

Abstract
Using transcriptome profiling to determine differential gene expression between the permanent mouse articular cartilage and the transient growth plate cartilage, we identified a highly expressed gene, Cilp2, which is expressed differentially by articular chondrocytes. CILP-2 is highly homologous to CILP-1 (cartilage intermediate layer protein 1), which is expressed in the intermediate zone of articular cartilage and has been linked to cartilage degenerative diseases. We demonstrated that Cilp2 has a restricted mRNA distribution at the surface of the mouse articular cartilage during development, becoming localized to the intermediate zone of articular cartilage and meniscal cartilage with maturity. Although the extracellular CILP-2 protein localization is broadly similar to CILP-1, CILP-2 appears to be more localized in the deeper intermediate zone of the articular cartilage extracellular matrix at maturity. CILP-2 was shown to be proteolytically processed, N-glycosylated, and present in human articular cartilage. In surgically induced osteoarthritis in mice, Cilp1 and Cilp2 gene expression was dysregulated. However, whereas Cilp1 expression was increased, Cilp2 gene expression was down-regulated demonstrating a differential response to mechanically induced joint destabilization. CILP-2 protein was reduced in the mouse osteoarthritic cartilage. Ultrastructural analysis also suggested that CILP-2 may be associated with collagen VI microfibrils and thus may mediate interactions between matrix components in the territorial and inter-territorial articular cartilage matrix. mRNA expression analysis indicated that whereas Cilp1 and Cilp2 are expressed most abundantly in cartilaginous tissues, expression can be detected in muscle and heart.
Keywords: Connective Tissue, Development, Extracellular Matrix, Extracellular Matrix Proteins, Gene Expression, Arthritis, Articular Cartilage, Cartilage, Cartilage Structure, Osteoarthritis
Introduction
During limb development, chondrocytes follow alternative developmental pathways. Chondrocytes that form at the epiphyseal surface of long bones develop into permanent articular chondrocytes and form the smooth articular cartilage necessary for effective weight-bearing and joint movement. The other group of chondrocytes, the transient epiphyseal chondrocytes, are organized into growth plates, and undergo a sequential maturation program from resting cells to proliferative, prehypertrophic, and ultimately hypertrophic end-stage chondrocytes that are replaced with bone during endochondral ossification. Both populations of chondrocytes are critical for the formation of the skeleton and disturbances to normal chondrocyte development and function results in chondrodysplasias (reviewed by Refs. 1–3).
One of the important determinants of cartilage function is extracellular matrix (ECM)3 structure and composition. To identify new ECM determinants of the articular cartilage, we compared the gene expression profile of mouse articular cartilage with growth plate cartilage. These studies revealed that the most highly differentially expressed cDNA on the array was a Riken clone that corresponded to Cilp2 (cartilage intermediate-layer protein, isoform 2), recently reported as a product of cartilage cells (4, 5). CILP-2 is highly homologous to a known cartilage protein, CILP-1 (5). CILP-1 is a large secreted glycoprotein that is thought to play a role in cartilage scaffolding (6). It was isolated from human articular cartilage and its expression has been reported to be localized to the intermediate zone of articular cartilage in the territorial matrix (6). CILP-1 is a pro-form of two polypeptides, and is cleaved into distinct N- and C-terminal fragments at a furin endoprotease consensus site (7). The N-terminal of CILP-1 has been shown to bind to and inhibit TGFβ1 in vitro (8), and CILP1 mRNA is induced by TGFβ1 (9). CILP-1 levels have been shown to increase with age (6, 10) and in patients with early stage osteoarthritis (11). The association of single nucleotide polymorphisms in the CILP1 gene with musculoskeletal disorders including osteoarthritis (12–14), and lumbar disc disease in a Japanese population (8) implies that CILP proteins may be important in cartilage structure and disease. In contrast to CILP-1, little is known about the mRNA and protein expression of CILP-2 in cartilage and noncartilaginous tissues. Our studies show that Cilp2 has a restricted mRNA distribution at the surface of the mouse articular cartilage during development, becoming localized to the intermediate zone of articular cartilage and meniscal cartilage with maturity. We show that CILP-2 is a glycoprotein and is proteolytically processed, similar to CILP-1. CILP-2 protein is present in human articular cartilage and ultrastructural studies demonstrated that CILP-2 may be associated with collagen VI containing suprastructures. Importantly, our studies show that in a mouse experimental model of mechanically induced osteoarthritis Cilp2 gene expression is down-regulated suggesting a role for loss of CILP-2 in the pathophysiology of arthritis. In addition, our studies show for the first time the expression of Cilp1 and Cilp2 in skeletal muscle, suggesting that the CILPs can have additional roles in noncartilaginous tissue ECM structure and function.
