

Background: OXA-58 is a carbapenem-hydrolyzing class D β-lactamase (CHDL) found in Acinetobacter baumannii.
Results: OXA-58 exploits a carbamylated lysine in its catalysis. The deacylating water molecule comes from the α-face.
Conclusion: CHDLs employ the same hydrolytic machinery as oxacillinases. Structural changes in the active site may lead to imipenem hydrolysis.
Significance: This study provides insights for the design of CHDL inactivators.
Keywords: Antibiotic Resistance, Carboxylation, Enzyme Inactivation, Enzyme Kinetics, Enzyme Mechanisms, beta-Lactamases, Carbapenem-hydrolyzing beta-Lactamases, OXA-58, Carbamylation, Hydroxyalkylpenicillanate
Abstract
Carbapenem-hydrolyzing class D β-lactamases (CHDLs) represent an emerging antibiotic resistance mechanism encountered among the most opportunistic Gram-negative bacterial pathogens. We report here the substrate kinetics and mechanistic characterization of a prominent CHDL, the OXA-58 enzyme, from Acinetobacter baumannii. OXA-58 uses a carbamylated lysine to activate the nucleophilic serine used for β-lactam hydrolysis. The deacylating water molecule approaches the acyl-enzyme species, anchored at this serine (Ser-83), from the α-face. Our data show that OXA-58 retains the catalytic machinery found in class D β-lactamases, of which OXA-10 is representative. Comparison of the homology model of OXA-58 and the recently solved crystal structures of OXA-24 and OXA-48 with the OXA-10 crystal structure suggests that these CHDLs have evolved the ability to hydrolyze imipenem, an important carbapenem in clinical use, by subtle structural changes in the active site. These changes may contribute to tighter binding of imipenem to the active site and removal of steric hindrances from the path of the deacylating water molecule.
Introduction
Carbapenems such as imipenem and meropenem are β-lactam antibiotics with a wide spectrum of activity, initially designed to overcome β-lactam resistance because of their stability toward most β-lactamases (1, 2). However, as a result of selective pressure, these carbapenems have succumbed to the hydrolytic power of β-lactamases. β-Lactamases are grouped into four classes. Classes A, C, and D are serine-active site β-lactamases. Class B β-lactamases are metalloenzymes (3, 4).
Following the clinical introduction of the carbapenems ∼30 years ago, the progressively increasing ability of all four classes of β-lactamases to hydrolyze carbapenems has been documented (5–7). New carbapenem-hydrolyzing class D β-lactamases (CHDLs)2 are directly associated with outbreaks of carbapenem-resistant Acinetobacter baumannii around the world (8–11). Carbapenems have been the drug of choice in treatment of multidrug-resistant A. baumannii. Almost 45 variants of class D β-lactamases are reported to possess carbapenemase activity (12).
Class D β-lactamases are also known as oxacillinases (OXA β-lactamases) due to their ability to hydrolyze oxacillin, a penicillin β-lactam. The OXA β-lactamases were first observed in the late 1980s as a resistance mechanism against imipenem, the first clinically used carbapenem (13). Sequence alignments indicate that CHDLs share 18% sequence identity with oxacillinases, and 40–90% sequence identity with each other (7, 12).
CHDLs are divided into nine distinct subgroups (7, 12). The main acquired and most widely spread carbapenemases in A. baumannii are OXA-23, OXA-24, and OXA-58, which represent three different CHDL groups and are encoded as either chromosomal or plasmid-borne genes (8, 14). Of these three group representatives, only the structure of OXA-24 has been elucidated (15–17). Structural analysis and mutagenesis studies of OXA-24 suggest that a hydrophobic cleft over the active site could be responsible for the widened substrate specificity of these enzymes; imipenem is hydrolyzed about 10-fold more efficiently than oxacillin (15).
Recently, the crystal structure of OXA-48, a CHDL prevalent in Klebsiella pneumoniae, was solved (18). The structure reveals that OXA-48 lacks the hydrophobic cleft over the active site that is seen in OXA-24 and also that it has a differently shaped active site (18). It was reported that OXA-48 might employ a different catalytic mechanism (18). These structural studies suggest that each of these CHDL subgroups have arisen via different evolutionary pathways. Given the ever-increasing diversity of CHDLs, and the direct relevance of this diversity to bacterial carbapenem resistance, there is an urgent need to investigate the mechanism of carbapenem hydrolysis employed by CHDLs and to translate this knowledge into the design of novel β-lactamase-resistant β-lactam antibiotics.
In this study, we evaluated the catalytic mechanism of OXA-58. This β-lactamase is the only member of a subgroup of CHDLs (7, 12) and shares less than 50% sequence identity with other CHDL subgroups (7, 12). OXA-58 was first identified in 2003 in France (19), and since then it has been isolated in different parts of the world (7, 8). Our study addresses the carbamylation state of its conserved lysine residue, and the role of the lysine-carbamate functional group in OXA-58 catalysis. In addition, we have probed the catalytic mechanism of OXA-58 with a series of 6α- and 6β-hydroxyalkyl penicillin derivatives (20, 21). Our study provides insights into the mechanism of inhibition of these enzymes by 6α-hydroxyalkyl penicillanate, and the mechanism of carbapenem hydrolysis by CHDLs.
EXPERIMENTAL PROCEDURES
All chemicals and antibiotics were purchased from Sigma or Fisher unless otherwise stated. Growth media were purchased from EMD Biosciences (Mississauga, Ontario, Canada). Chromatography media and columns were purchased from GE Healthcare. Escherichia coli Nova Blue and BL21(DE3) strains and cloning and expression plasmids were purchased from Novagen (Mississauga, Ontario, Canada). Restriction enzymes were purchased from New England Biolabs (Pickering, Ontario, Canada) or Agilent Technologies (Wilmington, DE). D2O (D, 99.9%) and sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) were manufactured by Cambridge Isotope Laboratories Inc. (Andover, MA) and purchased from Sigma.
Cloning of the blaOXA-58ΔSP Gene
The OXA-58 amino acid sequence was analyzed using the on-line software SignalP 3.0 offered via the ExPASy Proteomics Server of the Swiss Institute of Bioinformatics. The software predicted a 0.997 probability of a signal peptide in the first 24 amino acids at the N-terminal region. The DNA sequence of the gene (843 bp) that encodes for OXA-58 without the signal peptide (blaOXA-58ΔSP) was amplified from the A. baumannii genome using Pfu TurboTM DNA polymerase and the following primers: Oxa58F, 5′-ACGCCATGGCTAGTCGAGCAAAAACAAGTACAA-3′and Oxa58R, 5′-ACGGAATTCTTATTATAAATAATGAAAAACACCCAAC-3′ (the restriction sites for NcoI and EcoRI are in italics and underlined). This cloning scheme introduced an extra amino acid, an alanine, at the N terminus of the protein. The digested PCR amplicon was ligated using T4 DNA ligase to the pET24d(+) vector. Cloning was confirmed by sequencing of the cloned DNA using the T7 universal primer. The pET24d(+)::blaOXA-58ΔSP plasmid DNA, ∼6.1 kb, was then used to transform E. coli BL21(DE3) cells.
Site-directed Mutagenesis of Lys-86 to Ala in OXA-58ΔSP
The Lys-86 residue (numbering as in full-length OXA-58) of the SXXK motif was replaced by alanine using the QuikChangeTM site-directed mutagenesis protocol (Stratagene, La Jolla, CA). The recombinant DNA construct containing the mutation was generated using pET24d(+)::blaOXA-58ΔSP as the template, Pfu TurboTM DNA polymerase; and two corresponding mutagenic primers (the mutation site is underlined): MutOxa58F, 5′-CCTGCATCTACATTTGCAATTGCCAATGCAC-3′ and MutOxa58R, 5′-GTGCATTGGCAATTGCAAATGTAGATGCAGG-3′. The PCR product was treated with the restriction endonuclease DpnI, and the resulting mixture was used to transform E. coli DH5α cells. The presence of the desired mutation was verified by DNA sequencing, and the mutant DNA construct was used to transform E. coli BL21(DE3) cells.
