

Background: Experimental bullous pemphigoid is mediated by mast cells (MCs), which release proteases when activated.
Results: Mouse MC protease-4 (mMCP-4), the likely murine homolog of human MC chymase, activates the enzyme MMP-9 and directly injures the skin adhesion protein BP180.
Conclusion: MMCP-4 regulates experimental BP by two different mechanisms.
Significance: MMCP-4 is a potential therapeutic target for human BP treatment.
Keywords: Animal Models, Autoimmune Diseases, Basement Membrane, Mast Cell, Protease, Skin
Abstract
Mouse mast cell protease-4 (mMCP-4) has been linked to autoimmune and inflammatory diseases, although the exact mechanisms underlying its role in these pathological conditions remain unclear. Here, we have found that mMCP-4 is critical in a mouse model of the autoimmune skin blistering disease bullous pemphigoid (BP). Mice lacking mMCP-4 were resistant to experimental BP. Complement activation, mast cell (MC) degranulation, and the early phase of neutrophil (PMN) recruitment occurred comparably in mMCP-4−/− and WT mice. However, without mMCP-4, activation of matrix metalloproteinase (MMP)-9 was impaired in cultured mMCP-4−/− MCs and in the skin of pathogenic IgG-injected mMCP-4−/− mice. MMP-9 activation was not fully restored by local reconstitution with WT or mMCP-4−/− PMNs. Local reconstitution with mMCP-4+/+ MCs, but not with mMCP-4−/− MCs, restored blistering, MMP-9 activation, and PMN recruitment in mMCP-4−/− mice. mMCP-4 also degraded the hemidesmosomal transmembrane protein BP180 both in the skin and in vitro. These results demonstrate that mMCP-4 plays two different roles in the pathogenesis of experimental BP, by both activating MMP-9 and by cleaving BP180, leading to injury of the hemidesmosomes and extracellular matrix of the basement membrane zone.
Introduction
Mast cells (MCs)6 are key effector cells involved in innate and adaptive immunity, as well as allergic and anaphylactic reactions (1, 2). MCs are found in particularly high concentrations in the connective tissues just below the epithelial surfaces of the body, such as the submucosal tissues of the gastrointestinal and respiratory tracts and the dermis of the skin, positioning them as key sentinels against external challenges. Upon activation, MCs degranulate and release preformed bioactive compounds from their secretory granules, including histamine, cytokines, proteoglycans, and proteases (3). Together, these inflammatory mediators increase the permeability of local blood vessels and recruit effector cells to the site. In addition to their role in protection against environmental stimuli, MCs are also implicated in chronic inflammatory and autoimmune diseases. Mice lacking MCs are partially or completely protected from disease in animal models of multiple sclerosis, collagen-induced arthritis, and bullous pemphigoid (BP) (4–6).
BP is an autoimmune skin blistering disease triggered by autoantibodies against the hemidesmosomal transmembrane protein BP180 (also called BPAG2 or type XVII collagen) (7–13). Using an animal model of BP in which neonatal BALB/c mice are injected with anti-murine BP180 (mBP180) antibodies (14), we previously demonstrated that dermal-epidermal separation is triggered by anti-BP180 IgG and depends on complement activation, MC degranulation, and neutrophil (PMN) recruitment (15, 16). Activated PMNs release proteolytic enzymes such as the serine proteinase neutrophil elastase (NE) and gelatinase B/matrix metalloproteinase (MMP)-9, which in turn degrade extracellular matrix (ECM) components of the basement membrane zone (BMZ), leading to separation of the dermis from the epidermis (17–19).
MC proteases are a major class of inflammatory mediators stored in great quantities as fully active enzymes in the secretory granules of MCs (20, 21). MC proteases are broken into three classes as follows: chymases, tryptases, and carboxypeptidase A (22). In this study, we focus on one of several murine chymases, mouse MC protease-4 (mMCP-4), which is widely recognized as the likely homolog of the single identified human chymase (23–25). Mice deficient in mMCP-4 (mMCP4−/−) have normal numbers of MCs, no alteration in MC morphology, and no abnormalities in the storage of other MC proteases. However, mMCP-4−/− mice exhibit a complete loss in chymotryptic activity in ear tissue, indicating that mMCP-4 is the main source of stored chymotrypsin-like activity in the skin (25).
Using the mMCP-4−/− mice, mMCP-4 activity has recently been associated with inflammatory processes in allergic asthma, immune complex-mediated glomerulonephritis, autoimmune arthritis, and epidermal burn injury (26–29). Importantly, mMCP-4 can activate MMP-9 in vitro, a key mediator of experimental BP (30). These observations, in conjunction with the known role of MCs in BP, suggest that mMCP-4 may contribute to the pathogenesis of BP. Using mMCP-4−/− mice, we demonstrate two pathogenic roles for mMCP-4 in experimental BP as follows: mMCP-4 contributes to activation of MMP-9 and also directly damages the ECM by degrading BP180.
EXPERIMENTAL PROCEDURES
Reagents
Purified murine MMP-9 was obtained from Triple Point Biologics, Inc. Purified PMN myeloperoxidase (MPO) was purchased from Athens Research and Technology, Inc. (Athens, GA). Rabbit anti-mMCP-4 antibody was made in-house as described previously (31). mMCP-4 was purified as described previously (32). Protein concentration was determined with the RC DC protein assay purchased from Bio-Rad. The ECL Western blotting analysis kit was purchased from GE Healthcare.
Laboratory Animals
C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). mMCP4−/− mice were generated and backcrossed onto B6 background as described previously (25). The mice were maintained at the University of North Carolina Chapel Hill Animal Center. Neonatal mice (24–36 h old with body weights between 1.4 and 1.6 g) were used for passive transfer experiments, and adult mice (8–10 weeks old) were used for mast cell reconstitution experiments. Animal care and animal experiments were approved by the Animal Care Committee at the University of North Carolina, Chapel Hill.
Preparation of Pathogenic Anti-BP180 IgG
The preparation of recombinant murine BP180ABC and the immunization of rabbits were performed as described previously (14). Briefly, a segment of the ectodomain of the murine BP180 antigen (33) was expressed as a glutathione S-transferase (GST) fusion protein using the pGEX prokaryotic expression system (GE Healthcare). The murine BP180 fusion protein, designated GST-mBP180ABC containing NC14A and half of C13 domains, was purified to homogeneity by affinity chromatography using a glutathione-agarose column (34). New Zealand White rabbits were immunized with purified GST-mBP180ABC, and the IgG fraction from the serum (designated R530) was purified as described previously (14). The IgG fractions were concentrated and sterilized by ultrafiltration, and the protein concentrations were determined by A280 (E (1%, 1 cm) = 13.6). The titers of anti-murine BP180 antibodies in both the unfractionated rabbit serum and in the purified IgG fraction were assayed by indirect immunofluorescence (IF) using mouse skin cryosections as substrate. The antibody preparations were also tested by immunoblotting against the GST-mBP180ABC fusion protein. The IF and immunoblotting techniques have been reported elsewhere (14). The pathogenicity of these IgG preparations was tested by passive transfer experiments as described below.
