosphorylation of Ser-555 in Git1 was reported (16), we examined its potential effect on the hydrolysis. Substitution of the Ser at +1 with Ser(P) or acidic Asp significantly reduced the efficiency by 30∼40% (P6 and P7), indicating that the substrate property of Tyr(P)-554 is partly impaired by simultaneous phosphorylation of Ser at +1. Replacement of Ala at −2 with an acid residue had only a slight effect (P4).
Based on the findings that PtprzICR shows a strong preference for acidic residues at −4 and −3, we further examined the contribution of the acidic Glu residue at −5 to the catalysis, which is conserved in the Git1 Tyr-554 and p190RhoGAP Tyr-1105 sites. The addition of Glu on the N-terminal side of the Git1550–556 peptide (P1), namely Git1549–556 (P8), did increase the hydrolysis by ∼40% compared with that of the parental P1 peptide, whereas the addition of Leu at position −6 (P9) did not result in any further improvement. Substitution of the acidic Glu at −5 with the nonpolar Ala (P10) exerted an inhibitory effect on the parental P8 peptide, returning to the same level as the P1 peptide. Thus, acidic residues at positions −5 to −3 and hydrophobic or nonpolar residues at positions −1 and +1 were revealed to be important for the substrate recognition by the in vitro assay (Fig. 6C).
As mentioned above, the Magi1 Tyr-858 site has only one acidic residue at −3 position, and its dephosphorylation by Ptprz required the PDZ binding activity. To confirm the validity of the sequence preference for the conserved acidic residues located on the N-terminal side in a cellular context, we generated an additional construct by replacement of Gly residue at position −4 of Tyr-858 with Asp from Magi1 Y373F. We tested the possibility that this site can be dephosphorylated by the PDZ-binding motif mutant of Ptprz-B. Intriguingly, the introduction of Asp at position −4 changed the Tyr-858 site to be susceptible to dephosphorylation by Ptprz-B without the PDZ binding in the cell-based assay (Fig. 7). Thus, the sequence motif surrounding a phosphotyrosine residue is crucial for the direct substrate recognition by the catalytic PTP domain of Ptprz also in the cellular context, as in the in vitro context.
FIGURE 7.
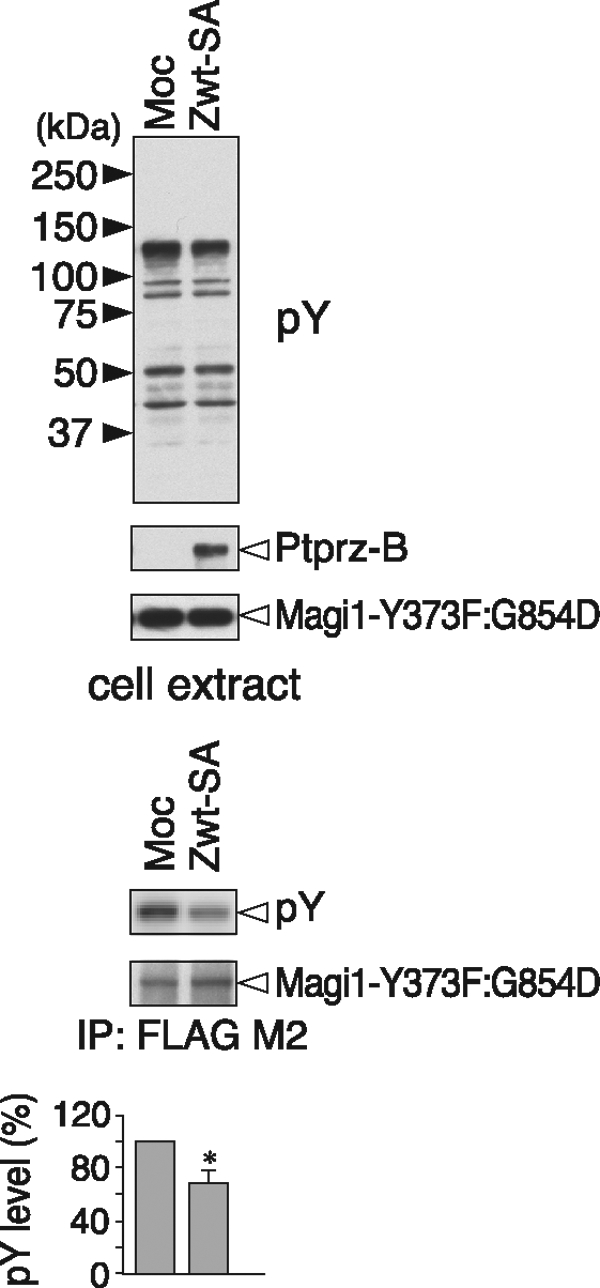
Relevance of the sequence motif for the substrate recognition of Ptprz in cells. The expression construct for FLAG-tagged Magi1-Y373F:G854D mutant was transfected into v-Src-HEK293T cells, together with the C-terminal PDZ-binding motif mutant of Ptprz-B (Zwt-SA), or control vector (Moc). The tyrosine phosphorylation and protein expression were analyzed as above. The data are presented as the mean ± S.E. (error bars; n = 3). *, p < 0.05 compared with control Moc cells.