Purification of OXA-58ΔSP and K86A Mutant
A seed culture of E. coli BL21(DE3) containing the appropriate expression vector was grown overnight at 37 °C in Luria Bertani medium containing 50 μg/ml kanamycin. A 2.5-ml aliquot of the seed culture was used to inoculate 800 ml of Terrific Broth medium containing 50 μg/ml kanamycin. The cell culture was grown at 37 °C until an absorbance at 600 nm (A600 nm) of 0.6 was reached, at which point overexpression was induced by the addition of isopropyl β-d-thiogalactopyranoside to a final concentration of 0.1 mm. The cells were allowed to overexpress the requisite protein for 16 h at 25 °C with shaking. The cells were then harvested by centrifugation at 7,000 × g for 10 min at 4 °C. The cell pellet (3 g) was resuspended in 90 ml of 10 mm sodium phosphate buffer, pH 6.4. The cytoplasmic contents were liberated by sonication, and the resulting cell extract was spun down at 18,000 × g to remove insoluble cell debris. The supernatant was then loaded onto an SP-Sepharose cation exchange column equilibrated with 10 mm sodium phosphate buffer, pH 6.4. The protein was eluted with a linear gradient of 10–200 mm sodium phosphate buffer, pH 6.4. Fractions containing the protein were concentrated and loaded onto a desalting column to exchange the buffer to 50 mm sodium phosphate buffer, pH 7.0. All purification steps were carried out at 4 °C.
To confirm the identity of the isolated protein, the suspected band in the SDS-PAGE was excised and subjected to trypsin digestion using the in-gel digestion kit (Sigma). The trypsin digestion mixture was sent for peptide mass fingerprinting analysis at the Advanced Protein Technology Centre, The Hospital for Sick Children (Toronto, Ontario, Canada).
Determination of the Kinetic Parameters by Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) experiments were conducted on a MicroCal VP-ITC instrument (GE Healthcare) using the protocol developed by Kotra and co-workers (22). The kinetic measurements were performed at 25 °C. Antibiotic stocks were prepared in 100 mm sodium phosphate buffer, pH 7.0. The working concentration of the enzyme in the micro-syringe was also prepared in 100 mm sodium phosphate buffer, pH 7.0. The reference cell contained 100 mm sodium phosphate buffer, pH 7.0. We used a single injection method for all enzyme kinetic experiments, in which the sample cell contained the antibiotic solution (substrate), and the injection syringe loaded the enzyme into the sample cell. The final concentration of OXA-58ΔSP in the sample cell was maintained at 20 or 200 nm following injection of a 5- or 50-μl aliquot. In all cases, the injection flow rate was 1 μl/s. The chloride anion inhibition experiments used NaCl at a final concentration of 25–200 mm.
Following initiation of the reaction by the single injection of the enzyme solution, the change in heat was measured until a stable base line was achieved, indicating the end of the enzymatic reaction. The reaction mixtures were stirred continuously. Data points were collected every 2 s. Progress curves, referred to as thermograms, were recorded in terms of microcalories versus time (thermal power). All reactions were carried out in triplicate. The effect of CO2 on the reaction kinetics was investigated by adding NaHCO3 to the sample cell at a final concentration of 50 mm.
Analysis of raw data were done using Origin version 7.0 software and as described previously (22). The data points were corrected for the heat of dilution of the enzyme and mixing. The data were then analyzed in the enzyme assay mode in Origin software, which fitted them using the Michaelis-Menten equation. To determine the kinetic parameters kcat and Km from the thermograms, we considered the most linear portion of the curves that best represents steady state conditions. When biphasic kinetics was observed, the data points of the second phase were used to determine steady state kinetics.
Determination of the Catalytic Parameters by UV-Visible Spectrophotometry
The catalytic parameters (Km and kcat) of OXA-58ΔSP with nitrocefin, and those of TEM-1 with penicillin G, were determined under steady state conditions at room temperature using a Cary 100 Bio UV-visible spectrophotometer (Varian Inc., Palo Alto, CA). Briefly, each β-lactam (20–100 μm) was mixed with enzyme (5 nm in the case of nitrocefin, ϵ486 = 20,500 m−1 cm−1; 120 nm in the case of penicillin G, ϵ240 = 570 m−1 cm−1). The initial rates were measured at 5% substrate turnover. All the kinetic reactions were carried out at room temperature in 50 mm sodium phosphate buffer, pH 7.0.
13C NMR Experiments
The water used to prepare the buffers described below was boiled, degassed, cooled, and purged with argon. Buffer components were added under argon atmosphere, except in the case of the NaH13CO3-supplemented buffers. The two enzymes, OXA-58ΔSP and K86A mutant (10-mg portions), were each decarbamylated by treatment with 25 mm sodium acetate buffer, pH 4.5, using an ultracentrifugation membrane (Ultracel 3K, Millipore-Amicon, Bedford, MA). Next, the protein solutions were diluted in 10 mm sodium phosphate buffer, pH 7.5. Finally, the proteins were exchanged into 10 mm sodium phosphate containing 20 mm NaH13CO3. The protein solutions were concentrated to a final concentration of 1 mm. D2O was added into 500 μl of the final sample, at up to 10% of the sample volume. The 13C (proton-decoupled) NMR spectra at 150.886 MHz were acquired at 25 °C on a Bruker DRX 600 NMR spectrometer with a 34,700 Hz spectral width, 64,000 data points, 30° pulse angle, and 1-s relaxation delay. Chemical shifts were referenced to an external sample of DSS in D2O. The data were processed with 2 Hz line broadening.
Inactivation Kinetics
A Cary 100 Bio UV-visible spectrophotometer was used to perform kinetic measurements. Inactivation experiments were carried out with compounds 1–4 (Scheme 1). In a typical inactivation experiment, the β-lactam compound at different concentrations (100, 200, 300, 500, or 700 μm) was incubated on ice with the enzyme (1.25 μm) in 50 mm sodium phosphate buffer, pH 7.0. At different time intervals, a 20-μl aliquot of the incubation mixture was diluted 12.5-fold (to a final assay volume of 250 μl), and the remaining enzyme activity was monitored immediately at 240 nm using penicillin G (1 mm) as the reporter substrate (ϵ240 = 570 m−1 cm−1). Data were analyzed using the Kitz-Wilson method (23). The rate of recovery of enzyme activity was determined by monitoring the enzyme activity of the above mixtures over 24 h at 4 °C.
SCHEME 1.

The β-lactam compounds 1–4 were also assessed as competitive inhibitors using nitrocefin as a reporter substrate. The steady state parameters for nitrocefin are kcat = 99 ± 8 s−1 and Km = 21 ± 2 μm. In a typical competitive inhibition experiment, the enzyme was added to the reaction mixture containing nitrocefin and inhibitor. The final concentration of the enzyme was maintained at 5 nm. Two concentrations of nitrocefin, 30 and 100 μm, were used. Enzyme activity was monitored at 486 nm (ϵ = 20,500 m−1 cm−1). The concentrations of the inhibitors in these assays were selected such that they would flank 50% inhibition of enzyme activity. The experiments were repeated at least three times. The dissociation constants (Ki) were determined by the Dixon plot.