Induction of Experimental BP and Clinical Evaluation of Animals
Neonates were given on the back one intradermal injection of a sterile solution of IgG in PBS (50 μl of IgG, 2.64 mg of IgG/g body weight) as described previously (14). The extent of cutaneous disease was scored as follows: 0, no detectable skin disease; 1, mild erythematous reaction with no evidence of the epidermal detachment sign (elicited by gentle friction of the mouse skin; when positive, this produced fine, persistent wrinkling of the epidermis); 2, intense erythema and epidermal detachment sign involving 10–50% of the epidermis in localized areas; 3, intense erythema with frank epidermal detachment sign involving more than 50% of the epidermis in the injection site. After clinical examination, the animals were sacrificed, and skin and serum specimens were obtained. Each skin section (∼6 × 6 mm in size) was analyzed by H&E staining and routine histological examination to localize the lesional site and PMN infiltration, by direct IF assays to detect rabbit IgG and mouse C3 deposition at the BMZ, and by MPO enzymatic assay to quantify the PMN accumulation at the skin injection site as described below. Direct and indirect IF studies were performed as described previously (14) using commercially available FITC-conjugated goat anti-rabbit IgG (Kirkegaard & Perry Laboratories Inc.). Monospecific goat anti-mC3 IgG was purchased from Cappel Laboratories.
Quantification of MCs and MC Degranulation
Skin sections (∼3 × 3 mm) of IgG-injected mice were fixed in 10% formalin. Paraffin sections (6 μm thick) were prepared and stained with toluidine blue and H&E staining. Dermal MCs were counted by two individuals in the laboratory in a blinded fashion and classified as degranulated (>10% of the granules exhibiting fusion or discharge) or normal in five random fields under a light microscope at ×400 magnification (4, 35). MCs with complete degranulation that may be missed by toluidine blue staining were identified by indirect IF with FITC-conjugated rat anti-mouse c-Kit monoclonal antibody (BD Biosciences). Results were expressed as percentage of degranulated MC (number of degranulating MCs per total number of MCs in five random fields × 100%).
Quantification of PMN Accumulation at Antibody Injection Sites
Tissue MPO activity was used as an indicator of PMNs within skin samples of experimental animals as described elsewhere (36). We previously showed that clinical skin blistering is directly correlated with the number of infiltrating PMNs in the IgG injection site (16). The mouse skin samples (∼3 × 6 mm) were extracted by homogenization in 500 μl of extraction buffer. MPO content was expressed as relative MPO activity (A460 nm/mg of protein).
MC Reconstitution
mMCP-4−/− mice were reconstituted selectively and locally by the injection of growth factor-dependent bone marrow-derived cultured MCs into the skin (4, 37, 38). Briefly, femoral bone marrow cells from WT and mMCP-4−/− mice were maintained in vitro for 4 weeks in RPMI 1640 complete medium (Invitrogen) supplemented with 20% WEHI-3-conditioned medium until MCs represented >95% of the total cells as determined by toluidine blue staining and flow cytometry analysis using antibodies specific for the MC cell surface markers FcϵRI, c-Kit, and CD13 (37). Murine IgE and rat anti-mouse IgE were purchased from Southern Biotechnology Associates (Birmingham, AL). FITC-labeled rat anti-mouse c-Kit and FITC-labeled rat anti-mouse CD13 were obtained from DB Pharmingen (San Diego, CA). MCs (1 × 106 in 20 μl of medium) were injected i.d. into the ears of 8–10-week-old MCP-4−/− mice. Medium alone (20 μl) was injected i.d. into the ears of mMCP-4−/− mice to serve as negative controls. This procedure selectively and locally reconstitutes the dermal MC population without systemic effects (38). MC reconstitution was confirmed by staining skin sections from MC-injected sites with toluidine blue. Ten weeks after the adoptive transfer of MCs, when the injected MCs were fully matured into functional cutaneous MCs (4, 37, 38), both ears of the mice were injected i.d. with pathogenic anti-BP180 IgG (2 mg/20 μl/site). 24 h later, ear skin biopsies were obtained and analyzed by H&E staining, toluidine blue staining, and MPO enzyme assay as described above.
Isolation of PMNs and Local PMN Reconstitution
PMNs were isolated from heparinized blood from WT and mMCP-4−/− mice by dextran sedimentation followed by separation on a density gradient (39). PMN purity of the final cell preparation was consistently greater than 96% as determined by cell cytospin and LeukoStat staining (Fisher Diagnostics). The viability of the PMNs was greater than 96% as determined by trypan blue exclusion. PMNs were kept in PBS with 10 mm glucose at 4 °C before use. For PMN reconstitution, neonatal mice were injected i.d. with pathogenic IgG and, 90 min later, 5 × 105 purified mouse PMNs in 50 μl of PBS were injected into the IgG injection site.
Activation of Pro-MMP-9 in Mast Cell Culture
Bone marrow-derived cultured MCs (1 × 106 MCs/ml) were stimulated with recombinant mouse C5a (50 ng/ml) at 37 °C for 1 h. Cells were centrifuged at 14,000 rpm at 4 °C for 10 min. The supernatant was incubated with purified mouse pro-MMP-9 (1 μg/ml) at 37 °C for 4 h in the absence or presence of rabbit anti-mMCP-4 antibody. The digestion mixtures were analyzed by gelatin zymography as described below.
Gelatin Zymography
Gelatinase profiles were determined by zymography as described previously (40). In brief, protein extracts of skin sections from injected animals were subjected to SDS-PAGE on gelatin-containing acrylamide gels (10% acrylamide and 1% gelatin) under nonreducing conditions. After electrophoresis, gels were washed twice with 2.5% Triton X-100 for 30 min to remove SDS. Gels were then rinsed briefly with water followed by incubation overnight at 37 °C in reaction buffer containing 50 mm Tris, pH 7.4, 150 mm NaCl, and 5 mm CaCl2. The gels were stained with 0.125% Coomassie Brilliant Blue. Areas of gelatinolytic activity appeared as clear zones against a dark blue background.
Colorimetric Assay
Levels of MMP-9 were measured with a MMP colorimetric assay kit following manufacturer's instructions (BIOMOL Research Laboratories, Plymouth Meeting, PA) with minor modifications (41). Briefly, supernatant samples were incubated with the MMP colorimetric substrate or treated with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid to activate MMP-9 and then incubated with the MMP colorimetric substrate Ac-PLG[2-mercapto-4-methyl]-LG-OC2H5 in reaction buffer (final substrate concentration = 100 μm) at 37 °C. MMP activity in protein extracts was measured by the change in absorbance at 412 nm and was expressed as relative MMP activity (A412 nm reading/min/mg of protein of mouse skin injected with pathogenic IgG − A412 nm reading/min/mg protein of mouse skin injected with normal control IgG).
Injection of α2-AP Plasmin Inhibitor
Neonatal mice were injected i.d. with 50 μl of human α2-AP (25 μg/g body weight) prior to injection of pathogenic IgG (2.64 mg/g body weight) as described previously (42). Endotoxin levels in α2-AP preparations were minimal; mice injected with α2-AP showed no inflammatory response in the skin as determined by MPO enzyme activity assay.
Dermal-Epidermal Separation by mMCP-4 in Vitro
Mouse skin sections were obtained from neonatal BALB/c mice (36–48 h old) and cut into 2 × 2-mm strips with a razor blade. The skin strips were then incubated in minimum essential medium either with or without 1 μg/ml mMCP-4 at 37 °C for various periods of time. At the end of the incubation, the skin strips were rinsed in fresh minimum essential medium, fixed in 10% formalin, and embedded in paraffin, sectioned and stained with H&E. To identify BP180 degradation, mouse skin sections were homogenized in Tris-EDTA buffer containing 2% SDS, 0.5% Nonidet P-40, pH 6.8, plus proteinase inhibitors. Protein extracts were analyzed for BP180 degradation by SDS-PAGE (7%) followed by immunoblotting (19), using rabbit anti-mBP180 IgG and monospecific FITC-conjugated goat anti-rabbit IgG (Kirkegaard & Perry Laboratories Inc., Gaithersburg, MD).
mMCP-4 Digestion of Recombinant mBP180
The GST portion of the GST-mBP180ABC fusion protein described above was removed by PreScission protease cleavage, followed by purification on a glutathione column. Purified mBP180ABC protein (2 μg) was incubated with mMCP-4 or purified human chymase (1.25 milliunits) in PBS. Reactions were carried out at 37 °C for 0–60 min and terminated by adding an equal volume of SDS-PAGE sample buffer and heating at 100 °C for 5 min. Reaction mixtures were then resolved by electrophoresis through 18% SDS-polyacrylamide gels. The mBP180ABC and its degraded fragments were detected by immunoblotting using rabbit anti-mBP180 IgG.