Dephosphorylation of Paxillin at Tyr-118 by Ptprz
The substrate specificity profile provided us with a rational basis for the prediction of other candidate substrate proteins. A search of the PhosphoSitePlus database with the consensus sequence shown in Fig. 6C produced five hits: the sequences surrounding Tyr-118 of mouse paxillin, Tyr-56 of mouse actin filament-associated protein 1-like 2 (AFAP1L2), Tyr-277 of mouse KIAA1486, Tyr-1284 of mouse ubiquitin protein ligase E3 component n-recognin 3 (UBR3), and Tyr-486 of mouse tight junction protein 2 (TJP2/ZO-2) (see Fig. 6A, lower five proteins; the five sites are conserved in mouse, rat, and human). Among them, we focused on paxillin because midkine, a ligand for Ptprz, induces haptotactic migration of UMR-106, osteoblast-like cells that express Ptprz (17). Upon midkine application, the tyrosine phosphorylation of paxillin is stimulated (17).
Paxillin is a substrate for the focal adhesion kinase-Src complex that functions as an adaptor for various signaling molecules and structural proteins in adhesions, in which paxillin interacts with several molecules including Git1 (for review, see Ref. 18). The phosphorylation of Tyr-118 in paxillin has been well characterized, and a specific antibody against Tyr(P)-118 paxillin is available. To clarify whether paxillin Tyr-118 is indeed a substrate site for Ptprz, we expressed FLAG-tagged paxillin in cells together with Ptprz-B and analyzed the phosphorylation of the target site of paxillin. Immunoblotting with the specific antibody against Tyr(P)-118 paxillin revealed that the phosphorylation at Tyr-118 of paxillin is clearly decreased by co-expression of the wild-type Ptprz-B, but not of the PTP-inactive CS mutant of Ptprz-B compared with that in mock-transfected cells (Fig. 8). This strongly suggests that paxillin is a physiological substrate of Ptprz.
FIGURE 8.
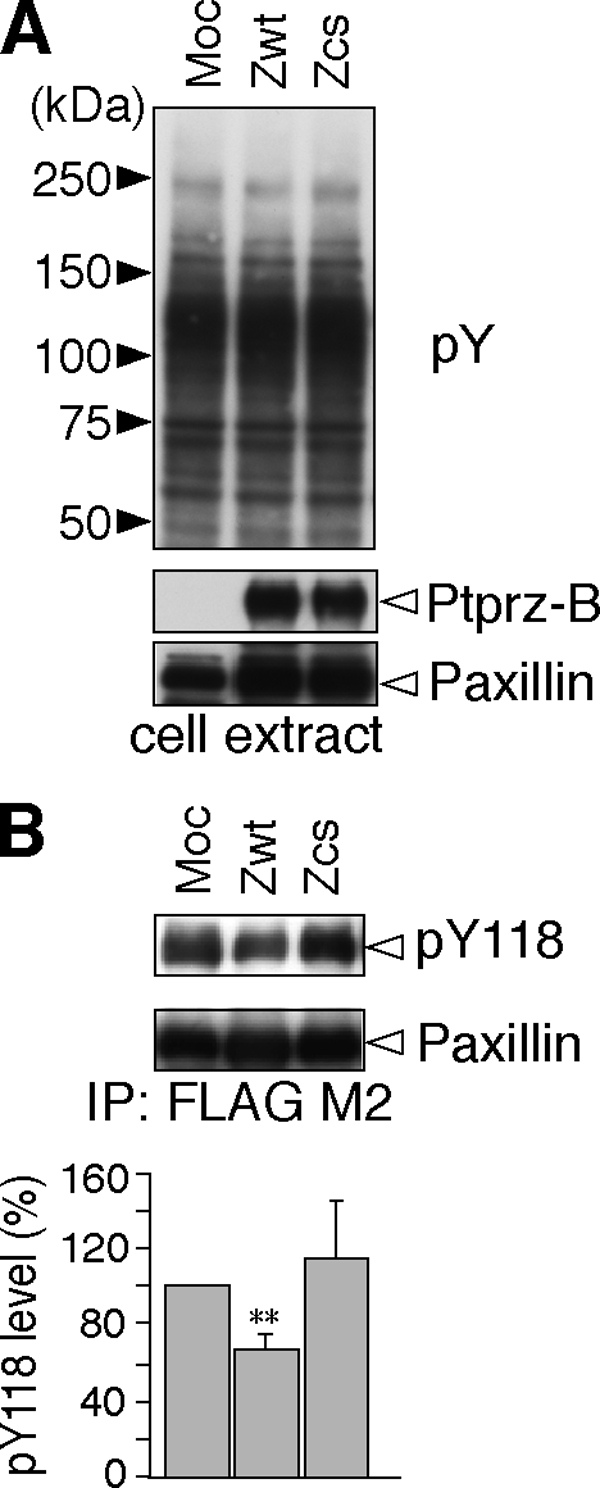
Dephosphorylation of the paxillin Tyr-118 site by Ptprz. A, expression construct for FLAG-tagged paxillin was transfected into v-Src-HEK293T cells with Ptprz-B (Zwt), Ptprz-B-CS mutant (Zcs), or control vector (Moc). After 3-h serum starvation, the tyrosine phosphorylation of cellular proteins and protein expression of Ptprz and paxillin were verified with PY20, anti-RPTPβ, and anti-FLAG, respectively. B, tyrosine phosphorylation of paxillin was analyzed as above with anti-pY118-paxillin. The amount of immunoprecipitated paxillin proteins was evaluated with anti-FLAG. The data are presented as the mean ± S.E. (error bars; n = 4). **, p < 0.01 compared with control Moc cells.