Circular Dichroism Spectroscopy
The far-UV CD spectra of OXA-58ΔSP(10 μm) in the absence and presence of a β-lactam (100 μm) were recorded using a Jasco J-810 instrument. In experiments with the β-lactam substrates, CD spectra were recorded immediately after substrate and enzyme were mixed together. All measurements were carried in 50 mm sodium phosphate buffer, pH 7.0, in a rectangular cuvette with a path length of 0.1 cm. Spectra were recorded from 260 to 200 nm at a scan rate of 10 nm/min and a 1.0-nm bandwidth, at 20 °C. All spectra were corrected for buffer and inhibitor as appropriate.
Electrospray Ionization Mass Spectrometry
OXA-58ΔSP at a concentration of 110 μm in 50 mm sodium phosphate, pH 7.0, was incubated individually with 5 mm of each of the compounds 1–4 for 1 h at room temperature. The mixture was then desalted using Zeba mini centrifugal columns (Pierce) equilibrated with water, concentrated using a SpeedVac, and frozen at −20 °C. Mass spectra were acquired on an Applied Biosystems/MDS Sciex API QSTAR XL Pulsar mass spectrometer in the Advanced Protein Technology Centre, The Hospital for Sick Children (Toronto, Ontario, Canada).
Homology Modeling of the OXA-58 Alone and in Complex with a 6α-(Hydroxyoctyl)penicillanate Derivative
The homology model of OXA-58 was built using the Swiss-Model program (24). The crystal structure of OXA-10 (PDB code 1K54) was used as the template in building the homology model.
Compound 3 was sketched in SYBYL. The acyl-enzyme species was geometry-optimized using a Gaussian (B3LYP/6–31+G(d)) basis function, and RESP charges on the inhibitor were generated using the antechamber module in the AMBER molecular modeling package. Compound 3 was manually docked into the OXA-58 active site, guided by the crystal structure of the 6α-(hydroxyisopropyl)penicillanate·OXA-10 complex (PDB code 1K54) and the crystal structure of OXA-24 (PDB code 2JC7). The resulting OXA-58·compound 3 complex was energy-minimized using the SANDER module in the AMBER package. Using the xLEaP module of AMBER, the complex structure was solvated in a TIP3P waterbox provided in xLEaP, such that any point on the protein was within 9 Å from the edges of the box. Within SANDER, the steepest descent method was used for the first 200 steps of the minimization to initially relax the structure, followed by 19,800 iterations using the conjugate gradient method for a total of 2 × 104 iterations. Further analysis of the acyl-enzyme complex and comparisons with other enzymes were conducted using various tools available in the SYBYL suite.
Determination of the OXA-58ΔSP Oligomerization State
A 20-μl aliquot of OXA-58ΔSP at a concentration of 110 μm was loaded onto a 7.8 × 300-mm TKSgel G2000 SW×I size exclusion column (TOSOH Bioscience Inc., San Francisco) equilibrated with 50 mm Tris, pH 7.4, buffer supplemented with 50 mm KCl and 5 mm MgCl2 at a flow rate of 0.5 ml/min. The column was connected to a ProStar HPLC instrument (Varian).
RESULTS
Cloning and Analysis of blaOXA-58ΔP
The DNA sequence of the full-length OXA-58 protein consists of an 843-bp open reading frame, referred to as blaOXA-58, which encodes a 280-amino acid protein. The OXA-58 amino acid sequence was analyzed using the on-line software SignalP 3.0 program (ExPASy Proteomics Server of the Swiss Institute of Bioinformatics). The software predicted a 0.997 probability of a signal peptide (SP) at the N-terminal region of the OXA-58 amino acid sequence, involving the first 24 residues. Accordingly, we deleted the first 72 nucleotides of the blaOXA-58 gene. The ∼0.8-kb fragment corresponding to the mature OXA-58, referred to as OXA-58ΔSP, was cloned into the overexpression vector pET24d. As a result of this cloning strategy, the sequence of OXA-58ΔSP carried one extra amino acid, an N-terminal alanine, and no fusion tags.
Purification of OXA-58ΔSP and K86A Mutant
The cloning strategy enabled high level expression of the protein in the cytoplasm (20 mg/liter), which in turn enabled isolation of >95% homogeneous protein (Fig. 1). A prior strategy involving the cloning of the full-length OXA-58 (including the signal peptide) had resulted in low protein expression levels. In addition, the isolated enzyme exhibited lower enzymatic activity, which might be due to a nonhomogeneous mixture of enzyme molecules with processed and nonprocessed signal peptide, and/or the improper protein folding in the periplasm.
FIGURE 1.

Isolation and purification of OXA-58ΔSP to homogeneity. 15% SDS-polyacrylamide gel stained with Coomassie Blue. Lane 1, cell extract after sonication. Lane 2, pellet collected after centrifugation. Lane 3, supernatant collected after centrifugation. Lanes 4–6, fractions containing OXA-58ΔSP after purification via a SP-Sepharose cation exchange column.
The identity of the OXA-58ΔSP protein was confirmed by peptide mass fingerprinting after in-gel trypsin digestion. The molecular mass determined by ESI-MS (28,948.1 Da) agreed with the expected mass (28,949.8 Da). OXA-58ΔSP is monomeric in solution as determined by size exclusion chromatography.
The K86A mutant was purified using the same protocol. Expression levels and purity were the same as for the wild-type protein.
Adaptation of ITC for Measurement of Enzyme Kinetics
The adaptation of the ITC protocol developed by Kotra and co-workers (22) was an important choice that allowed accurate determination of steady state kinetics parameters of OXA-58ΔSP against imipenem and other β-lactams with low extinction coefficients. The advantages of this method are high sensitivity, direct rate measurement, and easy data analysis. We determined experimentally that it was best to inject the enzyme, rather than substrate (as has been done in many reports), into the assay buffer, a strategy that mimics classical enzymatic assays performed using UV spectrophotometry. This method overcame the issue of high dilution heats that are observed upon injection of the β-lactam into buffer. We used the single injection method to determine the kcat and Km kinetic parameters of OXA-58ΔSP for several β-lactam antibiotics. To ensure that the heat released during the assays was due to the enzymatic reaction, we performed control experiments involving the injection of buffer into antibiotic solution in the sample cell, buffer into buffer, or OXA-58ΔSP into buffer. Typically, the heat released under these control conditions was no greater than 0.4 μcal/s. The heat released during enzymatic assays at 25 °C averaged 37 μcal/s. The performance of the method was verified by the agreement of the steady state kinetics of TEM-1 with benzylpenicillin obtained calorimetrically, with those obtained spectrophotometrically (supplemental Fig. S1).
ITC progress curves are reported in terms of microcalories per unit time, referred to as thermal power (Fig. 2A). The thermal power is proportional with the rate of substrate breakdown. As the substrate is depleted by the enzyme, the thermal power decreases until it reaches the base line. Equations 1–2 were used to convert thermal power into reaction rates (Fig. 2B), which were then plotted against substrate concentration (Equation 3) (22).
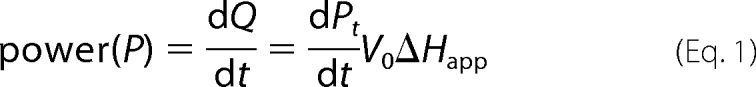 |
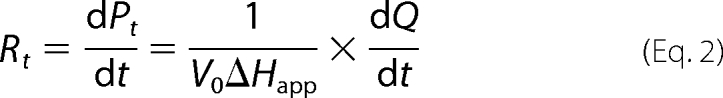 |
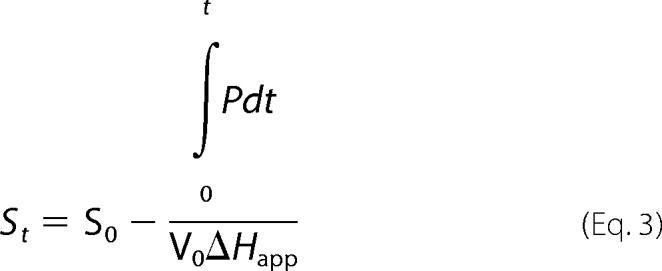 |
In the above equations, P is thermal power (μcal/s); Rt is the rate of the reaction at time t; St is the substrate concentration at time t; Q is the heat released during the enzymatic reaction; V0 is the volume of the ITC cell; ΔHapp is the molar enthalpy (calculated by dividing the net heat by the number of moles of substrate in the assay).