Statistical Analysis
For statistical analysis, the data were expressed as mean ± S.E. and were analyzed using the Exact Wilcoxon Two-sample Test or Student's t test. A p value less than 0.05 was considered significant.
RESULTS
mMCP-4-deficient Mice Are Resistant to Experimental BP
To determine whether mMCP-4 is critical for the formation of subepidermal blisters in experimental BP, neonatal C57BL/6J (WT) and mMCP-4-deficient (mMCP-4−/−) mice were injected with pathogenic anti-mBP180 IgG (2.64 mg/g body weight) and assessed for disease susceptibility. 24 h after injection, WT mice developed clinical blisters (Fig. 1A), although mMCP-4−/− mice were resistant to disease (Fig. 1E). By direct IF, the perilesional skin of both WT and mMCP-4−/− mice exhibited deposition of rabbit IgG (Fig. 1, B and F) and murine complement components (Fig. 1, C and G) along the dermal-epidermal junction, indicating that the lack of mMCP-4 did not affect antibody deposition or complement activation. H&E-stained skin sections confirmed that mMCP-4−/− mice had no loss of dermal-epidermal adhesion, whereas WT mice developed subepidermal separation (Fig. 1, D and H). Skin disease severity in mMCP-4−/− mice was significantly reduced as compared with WT mice (Fig. 1I).
FIGURE 1.

mMCP-4-deficient mice are resistant to experimental BP. Neonatal WT and mMCP-4-deficient (mMCP4−/−) mice were injected i.d. with pathogenic rabbit anti-mBP180 IgG (2.64 mg/g body weight) and examined 24 h post-injection. A, WT mice develop clinical blisters. Direct IF showed deposition of rabbit IgG (B) and mouse C3 (C) at the BMZ. H&E staining revealed dermal-epidermal separation (D). mMCP-4−/− mice receiving the same dose of pathogenic IgG showed no skin lesions clinically (E) and histologically (H), although BMZ deposition of IgG (F) and C3 (G) were seen in the skin. e, epidermis; d, dermis. Arrow, basal keratinocytes. 100× magnification. I, clinical examination of the IgG-injected mice showed a drastic reduction of skin disease severity (expressed as mean ± S.E.) in mMCP-4−/− mice as compared with WT control. Eight mice per group. *, p < 0.001 (bar 3 versus 4). The Exact Wilcoxon Two-sample Test was used. SE, standard error; Min, minimum; Max, maximum.
PMN Recruitment, but Not MC Degranulation, Is Impaired in mMCP-4-deficient Mice
Having determined that complement deposition is unaffected by the absence of mMCP-4, we next investigated MC degranulation and PMN recruitment, which occur downstream of complement activation in BP pathogenesis (4, 43). 2 h after injection with pathogenic IgG, toluidine blue staining revealed extensive MC degranulation in the dermis of both WT and mMCP-4−/− mice (Fig. 2, A and B). Quantification of MC degranulation revealed no significant difference between WT and mMCP-4−/− animals injected with pathogenic antibody (Fig. 2C). Thus, MC degranulation is not impaired by the absence of mMCP-4.
FIGURE 2.

mMCP-4 deficiency does not impair mast cell degranulation. Neonatal WT and mMCP4−/− mice were injected i.d. with pathogenic rabbit anti-mBP180 IgG (2.64 mg/g body weight) and examined at different time points post-injection. A and B, toluidine blue staining showed similar degrees of mast cell (MC) degranulation in pathogenic IgG-injected WT and mMCP-4−/− mice at 2 h post-injection, when MC degranulation reaches the peak level. Arrow, degranulating mast cells. C, quantification of MC degranulation (expressed as % of MC degranulation) further confirmed that there was no difference in the number of degranulating MCs in the dermis of IgG-injected WT and mMCP-4−/− mice. D, quantification of infiltrating PMNs in the skin by measuring MPO activity (expressed as relative MPO activity, A460 nm/mg of protein) showed similar levels of PMN recruitment at 4 h, but significantly reduced levels at 12 and 24 h between the two groups of mice. n = 6 for each group. *, p < 0.01.
Quantification of MPO activity demonstrated that 4 h post-injection, WT and mMCP-4−/− mice recruit comparable numbers of PMNs to the skin (Fig. 2D, bars 1 and 2). However, 12 and 24 h post-injection, the mMCP-4−/− mice exhibited significantly lower levels of PMN recruitment than WT mice (Fig. 2D, bars 3–6). These data indicate that mMCP-4 is not required for the initial phase of MC-mediated PMN recruitment, but it is critical for the secondary amplification phase of PMN accumulation.
mMCP-4 Activates MMP-9
mMCP-4 can process pro-MMP-9 to its active form in vitro (30), and MMP-9 is critical for experimental BP (17, 18). Therefore, we tested whether mMCP-4 is involved in activation of MMP-9 in experimental BP. We detected the levels of pro-MMP-9 and active MMP-9 present in the skin of WT and mMCP-4−/− mice at 4 and 24 h post-antibody injection. Both the pro- and active forms of MMP-9 were seen in lesional and nonlesional skin samples of pathogenic IgG-injected WT and mMCP-4−/− mice by gelatin zymography after 4 and 24 h (Fig. 3A, lanes 1–4), although MMP-9 band densities in mMCP-4−/− mice were often weaker than those in WT mice. No active MMP-9 was seen in skin samples of control IgG-injected mice (Fig. 3A, lanes 5 and 6). We used a highly sensitive MMP colorimetric assay to quantify the levels of active MMP-9 in the skin of WT and mMCP-4−/− animals at 0, 4, 12, and 24 h after antibody injection. In keeping with the PMN recruitment results, we observed that, although there was no difference in the levels of active MMP-9 between mMCP-4−/− and WT animals at 4 h post-injection, there was significantly less active MMP-9 in the skin of pathogenic IgG-injected mMCP-4−/− mice at 12 and 24 h as compared with MMP-9 levels in the lesional skin of the diseased WT mice 12 and 24 h post-injection (Fig. 3B). MMP-9−/− mice exhibit only background levels of MMP activity up to 24 h after pathogenic antibody injection using this colorimetric assay (data not shown and see Ref. 42).
FIGURE 3.