DISCUSSION
In this study, we found that there is considerable similarity in the dephosphorylation sites in the substrates of Ptprz, such as p190RhoGAP (7), Git1, and Magi1. We next determined the motif for the specificity of Ptprz by examining the contribution of respective amino acid side chains. The consensus motif for Ptprz thus identified is Glu/Asp-Glu/Asp-Glu/Asp-Xaa-Ile/Val-Tyr(P)-Xaa (Xaa is not an acidic residue): This sequence is quite different from Glu/Asp-Tyr(P)-Tyr(P)-Arg/Lys identified for the substrate site of PTP1B (2). Furthermore, we successfully discovered new substrates of Ptprz based on this motif. Although initially viewed as broad specificity “housekeeping” enzymes, PTPs are actually highly selective enzymes.
Consensus Substrate Motif for Ptprz
We used v-Src to phosphorylate the prey in the yeast substrate-trapping screening and isolated several proteins as substrates of Ptprz (9, 10). We have found in this screening that v-Src phosphorylated a variety of cellular proteins, but only a few clones were identified as substrates of Ptprz (10). As such, the substrate recognition site in physiological proteins appeared to be highly specific to the catalytic PTP domain of Ptprz.
The Git1 Tyr-554 and p190RhoGAP Tyr-1105 sites contain three, and the Magi1 Tyr-373 site two consecutive acidic residues at positions −5 to −3 on the N-terminal side. The peptide dephosphorylation assays in vitro revealed that additional replacement of the acidic residues at −5, −4, or −3 with nonpolar Ala residues proportionally reduced the hydrolysis by PtprzICR (Fig. 6B). In addition, the Magi1 Tyr-858 site that has only one acidic residue on the N-terminal side (Glu at −3) required the PDZ-mediated interaction for the dephosphorylation by Ptprz (Fig. 4C). However, the introduction of Asp at position −4 changed the Tyr-858 site to be susceptible to dephosphorylation by Ptprz without the PDZ-mediated interaction in the cellular context (Fig. 7). It thus seems that the number of acidic residues from −5 to −3 is an important factor, and at least two acidic residues are required for direct substrate recognition by the catalytic PTP domain of Ptprz.