EXPERIMENTAL PROCEDURES
Dissection and RNA Preparation
Dissection and RNA extraction from mouse articular cartilage was performed as previously described for growth plate cartilage (15). Briefly, 14-day-old (P14) Swiss white mice were sacrificed in accordance with Institutional Animal Ethics guidelines and femurs were dissected. The tissue was immersed in Tissue-Tek OCT embedding compound (Sakura Finetechnical), sectioned (5 μm) on a cryostat (Reichert-Jung), dehydrated in graded ethanol series, and air-dried. Slides were then immobilized on an inverted microscope (Leica) and the articular cartilage, and proliferative, prehypertrophic, and hypertrophic growth plate cartilage was dissected using an ophthalmic scalpel (Feather) (supplemental Fig. S1). Total RNA was extracted using the PicoPure RNA isolation kit (Arcturus Bioscience) and linearly amplified in two rounds using the MessageAmp aRNA kit (Ambion) following the manufacturer's instructions.
Mouse Osteoarthritis Model
Animal experimentation was approved by the Institutional Animal Ethics Committee. Osteoarthritis (OA) was induced in 10-week-old male C57BL6 mice by medial meniscal destabilization (DMM) of the right knee (16). Joints subjected to sham-operation (exposure of the medial menisco-tibial ligament but no transection) were used as controls. Animals were sacrificed at 2 and 6 weeks after surgery (n = 4 per time point). The joints were dissected to expose the articular cartilage, tibial epiphyses were isolated and placed in RNALater (Ambion) containing 20% EDTA, decalcified at 4 °C for 72 h, and then embedded in OCT and stored at −80 °C. Serial 7-μm coronal cryosections were fixed in ethanol, air-dried, and noncalcified medial tibial plateau articular cartilage from previously assigned areas of cartilage fibrillation and loss of toluidine blue staining were laser-microdissected (Arcturus). Total RNA was extracted and amplified as described above.
RT PCR Analysis
5 ng of aRNA from articular cartilage and growth plate cartilage was reverse transcribed (RT) in a reaction volume of 20 μl using SuperScript III reverse transcriptase (Invitrogen) at 50 °C for 1 h. PCR was performed as previously described (15) except that amplification consisted of 35 cycles of denaturation for 30 s at 94 °C, annealing for 30 s at 58 °C, and extension for 30 s at 72 °C. Primer sequences for Cilp1 and Cilp2 are listed under supplemental Table S1. For multitissue RT-PCR to compare Cilp1 and Cilp2 expression, tissues were dissected from outbred Swiss white mice and total was RNA isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer's directions. RNA quality was assessed on an RNA pico/nano labchip (Agilent Technology Bioanalyzer).
Microarray Expression Profiling
cDNA microarray analysis of the individually microdissected proliferative, prehypertrophic, and hypertophic zones of the growth plate cartilage has been previously described (15). Microdissected articular cartilage analysis was performed similarly using microarray slides printed with the 15k NIA cDNA mouse clone set (Australian Genome Research Facility). Microarray expression profiling was performed on amplified RNA from the DMM-operated and Sham-operated mouse cartilage using duplicate microarrays (Cy3/Cy5 dye-swap with replicate RNA samples). Labeled RNA was hybridized to 44k whole genome oligo microarrays (G4122A, Agilent Technologies). All arrays were scanned on an Axon 4000B scanner and features were extracted with GenePix Pro 4.1 software (Axon Instruments). Raw data were processed using a print-tip Loess normalization (17) using limmaGUI software (18, 19). Mean log2-transformed expression ratios and B-statistic values (log posterior odds ratio (17, 20) were calculated for all direct comparisons.