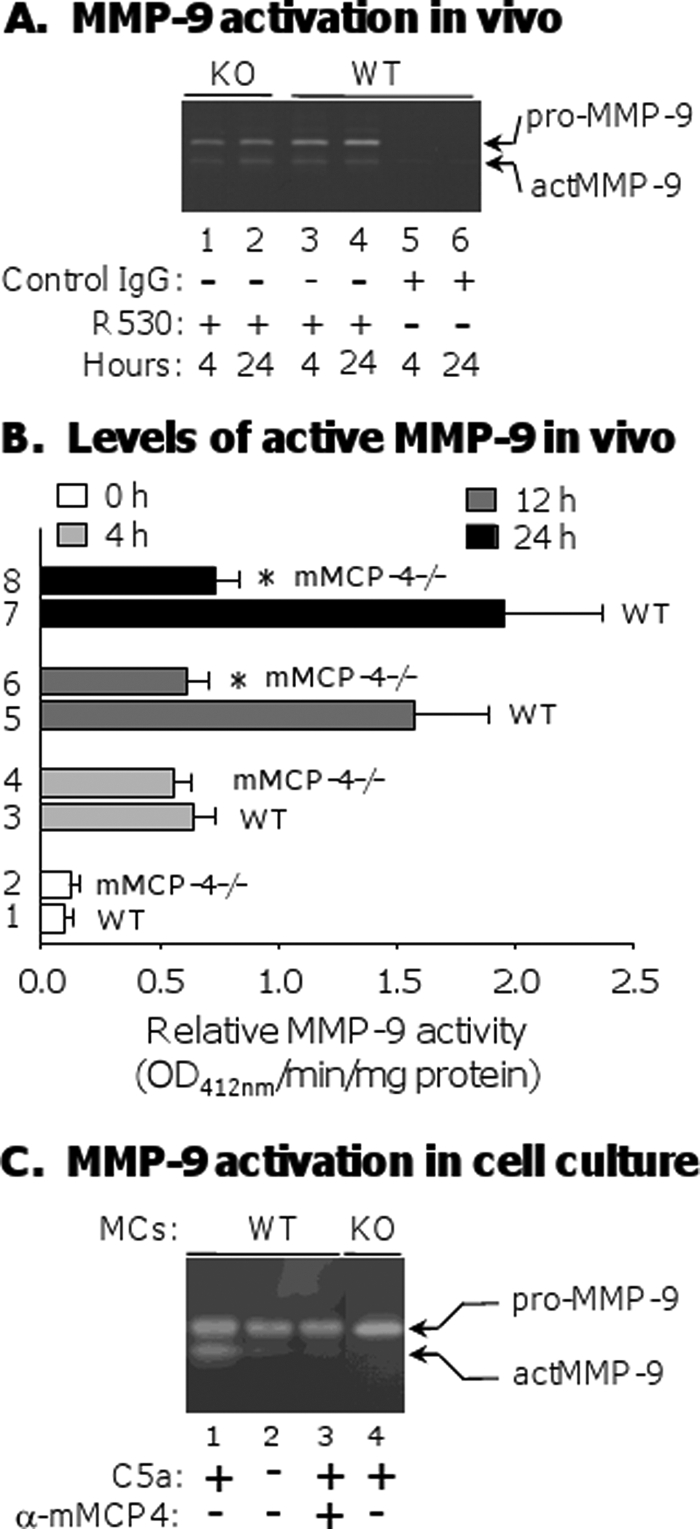
MMP-9 activation by mMCP-4 in vivo and in vitro. A, neonatal WT and mMCP-4−/− mice were injected i.d. with control IgG or pathogenic anti-mBP180 IgG. Skin samples were obtained at 4 and 24 h after IgG injection, and protein extracts (30 μg/lane) were analyzed by gelatin zymography. Both the pro- and active forms of MMP-9 were seen in skin samples of pathogenic IgG-injected WT (lanes 3 and 4) and mMCP-4−/− mice (lanes 1 and 2) at both time points. B, MMP colorimetric assay revealed a significant reduction of active MMP-9 in the skin samples of pathogenic IgG-injected mMCP-4−/− mice at 12 and 24 h as compared with active MMP-9 levels in the lesional skin of diseased WT mice (bars 6 and 8 versus 5 and 7). At 4 h, WT and mMCP-4−/− mice showed compatible levels of active MMP-9 (bars 3 and 4). MMP-9−/− mice exhibit background levels of activity at 0, 4, 12, and 24 h after injection with pathogenic antibodies (not shown). *, p < 0.01. n = 6 for each group. C, bone marrow-derived MCs (1 × 106) from WT or mMCP-4−/− mice were stimulated with recombinant C5a (50 ng/ml) at 37 °C for 1 h. The supernatants were then incubated with recombinant pro-MMP-9 (1 μg/ml) at 37 °C for 4 h. The digestion mixtures were analyzed by gelatin zymography. The supernatant of C5a-treated (lane 1) and not BSA control-treated (lane 2) MCs activated pro-MMP-9. Addition of rabbit anti-mMCP-4 antibody to the C5a-treated supernatant completely blocked MMP-9 activation (lane 3). C5a-activated mMCP-4−/− MC supernatants do not activate MMP-9 (lane 4).
To further establish that mMCP-4 released from MCs activates pro-MMP-9, we degranulated cultured bone marrow-derived mouse MCs from WT and mMCP-4−/− mice with recombinant C5a (rC5a) and collected the supernatant. Upon incubation with the rC5a-activated WT supernatant for 4 h at 37 °C, recombinant pro-MMP9 was converted into active MMP-9 (Fig. 3C, lane 1). Activation was completely blocked by addition of rabbit anti-mMCP-4 antibody to the supernatant (Fig. 3C, lane 3). Supernatants from cultured WT MCs treated with BSA instead of rC5a were unable to convert pro-MMP-9 to the active form (Fig. 3C, lane 2). Supernatants from cultured mMCP-4−/− MCs treated with rC5a also failed to activate MMP-9 (Fig. 3C, lane 4). Taken together, the in vitro and in vivo data strongly suggest that mMCP-4 released from degranulating MCs activates MMP-9.
PMN Reconstitution Restores Skin Blistering but Not MMP-9 Activation in mMCP-4−/− Mice
Infiltrating PMNs are a major source of MMP-9 in experimental BP (18). mMCP-4−/− mice fail to recruit the secondary wave of PMNs to the skin, which likely contributes to the reduction in active MMP-9 observed 12 and 24 h after pathogenic antibody injection. To determine the relative contributions of reduced PMN recruitment and mMCP-4 deficiency to the lower levels of active MMP-9 observed in mMCP-4−/− mice, we locally reconstituted mMCP-4−/− mice with PMNs to restore PMN levels. mMCP-4−/− mice injected with pathogenic antibodies alone were resistant to experimental BP (Fig. 4B). However, mMCP-4−/− mice locally injected with 5 × 105 WT or mMCP-4−/− PMNs 2 h after pathogenic antibody injection developed skin blisters (Fig. 4, C and D) equivalent to WT mice injected with pathogenic antibodies alone (Fig. 4A).
FIGURE 4.

Local PMN reconstitution restores levels of skin PMNs and skin disease but not levels of active MMP-9. Neonatal WT and mMCP-4−/− mice were injected i.d. with pathogenic antibodies and 2 h later locally reconstituted with 5 × 105 PMNs from WT or mMCP-4−/− mice. WT mice injected with pathogenic IgG (A) and mMCP-4−/− mice reconstituted with WT PMNs (C) or mMCP-4−/− PMNs (D) developed BP blisters. Unreconstituted mMCP-4−/− mice do not develop skin blisters (B). E, MPO activity assay of mouse skin samples revealed that at 4 h the PMN-reconstituted mMCP-4−/− mice (bars 7 and 8) had significantly increased number of PMNs compared with WT mice (bar 5). At 24 h, PMN-reconstituted mMCP-4−/− mice (bars 11 and 12) exhibited higher but not statistically significant numbers of PMNs as compared with WT (bar 9). F, MMP-9 activity assay revealed compatible levels of active MMP-9 at 4 h (bars 7 and 8 versus 5) and reduced levels of active MMP-9 at 24 h (bars 11 and 12 versus 9) between PMN-reconstituted mMCP-4−/− and WT mice. *, p < 0.05. n = 6 for each group.