Git1 Tyr-554, p190RhoGAP Tyr-1105, and paxillin Tyr-118 have a serine residue at +1 on the C-terminal side (Fig. 6A). When Ser-555 at position +1 in the Git1 sequence was replaced with a phosphoserine or acidic Asp, PtprzICR showed significantly less catalytic activity in the in vitro peptide assays (P6 and P7 in Fig. 6B). Interestingly, the phosphorylation of Ser-555 in Git1 (16) and the simultaneous phosphorylation of Tyr-118 and Ser-119 in paxillin (19) have been identified by mass spectrometry. It is thus likely that the phosphorylation of these serine residues at +1 serves a specific regulatory function in some physiological context by preventing Ptprz-mediated dephosphorylation.
Another Factor for Recognition of Substrates by Ptprz: Assisted Recognition
In addition to the sequence preference of substrates, other factors such as substrate localization, accessibility, and protein associations are also crucial for the substrate recognition. It should be noted that some groups including ours have already reported several proteins as Ptprz substrates, in which the typical consensus substrate motif proposed here is not found. We found several PDZ proteins among Ptprz substrates and Ptprz-interacting proteins in our screening (10). In the RPTP family, only the R5 subfamily members (Ptprz and Ptprg) have a canonical PDZ-binding tail that interacts with various PDZ proteins (9, 10, 20). It is conceivable that some PDZ proteins can provide substrates to Ptprz by setting them in close vicinity to the catalytic domain. Consistent with this view, the PDZ-binding activity of Ptprz is required for the dephosphorylation of the Magi1 Tyr-858 site (Fig. 4), also suggesting that the sequence surrounding Tyr-858 is not fully favorable for the catalytic PTP domain of Ptprz to recognize it as substrate directly (Fig. 7).
In addition, we reported that ErbB4 is a substrate for Ptprz (13). Ptprz pulled down ErbB4 together with PSD95 from rat brain lysate (13). ErbB4 is a member of the ErbB family of tyrosine kinases known as neuregulin receptors. Both Ptprz and ErbB4 bind to PSD95 in the second and the first/second PDZ domains, respectively. Of note is that expression of PSD95 is required for dephosphorylation of ErbB4 by Ptprz in HEK293T cells (13).
Furthermore, we recently found that TrkA is efficiently dephosphorylated by Ptprz at Tyr-674, Tyr-675, or both in the kinase domain (21). TrkA is a member of the Trk family of receptor tyrosine kinases and reportedly bound to the PDZ domain of GIPC (GAIP- interacting protein, C terminus) (22). The sequence of the substrate site in TrkA, Tyr-Ser-Thr-Asp-Tyr(674)-Tyr(675)-Arg, does not match the substrate motif demonstrated here well; however, several tyrosine residues including Tyr-670, Tyr-674, and Tyr-675 in TrkA are autophosphorylated upon NGF binding (23). It is tempting to speculate that a critical acidic residue is regularly provided by the phosphorylation at Tyr-670 located on the N-terminal side of the target Tyr(P) (674, 675, or both).
Other groups have reported that β-catenin (24) and the sodium channel (Nav1.2) α subunit (25) are substrates for Ptprz. Because β-catenin reportedly binds to the fifth PDZ domain of Magi1 (26), Ptprz and β-catenin may exhibit an enzyme-substrate relation on the Magi1 scaffold. Likewise, the interaction between the sodium channel and Ptprz is presumably mediated by syntrophin because sodium channels reportedly bind to the first pleckstrin homology domain or the syntrophin-unique domain of syntrophin (27), and Ptprz also binds to the PDZ domain of syntrophin (10). PDZ-mediated interaction is a general mechanism by which membrane-associated proteins are recruited to specific subcellular regions to form signaling complexes. Some proteins associated with PDZ proteins together with Ptprz are thus likely dephosphorylated by Ptprz without a strict substrate recognition structure.