Quantitative Real Time PCR
Quantitative real time PCR (qPCR) was used for validation of differential expression of Cilp2 in articular and on the pooled amplified aRNA of the different zones of the growth plate cartilage. Gene-specific primers and prevalidated probes were designed using online software (from Roche Applied Science) and are listed under supplemental Table S1. qPCR was performed using the Universal ProbeLibrary System (Roche Applied Science) as previously described (22). mRNA was quantitated using the comparative CT method (23), using Mapk1 as the internal control because it was not differentially expressed between the growth plate zones and articular cartilage. In expression studies on DMM mouse cartilage, gene expression values were normalized using the geometric mean expression of two housekeeping genes, Atp5b and Rpl10. These housekeeper genes were shown by microarray expression profiling to be unchanged during the onset and progression of OA in the DMM mouse model (data not shown).
In Situ Hybridization
Knee joints of 14-day-old Swiss white mice were embedded and 8-μm cryosections were mounted onto SuperFrost Plus slides and in situ hybridization was performed using antisense [35S]CTP-RNA probes as described previously (15). To prepare the probes PCR amplification was performed and consisted of 35 cycles of denaturation for 1 min at 94 °C, annealing for 1 min at 58 °C, and extension for 1 min at 72 °C using specific primer sets for Cilp1 and Cilp2 (supplemental Table S1). Hybridization and autoradiography was performed as previously described (15).
Antibodies
Polyclonal antisera against the CILP-2 mouse sequence TLLDRRQQGSPHELE was produced in rabbits (Millipore). The CILP-2 antisera bound to the peptide antigen in a dose-dependent manner by standard enzyme-linked immunosorbent assay and immunodot blots (data not shown). The CILP-2 polyclonal antibody was further affinity purified against the antigen using the 1-ml HiTrapTM NHS-activated pre-packed column (GE Healthcare) according to the manufacturer's instructions. The purified CILP-2 polyclonal antibody was tested by standard enzyme-linked immunosorbent assay and immunoblotting and was found to bind to antigen and CILP-2 protein in cartilage extracts (data not shown). Serum from the same rabbit, taken prior to immunization with the CILP-2 peptide antigen, was used as a negative control. The rabbit anti-human CILP-1 antibody raised against peptide sequences in the N-terminal region of CILP-1 was generously provided by Dr. Pilar Lorenzo, Lund University, Sweden.
SDS-PAGE and Immunoblotting
14-day-old Swiss white mice were sacrificed and the femoral heads were harvested from the hip joints. Protein extraction from cartilage was performed as previously described (24). Briefly, 10 femoral heads were solubilized in 1 ml of 4 m guanidine HCl, 50 mm sodium acetate, pH 5.8, containing 10 mm EDTA, and mini-protease inhibitor mixture (Roche Applied Science) at 4 °C for 24 h. Insoluble material was removed by centrifugation at 13,0006 × g for 20 min at 4 °C. The supernatant was precipitated overnight at −20 °C using 9 volumes of ice-cold ethanol, followed by one wash in 70% ethanol to remove residual guanidine HCl and other salts. The precipitate was resuspended and denatured in reducing Laemmli sample buffer (Bio-Rad), separated on a 10% polyacrylamide gel, and transferred to ImmobilonTM-P polyvinylidene difluoride membrane (Millipore). Immunoblotting was performed as previously described (25) with some modifications. The membrane was blocked in 5% skim milk powder in PBS for 1 h and then incubated in antibody buffer (0.5% skim-milk powder in PBS with 0.1% Tween 20) containing rabbit anti-mouse CILP-2 antibody (10 μg/ml) for 1 h. Following six washes in PBS with 0.1% Tween 20, anti-rabbit IgG horseradish peroxidase secondary antibody (Dako) was added at a dilution of 1:10,000 in antibody buffer and incubated for 1 h. Following washing, the signal was developed with the ECL Plus Western blotting detection system (GE Healthcare) and autoradiography using X-Omat film.
N-Glycosidase F Treatment
For N-glycosidase F digestion, 40 μg of protein extracted from cartilage (described above) was resuspended in 25 μl of phosphate buffer (0.02 m sodium phosphate, pH 7.0, 0.1% SDS, 0.05 m EDTA, and 0.5% Nonidet P-40) and boiled for 2 min. Then 5 units of N-glycosidase F (Roche Applied Science) enzyme was added to the sample. Deglycosylation was performed in a heating block at 37 °C for 8 h. Following deglycosylation, samples were denatured in reducing Laemmli sample buffer (Bio-Rad), separated on a 7.5% polyacrylamide gel, and transferred to Immobilon-P polyvinylidene difluoride membrane. Immunoblotting was performed as described above.