Four hours post-injection, WT and mMCP-4−/− mice given pathogenic antibodies alone recruited equivalent numbers of PMNs to the skin and exhibited comparable levels of MMP-9 activity (Fig. 4, E, bars 5 and 6, and F, bars 5 and 6; also see Figs. 2D and 3B). MPO analysis confirmed that within 2 h of PMN injection (4 h post-IgG injection), the PMN-reconstituted mice exhibited significantly higher levels of skin PMNs than the WT and mMCP-4−/− mice injected with antibody alone (Fig. 4E, bars 7 and 8 versus bars 5 and 6). However, MMP-9 activity is not significantly different between the four groups of mice (Fig. 4F, bars 5–8). These data suggest that MMP-9 activation at the early stage of disease is not dependent on either PMNs or mMCP-4.
24 h post-injection, skin PMN levels were not significantly different between the PMN-reconstituted mMCP-4−/− mice and the WT mice injected with pathogenic antibodies alone (Fig. 4E, bars 11 and 12 versus bar 9). However, MMP-9 activity was significantly lower in the reconstituted mMCP-4−/− mice than the WT mice (Fig. 4F, bars 11 and 12 versus bar 9). These data demonstrate that artificial recruitment of PMNs to the skin in the mMCP-4−/− animals does not completely restore MMP-9 activity to WT levels, indicating that differences in the numbers of skin PMNs alone cannot account for the difference in MMP-9 activation observed.
mMCP-4 Directly Degrades BP180
Our data thus far indicate that mMCP-4 mediates inflammatory destruction of the ECM indirectly by regulating the activity of MMP-9. However, many MC proteases are also capable of directly cleaving ECM components (21). To determine whether mMCP-4 is able to directly cause tissue damage, we incubated neonatal mouse skin sections with 1 μg/ml mMCP-4 or medium alone for 12 h at 37 °C. When examined by H&E staining, the sections incubated with mMCP-4, but not medium alone, exhibited dermal-epidermal separation (Fig. 5A). We extracted proteins from the sections and immunoblotted for BP180. A single strong 180-kDa band corresponding to intact BP180 was detected in the medium-incubated sections, although both intact and cleaved BP180 bands were detected in the lysates from mMCP-4-incubated sections (Fig. 5B). These data show that mMCP-4 is capable of cleaving BP180 in tissue directly and causing dermal-epidermal separation.
FIGURE 5.

mMCP-4 produces dermal-epidermal separation of the skin and cleaves BP180 in tissue and in vitro. A, neonatal mouse skin sections were incubated with culture medium alone or with mMCP-4 (1 μg/ml) at 37 °C for 12 h and were examined by H&E staining. Dermal-epidermal separation was seen in the skin incubated with mMCP-4 (right panel) but not the medium control (left panel). E, epidermis; D, dermis. Arrow, basal keratinocytes. 100× magnification. B, protein extracts from mouse skin incubated with culture medium alone or mMCP-4 above were analyzed by immunoblotting using an anti-mBP180 antibody. mMCP-4 (lane 2), but not culture medium (lane 1), degraded BP180. C, recombinant mBP180ABC was incubated with mMCP-4 at 37 °C for 0–60 min. The digestion products were resolved by SDS-PAGE and detected by immunoblotting using an anti-mBP180 antibody. Degradation products were seen after incubation with mMCP-4 for 5, 15, 30, and 60 min.
To confirm that mMCP-4 directly degrades BP180, we performed an in vitro degradation assay by incubating purified mMCP-4 with purified recombinant protein (mBP180ABC) as the substrate. mBP180ABC corresponds to a 20-kDa portion of the murine BP180 extracellular domain. Degradation products of mBP180ABC are detectable by immunoblotting within 5 min of incubation with mMCP-4 (Fig. 5C).
MC Reconstitution Restores BP in mMCP-4-deficient Mice
We next performed a series of MC reconstitution experiments to confirm that mMCP-4 released from MCs is crucial for dermal-epidermal separation, activation of MMP-9, and PMN recruitment. We injected the left ears of mMCP-4−/− mice with mMCP-4−/− MCs and the right ears of the same mice with mMCP-4+/+ MCs (1 × 106 cells/site). mMCP-4−/− mice were injected with PBS as controls. Ten weeks later, both ears of the mice were injected i.d. with pathogenic anti-BP180 IgG (2 mg/20-μl/site) and examined 24 h post-IgG injection. We observed similar numbers of MCs in the mMCP-4+/+ MC-reconstituted ears and the mMCP-4−/− MC-reconstituted ears. PBS-injected mMCP-4−/− controls and mMCP-4−/− mice reconstituted with mMCP-4−/− MCs were protected from blistering, although mMCP-4−/− mice reconstituted with mMCP-4+/+ mast cells developed dermal-epidermal separation (Fig. 6, A–C). The mMCP-4−/− ears reconstituted with mMCP-4+/+ MCs exhibited significantly higher levels of active MMP-9 and PMN infiltration than the mMCP-4−/− ears reconstituted with mMCP-4−/− MCs (Fig. 6, F, bars 3 and 4, and G, bars 3 and 4).
FIGURE 6.

MC reconstitution restores BP in mMCP-4-deficient mice. Pathogenic antibodies were injected into ears of WT mice, WT mice pretreated with anti-mMCP-4 antibody, mMCP-4−/− mice, and mMCP-4−/− mice reconstituted with 1 × 106 MCs from either mMCP-4−/− mice or WT mice. The ears were examined 24 h post-IgG injection. H&E staining showed that WT mice (D) and mMCP-4−/− mice reconstituted with WT MCs (C) developed dermal-epidermal junction separation following antibody injection. mMCP-4−/− mice reconstituted with PBS (A) or reconstituted with mMCP-4−/− MCs (B) failed to develop dermal-epidermal junction separation. Pretreatment with anti-mMCP-4 antibody abolished pathogenic IgG-induced skin lesions in WT mice (E). F, quantification of active MMP-9. MMP colorimetric assay revealed that lesional skin of WT MC-reconstituted mMCP-4−/− mice (bar 4) had significantly elevated levels of active MMP-9 compared with mMCP-4−/− mice reconstituted with mMCP-4−/− MCs (bar 3). Anti-mMCP-4 antibody treatment led to a significant decrease in active MMP-9 levels in WT mice (bar 7) as compared with WT mice without mMCP-4 blocking (bar 6). G, quantification of PMN infiltration. Pathogenic antibodies induced experimental BP with significantly increased PMN infiltration in WT mice (bar 6) and mMCP-4−/− mice reconstituted with WT MCs (bar 4) but not mMCP-4−/− mice reconstituted with mMCP-4−/− MCs (bar 3) and WT mice pretreated with anti-mMCP-4 antibody (bar 7). *, p < 0.01, six mice per group. Three independent experiments were done for each group of mice.
WT mice injected with pathogenic antibody developed dermal-epidermal separation as expected, and this blistering was completely blocked by pretreatment with an anti-mMCP-4 antibody (Fig. 6, D and E). Pretreatment with the anti-mMCP-4 antibody also significantly reduced the levels of active MMP-9 and PMN infiltration in the ears (Fig. 6, F, bar 6 versus 7, and G, bar 6 versus 7). Taken together, these data show that mMCP-4 released from MCs is critical for the activation of MMP-9, the secondary wave of PMN infiltration, and blister formation.
mMCP-4 and Plasmin Are Required for MMP-9 Activation and Skin Blistering
We previously showed that plasmin plays a critical role in the early stages of experimental BP by activating MMP-9 (42). In light of our data implicating mMCP-4 as an important mediator of MMP-9 activation, we next sought to clarify the relationship of plasmin and MMCP-4 in MMP-9 activation. We treated mMCP-4−/− mice with the plasmin inhibitor α2-antiplasmin (α2-AP) and injected the animals with pathogenic antibody. At 4, 24, and 48 h after antibody injection, the α2-AP-treated mMCP-4−/− mice had greater levels of PMN infiltration and activated MMP-9 than at time 0 (Fig. 7, A and B), indicating that MMP-9 is activated in the absence of both mMCP-4 and the plasmin cascade. Both MPO and MMP-9 activity were significantly lower in the α2-AP-treated mMCP-4 mice than in WT or untreated mMCP-4 mice 24 and 48 h post-injection (Fig. 7, A and B, bars 7 and 8 versus 9). Untreated mMCP-4−/− and α2-AP treated mMCP-4−/− mice are both protected from disease 24 h after pathogenic antibody injection (Fig. 7C, bars 2 and 3). 48 h after injection, the mMCP-4−/− and mMCP-4−/− mice treated with α2-AP exhibited modest increases in disease score but were still significantly protected from blistering as compared with WT animals (Fig. 7C, bars 5 and 6 versus 4). These data indicate that both mMCP-4 and plasmin play major roles in MMP-9 activation and skin blistering and that additional pathway(s) of MMP-9 activation exist in experimental BP.