Functional Regulation of Ptprz Substrates by Dephosphorylation
p190RhoGAP, Git1, and paxillin are key regulators of the actin cytoarchitecture important for eukaryotic cell migration and adhesion, acting through the regulation of cellular protrusive activity by targeting multiprotein signaling complexes. The phosphorylated Tyr-1105 in p190RhoGAP and the phosphorylated Tyr-118 in paxillin constitute binding sites for a SH2 domain of p120 RasGAP (28). In mammary gland epithelial cells, it was reported that the Tyr-31/Tyr-118-phosphorylated paxillin competes with p190RhoGAP for binding to p120RasGAP and that p190RhoGAP freed from p120RasGAP efficiently suppresses RhoA activity in cell detachment and migration (28). Git1 forms a stable complex with the p21-activated kinases interacting exchange factor (PIX) family, Rho guanine nucleotide exchange factors. One important function of Git1 is to link a complex of p21-activated kinase and p21-activated kinases interacting exchange factor to remodeling focal adhesions by interacting with paxillin or its homolog Hic-5 (29). By enhancing focal adhesion turnover, Git1 might facilitate cell migration and spreading (30). One should note that the phosphorylation of the Git1 Tyr-554 site attenuates the association with Hic-5.3
Magi1 constitutes the Magi family together with Magi2 and Magi3. Although the functional consequences of the phosphorylation at the Tyr-373 site in the second WW domain and at the Tyr-858 site in the third PDZ domain remain to be explored, the binding properties of each domain of Magi1 are probably affected. Magi2 is known as a synaptic scaffolding molecule (S-SCAM) interacting with a wide variety of molecules at excitatory and inhibitory synapses (31 and references cited therein). Interestingly, the sequence surrounding Tyr-361 of Magi2 is completely identical to the Magi1-Tyr-373 substrate site, and the sequence of Gly-Asp-Glu-Leu-Val-Tyr(826)-Val of Magi2 is better matched to the substrate motif than the Magi1-Tyr-858 site: The Magi2-Tyr-826 site contains two acidic residues at positions −4 and −3, whereas the Magi1-Tyr-858 site contains one acidic residue at position −3. The phosphorylation of Tyr-361 and Tyr-826 in Magi2 has been registered in the public databases mentioned above, suggesting that the two are substrate sites for Ptprz. Indeed, the tyrosine phosphorylation of Magi2 was equally decreased by co-expression of the wild-type Ptprz-B and its PDZ-binding motif mutant (supplemental Fig. S1). On the other hand, the sequences surrounding the corresponding tyrosine residues in Magi3 differ from those of Magi1 and Magi2, and there is no evidence of their phosphorylation.
Potential Physiological Roles of Ptprz Signaling
The physiological importance of Ptprz has been demonstrated through studies with knock-out mice (5–8). Adult Ptprz-deficient mice exhibit an enhancement of long term potentiation in the CA1 region of the hippocampus along with learning deficits (6, 7). We found that phosphorylation of the p190RhoGAP Tyr-1105 site is decreased by contextual fear conditioning in the hippocampus of wild-type mice, and the phosphorylation of Tyr-1105 positively regulates its GAP activity (7). Git1 is also expressed in hippocampal neurons (32). Very recently, two independent groups reported that Git1 knock-out mice showed deficits in conditioned fear (33) and impaired formation of dendritic spines in the hippocampus (34). As described above, Magi2 (S-SCAM) is also probable as a physiological substrate for Ptprz at central synapses. Primary cultured hippocampal neurons from mutant mice lacking S-SCAMα, the longest isoform of S-SCAM, harbor abnormally elongated dendritic spines (31). Thus, we consider Ptprz signaling mediated by these substrates to be the molecular basis of synaptic plasticity through actin-dependent morphological changes on the postsynaptic side (for review, see Ref. 35). In line with this view, we reported that Ptprz in the stomach functions as the receptor of VacA, a cytotoxin secreted by H. pylori, which induces gastric ulcers (8). The binding of VacA to Ptprz induces erroneous signaling in gastric epithelial cells, which likely impairs the actin cytoskeleton network and focal adhesion complex, leading to detachment from the basement membrane (8).