FIGURE 2.

A typical ITC thermogram and analysis of the data. A, ITC thermogram of benzylpenicillin hydrolysis; OXA-58ΔSP at 5 nm and benzylpenicillin at 100 μm. P, thermal power. B, nonlinear least squares fit (solid line) of the generated data (dotted line) to the Michaelis-Menten equation to determine the kinetic parameters of benzylpenicillin hydrolysis.
Kinetics of OXA-58ΔSP
ITC experiments indicated that OXA-58ΔSP readily hydrolyzed ampicillin, oxacillin, benzylpenicillin, amoxicillin, and cephalothin but not carbenicillin and cephalosporin C (Table 1). OXA-58ΔSP exhibited visible biphasic kinetics with ampicillin, amoxicillin, and cephalothin in the absence of carbon dioxide. For these substrates, a fast kinetic phase was followed by a slower phase before the completion of hydrolysis (Fig. 2). Changes in substrate concentration did not alter the biphasic character of the kinetics, indicating that substrate inhibition is not the source of the biphasicity. The reported kinetics parameters are derived from the second phase (Table 1). The first phase was too rapid to be determined accurately under our experimental conditions.
TABLE 1.
Steady state kinetic parameters of OXA-58ΔSP for different antibiotics
| Antibiotic | kcat | Km | kcat/Km × 103 |
|---|---|---|---|
| s−1 | μm | m−1s−1 | |
| Oxacillin | 83 ± 8 | 52 ± 5 | 1596 ± 217 |
| (NaHCO3)a | (122 ± 10) | (41 ± 7) | (2976 ± 499) |
| Ampicillin | 88 ± 14 | 77 ± 8 | 1143 ± 216 |
| (NaHCO3) | (556 ± 90) | (80 ± 19) | (6950 ± 1997) |
| Amoxicillin | 86 ± 15 | 46 ± 8 | 1869 ± 461 |
| (NaHCO3) | (132 ± 12) | (13 ± 2) | (10153 ± 1927) |
| Benzyl penicillin | 59 ± 8 | 11 ± 2 | 5363 ± 1216 |
| (NaHCO3) | (103 ± 13) | (8 ± 2) | (12875 ± 3180) |
| Carbenicillin | NHb | NH | NH |
| Imipenem | 0.22 ± 0.05 | 1.3 ± 0.4 | 169 ± 64 |
| (NaHCO3) | (2.6 ± 0.2) | (2.8 ± 0.4) | (928 ± 150) |
| Cephalothin | 19 ± 3 | 190 ± 30 | 100 ± 22 |
| (NaHCO3) | (37 ± 2) | (52 ± 13) | (712 ± 182) |
| Cephalosporin C | NH | NH | NH |
a In parenthesis are provided the kinetic parameters measured in the presence of 50 mm sodium bicarbonate.
b NH means not hydrolyzed; no turnover was detected with enzyme concentration up to 400 nm. The turnover rate constant could be smaller than 1 × 10−3 s−1.
Notably, penicillins are hydrolyzed better than cephalosporins. Imipenem has a slow turnover rate, but its low Km makes imipenem a moderate substrate (kcat/Km = 169 × 103 m−1s−1). Interestingly, carbenicillin is not hydrolyzed by OXA-58. Previous studies on other CHDLs did not examine carbenicillin as a substrate. To investigate the interaction mode of carbenicillin with OXA-58ΔSP, we carried out competition assays using nitrocefin as a reporter substrate. Carbenicillin competitively inhibited OXA-58ΔSP with a Ki = 78 ± 2 μm.
The kinetic parameters (but not the biphasic character of the hydrolysis) determined for OXA-58 in this study differ from those published by Poirel et al. (19), who reported generally smaller kcat and higher Km values for similar substrates. The differences may derive from the differences in experimental conditions and methods. Poirel et al. (19) cloned a full-length OXA-58, which included the signal peptide. In our hands, efforts toward a similar strategy led to the isolation of an enzyme that displayed poor activity. It is possible that the signal peptide may have been improperly processed by E. coli signal peptidases. In turn, this could affect protein translocation to, and folding in, the periplasm. Furthermore, the determination of kinetic parameters by Poirel et al. (19) was carried out spectrophotometrically.
Mutation of the Lys-86, which is located in the conserved motif SXXK, to Ala rendered the enzyme practically inactive. The activity of the K86A mutant enzyme was assessed by ITC and UV-visible spectrophotometry using benzylpenicillin. Similar observations have been made for OXA-1 and OXA-10 (25, 26). Vercheval et al. (26) reported that a K70A mutation in OXA-10 reduced its catalytic efficiency by 4,250-fold (kcat/Km = 4 × 10−3 s−1 μm−1).
Effect of CO2 on the Kinetics of OXA-58ΔSP
Class D oxacillinases, such as OXA-10, have been shown to adopt a lysine-carbamate species in their catalysis (also referred to as carboxylated lysine) (27). Hydrolysis progress curves for several β-lactams with OXA-10 displayed biphasic kinetics, which simplified to monophasic when buffers were supplemented with NaHCO3 (the source of CO2). These studies demonstrated that the biphasicity in kinetics was due to the decarbamylation of the lysine active site residue during the kinetics (26, 27).
We investigated the source of the biphasicity of the kinetics exhibited by OXA-58 in the turnover of β-lactams by supplementing the buffers with NaHCO3. The buffers used for purification of OXA-58 and for the kinetics were not supplemented with NaHCO3. The content of CO2 in these buffers could be less than 10 μm, because atmospheric CO2 content is 10 μm. Because of the low buffer CO2 concentration, not all the OXA-58 molecules would be carbamylated at Lys-86. Furthermore, during the reaction, the lysine might undergo decarbamylation (see “Discussion”); the low concentration of CO2 in the buffer would prevent re-carbamylation of the lysine. As a result, in the course of catalysis, the enzyme is predicted to lose activity, resulting in a slower phase following the initial rapid phase in the progress curves. An excess of CO2 in buffer would ensure a fully carbamylated enzyme and enable re-carbamylation of Lys-86, which would therefore result in a monophasic progress curve.
The addition of bicarbonate to the reaction buffer increased the rate of hydrolysis of all the β-lactams and simplified the thermograms for ampicillin, amoxicillin, and cephalothin to monophasic progress curves (Fig. 3). This indicates that supplementation of the buffers with bicarbonate enables all the enzyme molecules to have Lys-86 carbamylated even in the event when this species decomposes during the catalysis.
FIGURE 3.

Analysis of the effect of carbon dioxide in the turnover rates of β-lactams by OXA-58. ITC thermograms obtained for ampicillin and OXA-58ΔSP in the absence of sodium bicarbonate (A) and in the presence of sodium bicarbonate (50 mm) (B). P, thermal power.
The largest effect of CO2 on the turnover rate constant was observed for imipenem, for which a 12-fold increase in activity was observed. The other substrates experienced an increase in the kcat values by 1.5–6-fold. The effect of CO2 on the Km values was smaller, exhibiting either a slight increase or a decrease depending on the substrate. The kinetics of nitrocefin hydrolysis was not affected by bicarbonate concentration, as assessed by using UV-spectrophotometer. Similar observations were made by Vercheval et al. (26). Nitrocefin is intrinsically a chemically reactive molecule and as such partial loss or gaining of carbamylated species may not have an impact in the turnover rate of nitrocefin.