FIGURE 7.
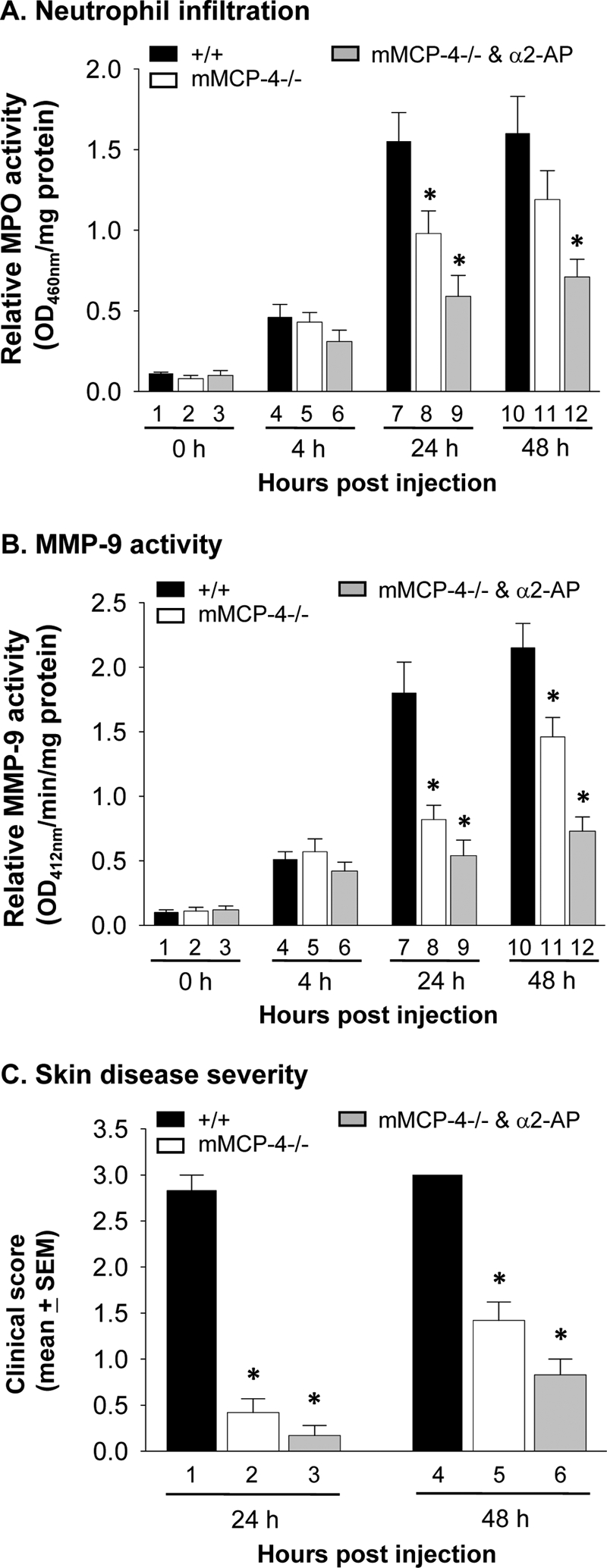
Both mMCP-4 and plasmin are required for MMP-9 activation and BP blistering. WT and mMCP-4−/− mice were pretreated with buffer control or plasmin inhibitor α2-AP and 2 h later were injected i.d. with pathogenic antibodies. The mice were examined at 0, 4, 24, and 48 h. A, quantification of PMN infiltration. MPO assay showed that at 4 h, compatible levels of PMN infiltration were seen in IgG-injected WT (bar 4) and mMCP-4−/− (bar 5) and α2-AP-treated mMCP-4−/− mice (bar 6). At 24 h, both mMCP-4−/− (bar 8) and α2-AP-treated mMCP-4−/− mice (bar 9) showed significantly reduced PMN infiltration as compared with WT mice (bar 7). PMN numbers in α2-AP-treated mMCP-4−/− mice were also significantly lower than mMCP-4−/− mice. At 48 h, α2-AP-treated mMCP-4−/− (bar 12) but not mMCP-4−/− mice (bar 11) showed significantly reduced PMN infiltration as compared with WT mice (bar 10). B, quantification of active MMP-9. MMP colorimetric assay revealed that at 4 h, compatible levels of active MMP-9 were present in IgG-injected WT (bar 4) and mMCP-4−/− (bar 5) and α2-AP-treated mMCP-4−/− mice (bar 6). At 24 and 48 h, both mMCP-4−/− (bars 8 and 11) and α2-AP-treated mMCP-4−/− mice (bars 9 and 12) showed significantly reduced active MMP-9 levels as compared with WT mice (bars 7 and 10). At 48 h, α2-AP-treated mMCP-4−/− mice (bar 12) also showed significantly reduced levels of active MMP-9 as compared with mMCP-4−/− mice (bar 11). C, clinical disease activity. WT mice (bars 1 and 4) developed extensive skin blisters, although mMCP-4−/− and α2-AP-treated mMCP-4−/− mice showed no skin blistering at 24 h (bars 2 and 3) and drastically reduced skin lesions at 48 h (bars 5 and 6). *, p < 0.01, 6 mice per group. The Exact Wilcoxon Two-sample Test was used.
DISCUSSION
The role of MC proteases in autoimmune and inflammatory diseases is currently a very active area of research. mMCP-4 has garnered particular interest in recent years because of its homology with human MC chymase and the recent generation of an mMCP-4-deficient line of mice (23–25). In this study, we provide the first evidence for the mechanisms by which mMCP-4 mediates an autoimmune disease. In experimental BP, mMCP-4 has two key functions. 1) It activates MMP-9 and indirectly drives the second phase of PMN infiltration, a critical step required to amplify disease activity for clinical skin blistering. 2) It directly cleaves the autoantigen BP180, a key component of the ECM.
We previously elucidated critical roles for NE and MMP-9 in experimental BP (17–19). These enzymes are released from the first wave of PMNs recruited to the inflamed skin by degranulated MCs (4 h post-injection). MMP-9 inactivates the serpin α1-proteinase inhibitor, the principal plasma inhibitor of NE (44). When relieved of α1-proteinase inhibitor inhibition, NE degrades components of the extracellular matrix, including BP180, leading to dermal-epidermal separation and a secondary wave (amplification) of PMN infiltration. Our study shows that BP180 is also a substrate for mMCP-4. mMCP-4 cleaves BP180 both in vitro and in vivo. Whether mMCP-4 degrades other critical hemidesmosomal and extracellular matrix proteins and whether mMCP-4 and NE act independently or in concert in experimental BP are currently under investigation in our laboratory.