Immunohistochemistry
Human articular cartilage from cadaveric donors aged 60 to 75 years were obtained from The International Institute of Advancement in Medicine (Jessup, PA; a division of the Musculoskeletal Foundation). Formalin-fixed paraffin-embedded tissue sections were de-paraffinized in xylene and rehydrated. To facilitate antibody penetration sections were treated with 30 μg/ml of Proteinase K in 50 mm Tris, pH 6.0, 5 mm CaCl2 at 37 °C for 30 min, and 0.2% hyaluronidase (bovine, type IV; Sigma) for 1 h at 37 °C. Sections were then treated with 0.3% H2O2 in PBS to inactivate endogenous peroxidases. After blocking with 1% goat serum in 1% BSA in PBS overnight at 4 °C, the sections were immunolabeled with rabbit anti-human CILP-2 antisera or nonimmune serum (dilution 1:200 in 1% BSA in PBS; antibodies a gift from Dr. Pilar Lorenzo, University of Lund, Sweden) and detected using the Vectastain Elite ABC kit (Vector Laboratories). Bound antibodies were visualized using Sigma FASTTM DAB tablets (Sigma), and sections were counterstained with hematoxylin. For immunohistochemistry on mouse tissue, serial sections of frozen, unfixed tissue were cut at 8-μm thickness on a Leica CM1580 cryostat at −20 °C, and thaw-mounted onto superfrost plus glass slides. The sections were fixed in HistoChoice® Tissue Fixative (Amersco) for 15 min at 4 °C then washed in 3 changes of PBS. Immunohistochemistry was performed as described for paraffin sections, except the sections were immunolabeled with rabbit anti-mouse CILP-2 antibody or preimmune serum diluted 1:500 in 1% BSA (w/v) in PBS.
Immunoelectron Microscopy
Fragments of suprastructural aggregates were obtained from human articular cartilage as previously described (26). In brief, slices of cartilage were twice homogenized in 15 volumes of 150 mm NaCl, 2 mm sodium phosphate buffer, pH 7.4 (PBS), containing a mixture of protease inhibitors, and centrifuged at low speed for clarification. This procedure was repeated twice. Aliquots of supramolecular fragments were spotted onto sheets of parafilm. Nickel grids covered with formvar and coated with carbon were floated on the drops for 10 min to allow adsorption of material, subsequently washed with PBS, and treated for 30 min with 2% (w/v) dried skim milk in PBS. Next, the adsorbed material was allowed to react for 2 h with polyclonal antibodies against CILP-2 and monoclonal antibodies against collagen VI (AF 6210, Medicorp) diluted 1:100 in PBS containing 0.2% dry milk. After washing five times with PBS, the grids were put on drops of 0.2% (w/v) milk solution containing colloidal gold particles coated with antibodies to rabbit (18-nm gold particles, Jackson ImmunoResearch) and mouse immunoglobulins (12-nm gold particles, Jackson ImmunoResearch). Finally, the grids were washed with distilled water and negatively stained with 2% uranyl acetate for 7 min. Control experiments were undertaken with the first antibody omitted. Electron micrographs were taken at 80 kV with a Philips EM 410 electron microscope.
RESULTS
Identification of Differential Expression of Cilp2 in Articular Cartilage in Vivo
Microarray analysis of the 15 K NIA mouse clone set revealed 50 genes that were up-regulated by 14-day-old mouse articular cartilage chondrocytes greater than 5-fold compared with growth plate chondrocytes (combined proliferative, pre-hypertrophic and hypertrophic zones) (Table S2). The most highly differentially expressed gene (>30-fold increase) in 14-day mouse articular cartilage compared with growth plate cartilage is Riken clone 1110031K21. This unannotated clone was identified as Cilp2. Differential expression of Cilp2 in articular cartilage was validated by PCR analyses where Cilp2 was detected in articular cartilage, but not in growth plate cartilage (Fig. 1A). Quantitative RT-PCR confirmed that the expression of Cilp2 was expressed ∼100-fold more highly in articular versus growth plate cartilage (data not shown). In situ hybridization analysis revealed localized expression of Cilp2 mRNA to the superficial and intermediate zones of 14-day-old mouse articular cartilage, as well as throughout the meniscus but was not detected in growth plate cartilage (Fig. 1B). Sequence analysis demonstrated that CILP-2 is highly homologous to a cartilage matrix molecule called CILP. CILP was originally isolated from human articular cartilage, is a large secreted glycoprotein, and is thought to play a role in cartilage scaffolding (6). CILP has been associated with osteoarthritis (12–14), and more recently with lumbar disc disease in a Japanese population (8), and will be referred to hereafter as CILP1 (5). We and others have shown CILP-1 and CILP-2 to be structurally similar (5) (supplemental Fig. S2), with both polypeptides containing a thrombospondin type 1 (TSP-1) repeat domain, and an immunoglobulin-like domain. Both proteins contain a signal peptide, putative N-glycosylation sites, cysteine residues forming disulfide bonds, and a furin endoprotease consensus site, but only CILP-1 has a putative phosphate-binding loop (P-loop) motif.