The 13C NMR spectra obtained for wild-type OXA-58ΔSP in the presence of 20 mm 13C-labeled sodium bicarbonate showed the signature peak for the carbamylated lysine at 164 ppm. This peak was absent in the 13C NMR spectrum of the K86A mutant (Fig. 4). Interestingly, the height of the 124.9 ppm peak, which corresponds to free CO2, increased upon removal of the carbamylation site in the K86A mutant. An additional peak was observed at 164.2 ppm for both wild-type and the mutant proteins. We believe that this might result from the binding of CO2 to a second site in the protein. The binding affinity of CO2 to the conserved lysine residue was calculated by measuring enzyme activity at different concentrations of CO2, as described previously (27). The calculated Kd is 16.5 ± 0.5 μm.
FIGURE 4.

13C NMR spectra of wild-type (A) and K86A OXA-58ΔSP mutant (B). The protein concentration in these experiments was 1 mm, prepared in 10 mm sodium phosphate buffer, pH 7.5, supplemented with 20 mm 13C-labeled sodium bicarbonate.
The inhibitory effect of chloride anions was investigated by injecting enzyme into a solution of NaCl and benzylpenicillin (1 mm). The end point of the thermogram progressively extended with increasing NaCl concentrations, and enzyme activity was completely abolished at 200 mm NaCl.
Mode of Interaction of OXA-58ΔSP with 6-Hydroxyalkylpenicillanate Derivatives
6β-Hydroxymethylpenicillanate (compound 2) was found to be a moderate OXA-58 substrate with kcat/Km = 79 × 104 m−1 s−1 (kcat = 32.2 ± 0.5 s−1; Km = 41 ± 1 μm). However, the 6β-hydroxyoctylpenicillanate derivative (compound 4) did not undergo hydrolysis as investigated by UV spectrophotometry, but this compound inhibited OXA-58ΔSP competitively (Table 2). Among the two 6α-hydroxyalkyl-penicillanate derivatives, 6α-[(2R)-2-hydroxyoctyl]penicillanate (compound 3) inactivated OXA-58ΔSP with kinact = 0.22 ± 0.01 s−1 (t½ = 3 min) at 4 °C (Fig. 5). The inactivation of OXA-58ΔSP (10 μm) persisted even after 24 h of incubation with this compound at concentrations as low as 100 μm at 4 °C. However, the 6α-(hydroxymethyl) penicillanate derivative (compound 1) inhibited OXA-58ΔSP only competitively.
TABLE 2.
Dissociation constants (Ki) of the β-lactams used in this study
The Ki values were determined using nitrocefin as a reporter substrate.
| Inhibitor | Ki |
|---|---|
| μm | |
| Compound 1 | 83 ± 3 |
| Compound 2 | 30 ± 4 |
| Compound 3 | 404 ± 10 |
| Compound 4 | 42 ± 1 |
| Carbenicillin | 78 ± 2 |
| Imipenem | 3.2 ± 0.3 |
FIGURE 5.

Examination of OXA-58ΔSP inactivation by compounds 3 and 4 with ESI-MS (deconvoluted ESI-MS spectra). In these experiments OXA-58ΔSP at 110 μm was incubated with compounds 3 or 4 at 5 mm for 1 h or 20 min, respectively, in 50 mm sodium phosphate, pH 7.0, at room temperature. The mixtures were desalted and subjected to ESI-MS for analysis.
The interaction modes of compounds 1–4 were also investigated by ESI-MS. These experiments show that compound 3 resulted in the formation of quantitative and stable acyl-enzyme species, whereas compound 4 resulted in the formation of a smaller amount of acyl-enzyme species (Fig. 5). The lack of measurable time-dependent inactivation of the enzyme by compound 4, as investigated by UV-visible spectrophotometry, suggests that the observed acyl-enzyme species could result from the formation of a short lived covalent complex. However, at the level of the detection limit of the UV-visible spectrophotometer, we were not able to measure any hydrolysis. It is worth noting that these β-lactam compounds have a low extinction coefficient between 235 and 240 nm, which confounds the detection of low levels of hydrolysis.
Structural Investigation of the Interactions between β-Lactams and OXA-58ΔSP β-Lactamase
Far-UV CD spectra of OXA-58 collected in the presence of different β-lactams show that benzylpenicillin and oxacillin introduce a conformational change in OXA-58 at the secondary structural level. By contrast, the effect of imipenem is smaller (supplemental Fig. S2).
OXA-58 Homology Model
The structural homology model of OXA-58 displayed an overall fold that was similar to that of OXA-24 and OXA-48, both carbapenemases (supplemental Fig. S3). The three conserved catalytic motifs found in all serine β-lactamases are also present in OXA-58 (Fig. 6). The Arg residue (Arg-244 in OXA-10) that interacts with the carboxylic group of β-lactams is conserved in OXA-58 (Arg-263). The side chains of the residues that form the hydrophobic pocket surrounding the carbamylated lysine, as seen in OXA-10, OXA-24, and OXA-48, are all conserved in OXA-58 (Phe-85, Val-132, Phe-135, Trp-169, and Leu-170).
FIGURE 6.

Sequence alignment of OXA-58 with OXA-10, OXA-24, and OXA-48. Highlighted in red are the three conserved motifs in serine β-lactamases. The green dots indicate the residues that form the hydrophobic pocket around the conserved lysine residue. The blue dots indicate the residues that form the hydrophobic cleft in OXA-24. The secondary structures correspond to those predicted by the OXA-58 homology model.
The most distinctive structural differences between the OXA-58 homology model and the OXA-10 crystal structure are found in the loop that connects the β4 and β5 strands and in the overall topology of the active site (supplemental Fig. S3). The loop that connects the β4 and β5 strands is predicted to be shorter in OXA-58 than in OXA-10. In addition, the OXA-58 loop adopts a closed conformation (Fig. 7 and supplemental Fig. S3), which is similar to the loop conformation observed in OXA-24 and OXA-48 (15, 18). The predicted topology of the active site in OXA-58 is narrower compared with the active site of OXA-10 and OXA-24. This results from the extension of hydrophobic side chains such as Phe-113, Phe-114, Ala-226, Gln-167, Met-225, and Ile-250 toward the active site (Fig. 7). Notably, the side chain of Met-225 of the β4-β5 loop (Met-223 in OXA-24 and Ser-244 in OXA-48) and those of Phe-113 (Tyr-112 in OXA-24 and Ile-104 in OXA-48) and Phe-114 of the α3-α4 loop are predicted to interact in the homology model of OXA-58 model. As a result, possibly aided by the flexibility of both loops, the active site in OXA-58 is predicted to have a more pronounced tunnel-like topology compared with the active sites of OXA-10 and OXA-24 (Fig. 7).
FIGURE 7.

Molecular surfaces of OXA-10, OXA-24, OXA-48, and the OXA-58 homology model. The surfaces are color-coded with brown indicating hydrophobicity and blue indicating hydrophilicity. The red star indicates the position of the hydrophobic pocket formed by the side chains of Leu-155 and Val-117 in OXA-10 (PDB code 1K54), Leu-168 and Val-130 in OXA-24 (PDB code 2JC7), Leu-158 and Val-120 in OXA-48 (PDB code 3HBR), and Leu-170 and Val-132 residues in the OXA-58 homology model.
The OXA-58 homology model also predicts that the side chains of Leu-170 and Val-132 in OXA-58 interact less with each other, which may result in a diminished hydrophobic pocket as seen in oxacillinases such as OXA-10 and OXA-13 (Leu-155 and Val-117) (Fig. 7). The same feature of a diminished hydrophobic pocket is also visible in the crystal structures of OXA-24 and OXA-48 (Fig. 7). The crystal structure of the OXA-13-meropenem acyl-enzyme complex shows that this hydrophobic pocket interacts with the methyl moiety of the 6α-hydroxyethyl group of meropenem (28).