In conclusion, the present study provides key insights into the molecular basis by which Ptprz recognizes substrates. Our knowledge of substrates for Ptprz suggests that Ptprz dephosphorylates multiple proteins associated with actin remodeling, which plays important roles in synaptic plasticity in the adult brain and cell adhesion/migration of epithelial cells.
Acknowledgments
We thank HiPep laboratories (Kyoto, Japan) for preparing the peptides used in this study, N. Nakanishi for technical assistance, and A. Kodama for secretarial assistance. A F-4500 spectrofluorometer was used at the National Institute for Basic Biology Center for Analytical Instruments.
This work was supported by grants-in-aid from the Ministry of Education, Science, Sports, and Culture of Japan (MEXT).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
A. Fujikawa and M. Noda, unpublished observation.
- PTK
- protein-tyrosine kinase
- bis-tris
- bis(2-hydroxyethyl)iminotris(hydroxymethyl)methane
- GAP
- GTPase-activating protein
- Git1
- G protein-coupled receptor kinase-interactor 1
- ICR
- Intracellular region
- Magi1
- membrane-associated guanylate kinase, WW and PDZ domain containing 1
- p190RhoGAP
- GAP for Rho GTPase
- pCAP
- phosphocoumaryl-aminopropionic acid
- PDZ
- PSD95/Disc large/zona occludens1
- PTP
- protein-tyrosine phosphatase
- Ptprz
- PTP receptor type Z
- RPTP
- receptor-like PTP
- S-SCAM
- synaptic scaffolding molecule.
REFERENCES
- 1. Tonks N. K. (2006) Nat. Rev. Mol. Cell. Biol. 7, 833–846 [DOI] [PubMed] [Google Scholar]
- 2. Myers M. P., Andersen J. N., Cheng A., Tremblay M. L., Horvath C. M., Parisien J. P., Salmeen A., Barford D., Tonks N. K. (2001) J. Biol. Chem. 276, 47771–47774 [DOI] [PubMed] [Google Scholar]
- 3. Nishiwaki T., Maeda N., Noda M. (1998) J. Biochem. 123, 458–467 [DOI] [PubMed] [Google Scholar]
- 4. Chow J. P., Fujikawa A., Shimizu H., Suzuki R., Noda M. (2008) J. Biol. Chem. 283, 30879–30889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shintani T., Watanabe E., Maeda N., Noda M. (1998) Neurosci. Lett. 247, 135–138 [DOI] [PubMed] [Google Scholar]
- 6. Niisato K., Fujikawa A., Komai S., Shintani T., Watanabe E., Sakaguchi G., Katsuura G., Manabe T., Noda M. (2005) J. Neurosci. 25, 1081–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tamura H., Fukada M., Fujikawa A., Noda M. (2006) Neurosci. Lett. 399, 33–38 [DOI] [PubMed] [Google Scholar]
- 8. Fujikawa A., Shirasaka D., Yamamoto S., Ota H., Yahiro K., Fukada M., Shintani T., Wada A., Aoyama N., Hirayama T., Fukamachi H., Noda M. (2003) Nat. Genet. 33, 375–381 [DOI] [PubMed] [Google Scholar]
- 9. Kawachi H., Fujikawa A., Maeda N., Noda M. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 6593–6598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fukada M., Kawachi H., Fujikawa A., Noda M. (2005) Methods 35, 54–63 [DOI] [PubMed] [Google Scholar]
- 11. Fukada M., Fujikawa A., Chow J. P., Ikematsu S., Sakuma S., Noda M. (2006) FEBS Lett. 580, 4051–4056 [DOI] [PubMed] [Google Scholar]
- 12. Nakamura K., Yano H., Uchida H., Hashimoto S., Schaefer E., Sabe H. (2000) J. Biol. Chem. 275, 27155–27164 [DOI] [PubMed] [Google Scholar]
- 13. Fujikawa A., Chow J. P., Shimizu H., Fukada M., Suzuki R., Noda M. (2007) J. Biochem. 142, 343–350 [DOI] [PubMed] [Google Scholar]