FIGURE 1.

Cilp2 is expressed in joint cartilage but not in growth plate cartilage. A, RT-PCR analysis demonstrates that Cilp2 is expressed in articular cartilage (AC) but not in proliferative (PR), prehypertrophic (PH), and hypertrophic (H) growth plate cartilage. Articular cartilage, and proliferative, prehypertrophic, and hypertrophic growth plate cartilage was microdissected from 2-week-old wild type mice. The RNA was isolated, linearly amplified, reverse transcribed, and subjected to 30 rounds of PCR. Gapdh was used as a positive control. B, an antisense probe was used to determine the distribution of Cilp2 mRNA in joint and growth plate cartilage. Darkfield images show that Cilp2 was expressed in at the surface and in the intermediate zone of joint cartilage and throughout meniscal cartilage. Cilp2 mRNA was not detected in growth plate cartilage. No hybridization with the sense probe was detected. MC, meniscal cartilage; R, resting zone; PR, proliferative zone; PH, prehypertrophic zone; H, hypertrophic zone; TB, trabecular bone. Scale bar, 100 μm.
Cilp1 and Cilp2 Have Distinct mRNA and Protein Expression in Mouse Articular Cartilage
The spatial distribution of Cilp1 and Cilp2 in the developing mouse knee joint was assessed during development at E16.5, newborn, and later in the 2- and 8-week-old knee joints (Fig. 2). In situ hybridization was performed to compare mRNA distribution, and immunohistochemistry was performed using the polyclonal peptide antibody raised to the amino acid sequence spanning residues 310–324 of CILP-2 with the corresponding preimmune sera used as a negative control.
FIGURE 2.

Expression of Cilp1 and Cilp2 mRNA during mouse cartilage development. In situ hybridization on embryonic (E16.5), newborn (P0), 2-week (P14) and 8-week (P56) postnatal mouse femurs. Articular cartilage morphology of mouse femur and tibia stained with toluidine blue (A) or hematoxylin and eosin (B–D) at different time points during development: E16.5 (A), P0 (B), P14 (C), and P56 (D). Dark and bright field images indicate that Cilp1 (E–H) and Cilp2 (I–L) are expressed in the articular and meniscal cartilage. No hybridization with the sense probe was detected (data not shown). AC, articular cartilage; MC, meniscal cartilage. Scale bar, 100 μm.
At E16.5, the anlage is mostly cartilaginous, the different zones of the growth plate are well defined, differentiation and ossification are underway and the process of joint cavitation has begun. At this stage of development, Cilp1 mRNA was also localized to both the femoral and tibial surface articular cartilage and absent from growth plate cartilage (Fig. 2E). Cilp2 expression during early chondrogenesis (E11.5–13.5) was not detected in microarray analysis (22) but was expressed after developing joints have begun to cavitate at E16.5 (Fig. 2I). Cilp2 mRNA was detected in the cells at the newly formed joint surface (superficial zone of articular cartilage). No expression was detected in the underlying epiphyseal cartilage. Newborn mouse knee joints have had minimal exposure to load bearing, and the secondary center of ossification has not begun to develop. Both Cilp1 and Cilp2 mRNA were confined to the surface of articular and meniscal cartilage (Fig. 2, F and J) and both were not detected in growth plate cartilage (data not shown). The most striking difference in mRNA expression between Cilp1 and Cilp2 occurs 2 weeks postnatal, after development of the secondary center of ossification. Cilp1 expression remains confined to the surface of articular and meniscal cartilage of both the femur and tibia (Fig. 2G). This is in contrast to the mRNA localization of Cilp2, which is expressed throughout the intermediate zone of articular cartilage of the femur and tibia, and widely expressed throughout meniscal cartilage (Fig. 2K). However, by 8 weeks postnatal development, when mice are approaching skeletal maturity, the expression of Cilp1 is detected in the intermediate zone of articular cartilage (Fig. 3H), which is comparable with that of Cilp2 (Fig. 2L). Likewise the expression of Cilp1 and Cilp2 in the meniscal cartilage are similar by 8 weeks of age (Fig. 4, A and C), confirmed at the protein level by immunohistochemistry (Fig. 4, B and D).