Computational docking of the 6α-(2-hydroxyoctyl)penicillanate derivative compound 3 into the OXA-58 active site showed intimate contacts between compound 3 and the active site residues of OXA-58, resulting in snug binding of compound 3 within the active site (Fig. 8). Furthermore, the 6α-octyl group of compound 3 extends well into the hydrophobic tunnel formed by the side chains of Trp-223, Met-225, Ala-226, Trp-117, Phe-113, Phe-114, Ala-72, Val-132 and Leu-170. The hydroxyl substituent at C-6 position of compound 3 forms a hydrogen bond (2.86 Å) with the amino group of Lys-86. The 2-carboxylate groups of compound 3 interact with the side chains of Arg-263, Lys-264, and Ser-221. The two C-3 methyl groups interact with the Phe-114 and Ile-260 side chains (Fig. 8).
FIGURE 8.

Close-up view of the active site of the OXA-58 structural homology model in complex with compound 3. A, predicted interactions between compound 3 and the active site residues in OXA-58. B, active site is partially presented in molecular surface view to highlight the predicted positioning of the alkyl chain of compound 3 in the active site of OXA-58 (the surface is color-coded as in Fig. 9).
DISCUSSION
Mechanism of β-Lactam Hydrolysis by OXA-58
Serine β-lactamases hydrolyze β-lactams in two steps as shown in Equation 4,
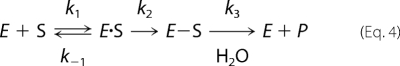 |
In the first step, the enzyme (E) employs a serine active site residue to form a transient acyl-enzyme species (E-S) by opening the β-lactam moiety. In the second step, this acyl-enzyme species is hydrolyzed by a water molecule. The formation of the acyl-enzyme species in the first step is made possible by the action of a general base within hydrogen-bonding proximity to the serine residue, thus transforming the weakly nucleophilic alcohol of the serine into a strong nucleophile. In class A β-lactamases, Lys-73 and Glu-166 play this role (29–33). In class C, activation of the serine may occur by either Lys-67 or Tyr-105 as the general base (34–36). In class D β-lactamases, a carbamylated lysine serves as the general base in both steps (26, 27, 37).
The steady state kinetic parameters of OXA-58ΔSP, determined in the presence or absence of CO2, establishes the role of CO2 in the creation of the catalytic machinery of the active site. Our 13C NMR and Lys-86 mutagenesis studies unambiguously correlate the catalytic activity with the carbamylation of this lysine (Fig. 4).
We measured a 2–7-fold increase in the catalytic efficiency of OXA-58ΔSP in the presence of CO2 with different β-lactams. This increase was mainly due to an increase in kcat (Table 1). In particular, the effect of bicarbonate on the catalytic efficiency of OXA-58ΔSP was dramatic for imipenem (Table 1), for which a 12-fold increase in kcat was measured. The observed enhancement of the turnover kinetics of imipenem in the presence of CO2 implies that the catalytic machinery of OXA-58, and of CHDLs in general, has evolved to hydrolyze imipenem as efficiently as do the class A carbapenemases (38). This catalytic role of a CO2 molecule resolves the apparent discrepancy between the low catalytic efficiency reported for imipenem in literature and the measured resistance levels (39). Recent structural studies on two CHDLs, OXA-24 and OXA-48, show the conserved lysine residue to be carbamylated (16, 18). It is plausible that the carbamylated lysine residue is mechanistically conserved among all OXA β-lactamases.
A schematic representation of the role of the carbamylated lysine in the hydrolysis reaction is presented in Fig. 9. Upon activation of Ser-83, the proton on the carbamate species might be abstracted by the N-4 of the penam (16). At the end of hydrolysis, the carbamylated lysine returns to its basic form, probably by the abstraction of the proton from the protonated species of carbamylated lysine by the N-4 amine group of the penam (16). It is possible that, during the activation of Ser-83 or the deacylating water molecule, it is the carbamate nitrogen of the Lys-86 that abstracts the proton. This would result in decarbamylation of the lysine (40) and hence inactivation of the enzyme in mid-catalysis. For the enzyme to become active, the lysine has to re-carbamylate. This would explain the biphasic kinetics observed with OXA-58 and oxacillinases (27).
FIGURE 9.

Mechanism of β-lactam hydrolysis by OXA-58.
In the second step of the β-lactam hydrolysis mechanism, β-lactamases strategically position the deacylating water molecule between the general bases and the acyl-enzyme to increase the efficiency of hydrolysis. In classes A and C β-lactamases, the catalytic water molecule is anchored through a hydrogen bond network in an optimum position for nucleophilic attack on the carbonyl carbon of the acyl-enzyme species. The position of this water molecule is crystallographically conserved (31, 36, 41) unlike in class D enzymes (42–44). Perturbation of the hydrogen bonding network of this water molecule or the physical displacements of the water molecule by the steric bulk of the C-6 side chains of a β-lactam are possible mechanisms to render the β-lactamase deacylation-deficient (20, 45–47). This is the goal in designing new generations of β-lactams that are stable toward β-lactamases (47).
Here, we use a set of 6α- and 6β-hydroxyalkylpenicillanate derivatives (compounds 1–4) to probe the face used in the approach of the deacylating water molecule to the ester carbonyl of the acyl-enzyme species (47, 48). Mobashery and co-workers (47) showed that compound 1 is an effective inhibitor of TEM-1 β-lactamase, a class A β-lactamase. Based on their study, it was proposed that the deacylating water approaches from the α-face in TEM-1. In the case of OXA-10, it was reported that 6β-hydroxymethylpenicillanate (compound 2) is a substrate for this class D oxacillinase, and hence it was suggested that the deacylating water molecule approaches from the α-face (42). The α- and β-face refer to the stereochemistry of the hydroxyalkyl group at the C-6 position of the β-lactam.
Our investigation of the interactions of these penicillanate derivatives with OXA-58 revealed that compound 2 is a substrate for this enzyme, and compound 3 is an inactivator. The lack of substantial hydrolysis of compound 4 by OXA-58 suggests that the increase in the bulkiness of the R group at the C-6 position could perturb the catalytic machinery of OXA-58, for example by causing decarbamylation of the enzyme (44). This is supported well by results from ESI-MS and competitive experiments, which reveal that compound 4 is able to bind to OXA-58, but only a small percentage of the noncovalent complex formed between compound 4 and the enzyme undergoes acylation. Time-dependent inactivation experiments show that compound 4 does not form a covalent species with the enzyme even after 24 h.
By contrast, the 6α-(hydroxyoctyl)penicillanate derivative compound 3 rapidly inactivates the enzyme (t½ = 3 min at 4 °C) forming a very stable acyl-enzyme complex (Fig. 5). This suggests that the water molecule approaches the acyl-enzyme species from the α-face (that of the side chain of compound 3). Indeed, the predicted structure of the OXA-58-3 acyl-enzyme complex (Fig. 8) shows that the hydroxyl group of compound 3 fits tightly into the active site. The close proximity of the hydroxyl group of compound 3 with the Nϵ of Lys-86 (2.86 Å apart) suggests a direct physical hindrance for the approach of the water molecule to the acyl-enzyme species. In this model, we have not considered the Lys-86 as carbamylated. It is clear from the model that the carbamate species will not accommodate the approach of the deacylating water molecule. In addition, the predicted close proximity between the hydroxyl group of compound 3 and the Nϵ-amino group of Lys-86 suggests that the hydroxyl group of compound 3 could destabilize the carbamate species of Lys-86 as a result of hydrogen bonding with the carbamate nitrogen. This has been shown to be the case with OXA-10 (44).