Extracellular proteolysis is critical for development, tissue repair, and progression of diseases in vivo (45). The processes of activating the zymogen forms of proteinases are strictly regulated and locally confined to prevent unwanted cascades of proteinase activation. MMP-9 is stored in inflammatory cells as a zymogen and is activated extracellularly. Our present results show that mMCP-4 secreted by degranulated MCs activates MMP-9 in experimental BP. mMCP-4 is produced only by MCs (46, 47), and WT MC reconstitution restores the blister phenotype in mMCP-4−/− mice (Fig. 6).
Pro-MMP-9 also can be activated by plasmin, uPA (48–52), chymase (53), tissue kallikrein (54), and other active MMPs (55–59). We previously demonstrated that MMP-9 is activated by plasmin or an indirect plasmin-dependent pathway and not by uPA, tissue plasminogen activator, and MMP-3 during the early stages of experimental BP disease development (42). Mice lacking plasmin activity (because of a lack of plasminogen or both tissue plasminogen activator and uPA) show minimal levels of MMP-9 activation and no skin lesions at 12 h post-pathogenic IgG injection. At a later time point (24 h), however, pathogenic IgG induces subepidermal blisters in these knock-out mice (34). In contrast, mMCP-4−/− mice are resistant to BP at both early (12 h) and late (24 h) stages of the disease process. These findings suggest that plasmin activates MMP-9 mainly at the early stage of the disease development, although mMCP-4 activates MMP-9 in the later stage of disease progression. It is not yet clear how these pathways of MMP-9 activation overlap or synergize. Our data demonstrating that mMCP-4−/− mice do not develop clinical or histological blistering indicate that plasmin alone is not sufficient to activate MMP-9 and induce disease. Mice lacking either mMCP-4 or plasmin activity are protected from disease, indicating that both of these pathways play a critical role in disease development. Interestingly, we demonstrated that mice lacking both mMCP-4 and plasmin activity still exhibit modest MMP-9 activation and develop mild disease over 48 h, strongly suggesting that at least one additional pathway of MMP-9 activation exists. This unidentified pathway of activation likely contributes to both the early and late stages of experimental BP pathogenesis, because mice lacking both mMCP-4 and plasmin activity exhibit MMP-9 activation within 4 h of antibody injection and increased disease severity over the course of 48 h. We hypothesize that infiltrating PMN-released MMP-9 is initially activated by both a plasmin-dependent mechanism and unidentified other activators, followed by mMCP-4. Further investigation is needed to identify the functional interplay between plasmin- and mMCP-4-dependent pathways for MMP-9 activation in experimental BP, as well as unidentified plasmin- and mMCP-4-independent pathways.
The discovery of the dual targets of mMCP-4 further elucidates a complex net of interactions that control local inflammation and tissue injury in experimental BP (Fig. 8). Because mMCP-4 is the functional homolog of human chymase, our current findings give us new insights into the immunopathogenesis of human autoimmune diseases and implicate this chymase as a promising potential therapeutic target in human disease.
FIGURE 8.

Working model of the role of mMCP-4 in experimental BP. Experimental BP is initiated by pathogenic anti-BP180 and depends on complement activation, mast cell degranulation, and PMN infiltration. Upon activation, infiltrating PMNs release neutrophil elastase (NE), MMP-9 zymogen (pro-MMP-9), and other proteinases. Plasmin is generated from plasminogen by tissue plasminogen activator and/or uPA and activates an unidentified MMP (?). In the early stages of blistering, pro-MMP-9 is mainly activated by the plasmin-activated MMP and mMCP-4. mMCP-4 and plasmin work together with other activators of MMP-9 to activate pro-MMP-9. Active MMP-9 (actMMP-9) cleaves α1-proteinase inhibitor (α1-PI) to release NE inhibition. Unchecked NE degrades BP180 and other ECM components, resulting in dermal-epidermal separation. mMCP-4 is also capable of directly cleaving BP180, further amplifying skin blistering.
Acknowledgments
We thank Dr. Pamela Groben and Joy Miller for routine histology.
This work was supported, in whole or in part, by National Institutes of Health Grants AI40768 and AI61430 (to Z. L.), AR052109 and AR053313 (to N. L.), AR32599 and AR32081 (to L. A. D.), and CA057621 (to Z. W.) from the USPHS. This work was also supported by grants from the Swedish Medical Research Council (to M. A. and G. P).
- MC
- mast cell
- BMZ
- basement membrane zone
- BP
- bullous pemphigoid
- ECM
- extracellular matrix
- mMCP
- mouse mast cell protease
- MMP
- matrix metalloproteinase
- MPO
- myeloperoxidase
- NE
- neutrophil elastase
- PMN
- neutrophil
- IF
- immunofluorescence
- i.d.
- intradermal
- α2-AP
- α2-antiplasmin
- uPA
- urokinase-type plasminogen activator.
REFERENCES
- 1. Galli S. J., Tsai M. (2010) Eur. J. Immunol. 40, 1843–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gurish M. F., Austen K. F. (2001) J. Exp. Med. 194, F1–F5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stevens R. L., Adachi R. (2007) Immunol. Rev. 217, 155–167 [DOI] [PubMed] [Google Scholar]
- 4. Chen R., Ning G., Zhao M. L., Fleming M. G., Diaz L. A., Werb Z., Liu Z. (2001) J. Clin. Invest. 108, 1151–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee D. M., Friend D. S., Gurish M. F., Benoist C., Mathis D., Brenner M. B. (2002) Science 297, 1689–1692 [DOI] [PubMed] [Google Scholar]
- 6. Secor V. H., Secor W. E., Gutekunst C. A., Brown M. A. (2000) J. Exp. Med. 191, 813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Diaz L. A., Ratrie H., 3rd, Saunders W. S., Futamura S., Squiquera H. L., Anhalt G. J., Giudice G. J. (1990) J. Clin. Invest. 86, 1088–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Giudice G. J., Emery D. J., Diaz L. A. (1992) J. Invest. Dermatol. 99, 243–250 [DOI] [PubMed] [Google Scholar]
- 9. Hopkinson S. B., Riddelle K. S., Jones J. C. (1992) J. Invest. Dermatol. 99, 264–270 [DOI] [PubMed] [Google Scholar]
- 10. Jordon R. E., Beutner E. H., Witebsky E., Blumental G., Hale W. L., Lever W. F. (1967) JAMA 200, 751–756 [PubMed] [Google Scholar]
- 11. Lever W. F. (1953) Medicine 32, 1–123 [DOI] [PubMed] [Google Scholar]
- 12. Li K., Guidice G. J., Tamai K., Do H. C., Sawamura D., Diaz L. A., Uitto J. (1992) J. Invest. Dermatol. 99, 258–263 [DOI] [PubMed] [Google Scholar]
- 13. Stanley J. R., Hawley-Nelson P., Yuspa S. H., Shevach E. M., Katz S. I. (1981) Cell 24, 897–903 [DOI] [PubMed] [Google Scholar]