- 14. Sabe H., Okada M., Nakagawa H., Hanafusa H. (1992) Mol. Cell. Biol. 12, 4706–4713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mitra S., Barrios A. M. (2005) Bioorg. Med. Chem. Lett. 15, 5142–5145 [DOI] [PubMed] [Google Scholar]
- 16. Webb D. J., Mayhew M. W., Kovalenko M., Schroeder M. J., Jeffery E. D., Whitmore L., Shabanowitz J., Hunt D. F., Horwitz A. F. (2006) J. Cell Sci. 119, 2847–2850 [DOI] [PubMed] [Google Scholar]
- 17. Muramatsu H., Zou P., Suzuki H., Oda Y., Chen G. Y., Sakaguchi N., Sakuma S., Maeda N., Noda M., Takada Y., Muramatsu T. (2004) J. Cell Sci. 117, 5405–5415 [DOI] [PubMed] [Google Scholar]
- 18. Deakin N. O., Turner C. E. (2008) J. Cell Sci. 121, 2435–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Webb D. J., Schroeder M. J., Brame C. J., Whitmore L., Shabanowitz J., Hunt D. F., Horwitz A. R. (2005) J. Cell Sci. 118, 4925–4929 [DOI] [PubMed] [Google Scholar]
- 20. Kawachi H., Tamura H., Watakabe I., Shintani T., Maeda N., Noda M. (1999) Mol. Brain Res. 72, 47–54 [DOI] [PubMed] [Google Scholar]
- 21. Shintani T., Noda M. (2008) J. Biochem. 144, 259–266 [DOI] [PubMed] [Google Scholar]
- 22. Lou X., Yano H., Lee F., Chao M. V., Farquhar M. G. (2001) Mol. Biol. Cell 12, 615–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cunningham M. E., Stephens R. M., Kaplan D. R., Greene L. A. (1997) J. Biol. Chem. 272, 10957–10967 [DOI] [PubMed] [Google Scholar]
- 24. Meng K., Rodriguez-Peña A., Dimitrov T., Chen W., Yamin M., Noda M., Deuel T. F. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 2603–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ratcliffe C. F., Qu Y., McCormick K. A., Tibbs V. C., Dixon J. E., Scheuer T., Catterall W. A. (2000) Nat. Neurosci. 3, 437–444 [DOI] [PubMed] [Google Scholar]
- 26. Dobrosotskaya I. Y., James G. L. (2000) Biochem. Biophys. Res. Commun. 270, 903–909 [DOI] [PubMed] [Google Scholar]
- 27. Gee S. H., Madhavan R., Levinson S. R., Caldwell J. H., Sealock R., Froehner S. C. (1998) J. Neurosci. 18, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsubouchi A., Sakakura J., Yagi R., Mazaki Y., Schaefer E., Yano H., Sabe H. (2002) J. Cell Biol. 159, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schmalzigaug R., Garron M. L., Roseman J. T., Xing Y., Davidson C. E., Arold S. T., Premont R. T. (2007) Cell. Signal. 19, 1733–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Nieuw Amerongen G. P., Natarajan K., Yin G., Hoefen R. J., Osawa M., Haendeler J., Ridley A. J., Fujiwara K., van Hinsbergh V. W., Berk B. C. (2004) Circ. Res. 94, 1041–1049 [DOI] [PubMed] [Google Scholar]
- 31. Iida J., Ishizaki H., Okamoto-Tanaka M., Kawata A., Sumita K., Ohgake S., Sato Y., Yorifuji H., Nukina N., Ohashi K., Mizuno K., Tsutsumi T., Mizoguchi A., Miyoshi J., Takai Y., Hata Y. (2007) Mol. Cell. Biol. 27, 4388–4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang H., Webb D. J., Asmussen H., Niu S., Horwitz A. F. (2005) J. Neurosci. 25, 3379–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmalzigaug R., Rodriguiz R. M., Bonner P. E., Davidson C. E., Wetsel W. C., Premont R. T. (2009) Neurosci. Lett. 458, 79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Menon P., Deane R., Sagare A., Lane S. M., Zarcone T. J., O'Dell M. R., Yan C., Zlokovic B. V., Berk B. C. (2010) Brain Res. 1317, 218–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lamprecht R., LeDoux J. (2004) Nat. Rev. Neurosci. 5, 45–54 [DOI] [PubMed] [Google Scholar]