The predicted binding interaction of compound 3 with OXA-58 also provides structural insight to guide the design of potential inhibitors of these class D enzymes. For example, the use of a bulky side chain at the C-6 position might enhance the binding affinity of β-lactams, and the presence of a 6α-hydroxylalkyl substituent as displayed by compound 3 might provide a barrier for the approach of the deacylating water molecule. Incidentally, a recent study by Bou et al. (16) has shown that the presence of a bulky and rigid substituent at the C-6 position of β-lactams causes rapid inactivation of the OXA-24 CHDL.
Insights into the Mechanism of Imipenem Hydrolysis
The basis for the acquisition of carbapenemase activity by oxacillinases remains largely unknown, despite the recent high resolution structures obtained for OXA-24 and OXA-48. Structural studies on OXA-24 revealed a hydrophobic cleft on the active site that might account for the preference of this enzyme for imipenem over oxacillin (15). However, this structural feature is not conserved in OXA-48 (18).
Sequence alignment of OXA-58 with OXA-24 and OXA-48 and comparison with the primary sequence of OXA-10 show that the residues that are intimately involved with catalysis in OXA-10 are conserved in these CHDLs (Fig. 6). Our study strongly suggests that these CHDLs use the same catalytic mechanism as OXA-10; yet CHDLs are able to hydrolyze imipenem, although OXA-10 cannot.
A close comparison of the structures of OXA-24, OXA-48, and OXA-58 (homology model) to the structure of OXA-10 indicates that the most visible differences are a shorter β4-β5 loop in CHDLs, which adopts a closed conformation (in OXA-48, a salt bridge between Asp-159 and Arg- 214 is behind this structural difference), and movement of the β6-α11 loop in CHDLs toward the active site (supplemental Fig. S3). As a result, the active sites of OXA-24, OXA-58, and OXA-48 become narrower and more hydrophobic (especially toward the β-face of the β-lactam ring) compared with that of OXA-10. These structural features may reposition the carbapenem-derived acyl-enzyme species to enable successful nucleophilic attack by the deacylating water molecule. Furthermore, the observed diminishing of the hydrophobic pocket in the active sites of CHDLs (OXA-24, OXA-48, and predicted for OXA-58), formed by the interactions between Leu-170 and Val-132 (numbering as per OXA-58), may re-orient the hydroxyethyl group of imipenem so as to remove the steric hindrance of this group toward the deacylating water. This mechanism was shown to be plausible by a simulation study carried out with OXA-48 (18).
The steady state kinetics of OXA-58ΔSP shows that this carbapenemase retains the ability to hydrolyze oxacillin 9.4-fold better than imipenem. By contrast, the hydrolytic activity of OXA-24 against oxacillin is poor but that against imipenem is moderately high (kcat/KmIMP/kcat/KmOXA = 13.8) (15). This has been attributed to the hydrophobic cleft that the side chains of Tyr-112 and Met-223 create in the OXA-24 active site (15). In the case of OXA-58, the structural homology model suggests that the hydrophobic side chains of Phe-113 and Phe-114 in the α3-α4 loop and Met-225 in the β4-β5 loop form a more extensive hydrophobic structure compared with OXA-24 (Fig. 7), and yet OXA-58ΔSP does not show a preference for imipenem over oxacillin. Hence, the implication of this structural feature in the activity of OXA-58ΔSP is not obvious.
The CD studies showing that the binding of benzylpenicillin, oxacillin, or imipenem induces a conformational change in the protein, possibly resulting from the adjustment of the above loops with respect to the side chain at the C-6 position in the β-lactam, indicated flexibility in this substructure. This structural flexibility may also result in an increase in the binding affinity of β-lactams to these CHDLs. Indeed, removal of a similar hydrophobic cleft in OXA-24 resulted in the decrease of binding affinity to β-lactams, from 1.6-fold for oxacillin to 5- and 15-fold for ampicillin and imipenem, respectively (15). Overall, the structural analysis of these CHDLs suggests that they have conserved the catalytic machinery of oxacillinases through their evolution and have acquired structural changes in their active sites to facilitate efficient hydrolysis of imipenem.
In conclusion, we have used ITC to analyze the catalytic mechanism of OXA-58. The independence of this technique from the chromophoric properties of the substrate allows for direct analysis of a wide range of substrates. Our study shows that OXA-58 uses a carbamylated lysine in β-lactam hydrolysis and that the deacylating water molecule approaches the acyl-enzyme species, anchored at the Ser-83, from the α-face. Furthermore, our data show that CHDLs, despite their different evolutionary pathways, have retained the catalytic machinery of oxacillinases, represented by OXA-10. A structural analysis of OXA-24, OXA-48, and the OXA-58 homology model, and their comparison with the structure of OXA-10, suggests that the hydrolysis of imipenem by CHDLs could be a result of accumulation of structural changes in the active site of CHDLs that allow the molecule to bind imipenem tightly and increase the probability of attack on the acyl-enzyme species by water molecules. Class D β-lactamases are in a catalytically disadvantageous position when it comes to the second step of catalysis, because the deacylating water molecule is not present in the active site as seen in β-lactamases of classes A and C. Thus, the removal of any steric hindrance in the path of a bulk water molecule toward the carbonyl carbon of the acyl-enzyme species will increase the chances of hydrolysis of this complex, resulting in a more efficient β-lactamase.
Acknowledgements
We are grateful to Professor Patrice Nordmann at Service de Bactériologie-Virologie, Hôpital de Bicêtre (Toulouse, France), for providing the OXA-58 gene and to Professor Shahriar Mobashery at University of Notre Dame (Notre Dame, IN) for providing the penicillanate derivatives. We thank Dr. Li Zhang at the Advanced Protein Technology Centre, The Hospital for Sick Children (Toronto, Ontario, Canada) for analysis of the acyl-enzyme complexes by ESI-MS. We thank Dr. Howard Hunter at the Department of Chemistry (York University, Toronto, Ontario, Canada) for assistance with the NMR experiments. We thank Dr. Jed F. Fisher, University of Notre Dame, for critical discussion of the manuscript.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- CHDL
- carbapenem-hydrolyzing class D β-lactamase
- ESI-MS
- electrospray ionization mass spectrometry
- ITC
- isothermal titration calorimetry
- MALDI-TOF
- matrix assisted laser desorption/ionization time-of-flight
- PDB
- Protein Data Bank.