- 14. Liu Z., Diaz L. A., Troy J. L., Taylor A. F., Emery D. J., Fairley J. A., Giudice G. J. (1993) J. Clin. Invest. 92, 2480–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Z., Giudice G. J., Swartz S. J., Fairley J. A., Till G. O., Troy J. L., Diaz L. A. (1995) J. Clin. Invest. 95, 1539–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Z., Giudice G. J., Zhou X., Swartz S. J., Troy J. L., Fairley J. A., Till G. O., Diaz L. A. (1997) J. Clin. Invest. 100, 1256–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Z., Shapiro S. D., Zhou X., Twining S. S., Senior R. M., Giudice G. J., Fairley J. A., Diaz L. A. (2000) J. Clin. Invest. 105, 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Z., Shipley J. M., Vu T. H., Zhou X., Diaz L. A., Werb Z., Senior R. M. (1998) J. Exp. Med. 188, 475–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Z., Zhou X., Shapiro S. D., Shipley J. M., Twining S. S., Diaz L. A., Senior R. M., Werb Z. (2000) Cell 102, 647–655 [DOI] [PubMed] [Google Scholar]
- 20. Schwartz L. B., Irani A. M., Roller K., Castells M. C., Schechter N. M. (1987) J. Immunol. 138, 2611–2615 [PubMed] [Google Scholar]
- 21. Schwartz L. B., Lewis R. A., Austen K. F. (1981) J. Biol. Chem. 256, 11939–11943 [PubMed] [Google Scholar]
- 22. Pejler G., Rönnberg E., Waern I., Wernersson S. (2010) Blood 115, 4981–4990 [DOI] [PubMed] [Google Scholar]
- 23. Andersson M. K., Karlson U., Hellman L. (2008) Mol. Immunol. 45, 766–775 [DOI] [PubMed] [Google Scholar]
- 24. Reynolds D. S., Stevens R. L., Lane W. S., Carr M. H., Austen K. F., Serafin W. E. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 3230–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tchougounova E., Pejler G., Abrink M. (2003) J. Exp. Med. 198, 423–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Magnusson S. E., Pejler G., Kleinau S., Abrink M. (2009) FASEB J. 23, 875–882 [DOI] [PubMed] [Google Scholar]
- 27. Scandiuzzi L., Beghdadi W., Daugas E., Abrink M., Tiwari N., Brochetta C., Claver J., Arouche N., Zang X., Pretolani M., Monteiro R. C., Pejler G., Blank U. (2010) J. Immunol. 185, 624–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Waern I., Jonasson S., Hjoberg J., Bucht A., Abrink M., Pejler G., Wernersson S. (2009) J. Immunol. 183, 6369–6376 [DOI] [PubMed] [Google Scholar]
- 29. Younan G., Suber F., Xing W., Shi T., Kunori Y., Abrink M., Pejler G., Schlenner S. M., Rodewald H. R., Moore F. D., Jr., Stevens R. L., Adachi R., Austen K. F., Gurish M. F. (2010) J. Immunol. 185, 7681–7690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tchougounova E., Lundequist A., Fajardo I., Winberg J. O., Abrink M., Pejler G. (2005) J. Biol. Chem. 280, 9291–9296 [DOI] [PubMed] [Google Scholar]
- 31. Forsberg E., Pejler G., Ringvall M., Lunderius C., Tomasini-Johansson B., Kusche-Gullberg M., Eriksson I., Ledin J., Hellman L., Kjellén L. (1999) Nature 400, 773–776 [DOI] [PubMed] [Google Scholar]
- 32. Pejler G., Karlström A. (1993) J. Biol. Chem. 268, 11817–11822 [PubMed] [Google Scholar]
- 33. Li K., Tamai K., Tan E. M., Uitto J. (1993) J. Biol. Chem. 268, 8825–8834 [PubMed] [Google Scholar]
- 34. Liu Z., Diaz L. A., Haas A. L., Giudice G. J. (1992) J. Biol. Chem. 267, 15829–15835 [PubMed] [Google Scholar]
- 35. Wershil B. K., Wang Z. S., Gordon J. R., Galli S. J. (1991) J. Clin. Invest. 87, 446–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bradley P. P., Priebat D. A., Christensen R. D., Rothstein G. (1982) J. Invest. Dermatol. 78, 206–209 [DOI] [PubMed] [Google Scholar]
- 37. Ryan J. J., DeSimone S., Klisch G., Shelburne C., McReynolds L. J., Han K., Kovacs R., Mirmonsef P., Huff T. F. (1998) J. Immunol. 161, 6915–6923 [PubMed] [Google Scholar]
- 38. Wershil B. K., Mekori Y. A., Murakami T., Galli S. J. (1987) J. Immunol. 139, 2605–2614 [PubMed] [Google Scholar]
- 39. Clark R. A., Nauseef W. M. (2001) Curr. Protoc. Immunol. Chapter 7, Unit 7.23 [DOI] [PubMed] [Google Scholar]
- 40. Twining S. S., Zhou X., Schulte D. P., Wilson P. M., Fish B., Moulder J. (1996) Invest. Ophthalmol. Vis. Sci. 37, 511–522 [PubMed] [Google Scholar]
- 41. Metcalf J. A., Nauseef W. M., Root R. K. (1985) Laboratory Manual of Neutrophil Function, pp. 2–10, Raven Press, Ltd., New York [Google Scholar]
- 42. Liu Z., Li N., Diaz L. A., Shipley M., Senior R. M., Werb Z. (2005) J. Clin. Invest. 115, 879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Heimbach L., Li Z., Berkowitz P., Zhao M., Li N., Rubenstein D. S., Diaz L. A., Liu Z. (2011) J. Biol. Chem. 286, 15003–15009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jiang H., Kanost M. R. (1997) J. Biol. Chem. 272, 1082–1087 [DOI] [PubMed] [Google Scholar]
- 45. Werb Z. (1997) Cell 91, 439–442 [DOI] [PubMed] [Google Scholar]
- 46. Jippo T., Tsujino K., Kim H. M., Kim D. K., Lee Y. M., Nawa Y., Kitamura Y. (1997) Am. J. Pathol. 150, 1373–1382 [PMC free article] [PubMed] [Google Scholar]
- 47. Miller H. R., Pemberton A. D. (2002) Immunology 105, 375–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Davis G. E., Pintar Allen K. A., Salazar R., Maxwell S. A. (2001) J. Cell Sci. 114, 917–930 [DOI] [PubMed] [Google Scholar]
- 49. Lijnen H. R., Van Hoef B., Lupu F., Moons L., Carmeliet P., Collen D. (1998) Arterioscler. Thromb. Vasc. Biol. 18, 1035–1045 [DOI] [PubMed] [Google Scholar]
- 50. Mazzieri R., Masiero L., Zanetta L., Monea S., Onisto M., Garbisa S., Mignatti P. (1997) EMBO J. 16, 2319–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ramos-DeSimone N., Hahn-Dantona E., Sipley J., Nagase H., French D. L., Quigley J. P. (1999) J. Biol. Chem. 274, 13066–13076 [DOI] [PubMed] [Google Scholar]
- 52. Woessner J. F., Nagase H. (2000) Matrix Metalloproteinases and TIMPs, pp. 72–86, Oxford University Press, Oxford [Google Scholar]
- 53. Fang K. C., Raymond W. W., Blount J. L., Caughey G. H. (1997) J. Biol. Chem. 272, 25628–25635 [DOI] [PubMed] [Google Scholar]
- 54. Desrivières S., Lu H., Peyri N., Soria C., Legrand Y., Ménashi S. (1993) J. Cell Physiol. 157, 587–593 [DOI] [PubMed] [Google Scholar]
- 55. Fridman R., Toth M., Peña D., Mobashery S. (1995) Cancer Res. 55, 2548–2555 [PubMed] [Google Scholar]
- 56. Imai K., Yokohama Y., Nakanishi I., Ohuchi E., Fujii Y., Nakai N., Okada Y. (1995) J. Biol. Chem. 270, 6691–6697 [DOI] [PubMed] [Google Scholar]
- 57. Knäuper V., Smith B., López-Otin C., Murphy G. (1997) Eur. J. Biochem. 248, 369–373 [DOI] [PubMed] [Google Scholar]
- 58. Murphy G., Atkinson S., Ward R., Gavrilovic J., Reynolds J. J. (1992) Ann. N.Y. Acad. Sci. 667, 1–12 [DOI] [PubMed] [Google Scholar]
- 59. von Bredow D. C., Cress A. E., Howard E. W., Bowden G. T., Nagle R. B. (1998) Biochem. J. 331, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]