REFERENCES
- 1. Bonfiglio G., Russo G., Nicoletti G. (2002) Expert Opin. Investig. Drugs 11, 529–544 [DOI] [PubMed] [Google Scholar]
- 2. Testero A., Fisher J. F., Mobashery S. (2010) in Burger Medicinal Chemistry, Drug Discovery and Development (Abraham D. J., Rotella D. P. eds) 7th Ed., pp. 259–404, John Wiley & Sons, Inc., New York [Google Scholar]
- 3. Livermore D. M. (1998) J. Antimicrob. Chemother. 41, Suppl. D, 25–41 [DOI] [PubMed] [Google Scholar]
- 4. Bush K., Jacoby G. A. (2010) Antimicrob. Agents Chemother. 54, 969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walsh T. R. (2008) Curr. Opin. Infect. Dis. 21, 367–371 [DOI] [PubMed] [Google Scholar]
- 6. Poirel L., Pitout J. D., Nordmann P. (2007) Future Microbiol. 2, 501–512 [DOI] [PubMed] [Google Scholar]
- 7. Queenan A. M., Bush K. (2007) Clin. Microbiol. Rev. 20, 440–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Poirel L., Nordmann P. (2006) Clin. Microbiol. Infect. 12, 826–836 [DOI] [PubMed] [Google Scholar]
- 9. Marqué S., Poirel L., Héritier C., Brisse S., Blasco M. D., Filip R., Coman G., Naas T., Nordmann P. (2005) J. Clin. Microbiol. 43, 4885–4888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Héritier C., Dubouix A., Poirel L., Marty N., Nordmann P. (2005) J. Antimicrob. Chemother. 55, 115–118 [DOI] [PubMed] [Google Scholar]
- 11. Zarrilli R., Giannouli M., Tomasone F., Triassi M., Tsakris A. (2009) J. Infect. Dev. Ctries. 3, 335–341 [DOI] [PubMed] [Google Scholar]
- 12. Walther-Rasmussen J., Høiby N. (2006) J. Antimicrob. Chemother. 57, 373–383 [DOI] [PubMed] [Google Scholar]
- 13. Paton R., Miles R. S., Hood J., Amyes S. G., Miles R. S., Amyes S. G. (1993) Int. J. Antimicrob. Agents 2, 81–87 [DOI] [PubMed] [Google Scholar]
- 14. Higgins P. G., Dammhayn C., Hackel M., Seifert H. (2010) J. Antimicrob. Chemother. 65, 233–238 [DOI] [PubMed] [Google Scholar]
- 15. Santillana E., Beceiro A., Bou G., Romero A. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 5354–5359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bou G., Santillana E., Sheri A., Beceiro A., Sampson J. M., Kalp M., Bethel C. R., Distler A. M., Drawz S. M., Pagadala S. R., van den Akker F., Bonomo R. A., Romero A., Buynak J. D. (2010) J. Am. Chem. Soc. 132, 13320–13331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schneider K. D., Ortega C. J., Renck N. A., Bonomo R. A., Powers R. A., Leonard D. A. (2011) J. Mol. Biol. 406, 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Docquier J. D., Calderone V., De Luca F., Benvenuti M., Giuliani F., Bellucci L., Tafi A., Nordmann P., Botta M., Rossolini G. M., Mangani S. (2009) Chem. Biol. 16, 540–547 [DOI] [PubMed] [Google Scholar]
- 19. Poirel L., Marqué S., Héritier C., Segonds C., Chabanon G., Nordmann P. (2005) Antimicrob. Agents Chemother. 49, 202–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miyashita K., Massova I., Taibi P., Mobashery S. (1995) J. Am. Chem. Soc. 117, 11055–11059 [Google Scholar]
- 21. Testero S. A., O'Daniel P. I., Shi Q., Lee M., Hesek D., Ishiwata A., Noll B. C., Mobashery S. (2009) Org. Lett. 11, 2515–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Poduch E., Bello A. M., Tang S., Fujihashi M., Pai E. F., Kotra L. P. (2006) J. Med. Chem. 49, 4937–4945 [DOI] [PubMed] [Google Scholar]
- 23. Kitz R. J., Ginsburg S., Wilson I. B. (1967) Biochem. Pharmacol. 16, 2201–2209 [DOI] [PubMed] [Google Scholar]
- 24. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 25. Schneider K. D., Bethel C. R., Distler A. M., Hujer A. M., Bonomo R. A., Leonard D. A. (2009) Biochemistry 48, 6136–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vercheval L., Bauvois C., di Paolo A., Borel F., Ferrer J. L., Sauvage E., Matagne A., Frère J. M., Charlier P., Galleni M., Kerff F. (2010) Biochem. J. 432, 495–504 [DOI] [PubMed] [Google Scholar]
- 27. Golemi D., Maveyraud L., Vakulenko S., Samama J. P., Mobashery S. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14280–14285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pernot L., Frénois F., Rybkine T., L'Hermite G., Petrella S., Delettré J., Jarlier V., Collatz E., Sougakoff W. (2001) J. Mol. Biol. 310, 859–874 [DOI] [PubMed] [Google Scholar]
- 29. Golemi-Kotra D., Meroueh S. O., Kim C., Vakulenko S. B., Bulychev A., Stemmler A. J., Stemmler T. L., Mobashery S. (2004) J. Biol. Chem. 279, 34665–34673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Herzberg O., Moult J. (1987) Science 236, 694–701 [DOI] [PubMed] [Google Scholar]
- 31. Strynadka N. C., Adachi H., Jensen S. E., Johns K., Sielecki A., Betzel C., Sutoh K., James M. N. (1992) Nature 359, 700–705 [DOI] [PubMed] [Google Scholar]
- 32. Lamotte-Brasseur J., Dive G., Dideberg O., Charlier P., Frère J. M., Ghuysen J. M. (1991) Biochem. J. 279, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nukaga M., Mayama K., Hujer A. M., Bonomo R. A., Knox J. R. (2003) J. Mol. Biol. 328, 289–301 [DOI] [PubMed] [Google Scholar]
- 34. Oefner C., D'Arcy A., Daly J. J., Gubernator K., Charnas R. L., Heinze I., Hubschwerlen C., Winkler F. K. (1990) Nature 343, 284–288 [DOI] [PubMed] [Google Scholar]
- 35. Dubus A., Ledent P., Lamotte-Brasseur J., Frère J. M. (1996) Proteins 25, 473–485 [DOI] [PubMed] [Google Scholar]
- 36. Chen Y., McReynolds A., Shoichet B. K. (2009) Protein Sci. 18, 662–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sun T., Nukaga M., Mayama K., Braswell E. H., Knox J. R. (2003) Protein Sci. 12, 82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walther-Rasmussen J., Høiby N. (2007) J. Antimicrob. Chemother. 60, 470–482 [DOI] [PubMed] [Google Scholar]
- 39. Afzal-Shah M., Woodford N., Livermore D. M. (2001) Antimicrob. Agents Chemother. 45, 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li J., Cross J. B., Vreven T., Meroueh S. O., Mobashery S., Schlegel H. B. (2005) Proteins 61, 246–257 [DOI] [PubMed] [Google Scholar]
- 41. Swarén P., Golemi D., Cabantous S., Bulychev A., Maveyraud L., Mobashery S., Samama J. P. (1999) Biochemistry 38, 9570–9576 [DOI] [PubMed] [Google Scholar]
- 42. Golemi D., Maveyraud L., Vakulenko S., Tranier S., Ishiwata A., Kotra L. P., Samama J. P., Mobashery S. (2000) J. Am. Chem. Soc. 122, 6132–6133 [Google Scholar]
- 43. Maveyraud L., Golemi D., Kotra L. P., Tranier S., Vakulenko S., Mobashery S., Samama J. P. (2000) Structure 8, 1289–1298 [DOI] [PubMed] [Google Scholar]
- 44. Maveyraud L., Golemi-Kotra D., Ishiwata A., Meroueh O., Mobashery S., Samama J. P. (2002) J. Am. Chem. Soc. 124, 2461–2465 [DOI] [PubMed] [Google Scholar]
- 45. Maveyraud L., Massova I., Birck C., Miyashita K., Samama J. P., Mobashery S. (1996) J. Am. Chem. Soc. 118, 7435–7440 [Google Scholar]
- 46. Mourey L., Miyashita K., Swaren P., Bulychev A., Samama J. P., Mobashery S. (1998) J. Am. Chem. Soc. 120, 9382–9383 [Google Scholar]
- 47. Golemi D., Maveyraud L., Ishiwata A., Tranier S., Miyashita K., Nagase T., Massova I., Mourey L., Samama J. P., Mobashery S. (2000) J. Antibiot. 53, 1022–1027 [DOI] [PubMed] [Google Scholar]
- 48. Nagase T., Golemi D., Ishiwata A., Mobashery S. (2001) Bioorg. Chem. 29, 140–145 [DOI] [PubMed] [Google Scholar]