FIGURE 3.

Distribution of CILP-1 and CILP-2 protein in articular cartilage. Immunohistochemistry was performed on newborn (P0), 2-week (P14), 8-week (P56), 3-month (P84), and 6-month (P182) old mouse femurs. Control sections were probed with preimmune sera (A–D). CILP-1 protein was not detected in newborn (E) or 2-week-old mouse femurs (F), but was detected in 8-week-old mouse femurs (G) where CILP-1 immunostaining was pericelluar throughout the superficial to intermediate zones of articular cartilage. CILP-2 protein was unable to be detected in newborn femurs (I), was present at the surface of 2-week-old mouse femurs (J), and in the deep zone of 8-week-old mouse femurs (K). CILP-2 protein remained present in the deep zone of articular cartilage in 3- (H) and 6-month (L) old mouse femurs. Scale bar, 50 μm.
FIGURE 4.

Distribution of CILP-1 and CILP-2 mRNA and protein expression in meniscal cartilage. Bright field images of in situ hybridization for Cilp1 (A) and Cilp2 (C) mRNA expression in adult mouse meniscal cartilage shows mRNA expression in the surface zones. No hybridization with the sense probe was detected (data not shown). Meniscal cartilage was also probed with antibodies against CILP-1 (B) or CILP-2 (D). Both CILP-1 and CILP-2 protein were strongly present in the fibrocartilage matrix of the meniscus in 8-week-old mice. CILP-1 had a pericellular localization in the center of the meniscus (B), and CILP-2 was apparent not only in the center of the tissue, but toward the outer zones of the meniscus (C). No staining was observed with the preimmune sera (E). Scale bar, 50 μm.
Immunostaining of CILP-1 and CILP-2 was unable to detect protein in newborn mouse knee joints (Fig. 3, E and I). In 2-week-old knee joints, CILP-2 protein was detected in the surface layer of the cartilage matrix of articular cartilage, and also in the inter-territorial matrix of articular cartilage in the intermediate zone (Fig. 3J). CILP-1 was unable to be detected at this stage (Fig. 3F). With maturity (by 8 weeks of age), CILP-2 protein was present in the intermediate to deep zone of mouse articular cartilage in the interterritorial matrix (Fig. 3K). This is in contrast to CILP-1, which at 8 weeks of age was detected predominantly in the pericellular zone of the cartilage matrix, extending from the superficial zone to the deep zones (Fig. 3G). Specific localization of CILP-2 to the deeper zones of mature cartilage was further confirmed in 3- and 6-month-old mice (Fig. 3, H and L). The localization of CILP-1 and CILP-2 to articular and meniscal cartilage suggests that these ECM proteins may be components of such permanent cartilages, rather than the transient growth plate cartilage.
CILP-2 Is Proteolytically Processed, Glycosylated, and Integrated into the Articular Cartilage ECM
Extraction of human articular cartilage with a strong denaturant, 4 m guanidine HCl, had previously been shown to solubilize the CILP-1 protein (6). This was found to be the same for the CILP-2 protein. Extraction of mouse femoral cartilage with 4 m guanidine HCl solubilized the CILP-2 protein, which was detected as a major band on SDS-polyacrylamide gels migrating at ∼90 kDa under reducing conditions (Fig. 5A). Given that the full-length molecular mass of CILP-2 is 125 kDa, this indicates that most CILP-2 protein in articular cartilage is cleaved, mostly likely at the furin cleavage site. This is also true for CILP-1 (6). That both CILP-1 and CILP-2 need to be solubilized by such a strong denaturant indicated that both proteins are interacting components of the cartilage ECM, which is similar to other cartilage ECM proteins, such as WARP (25) and matrilin-1 (27).
FIGURE 5.
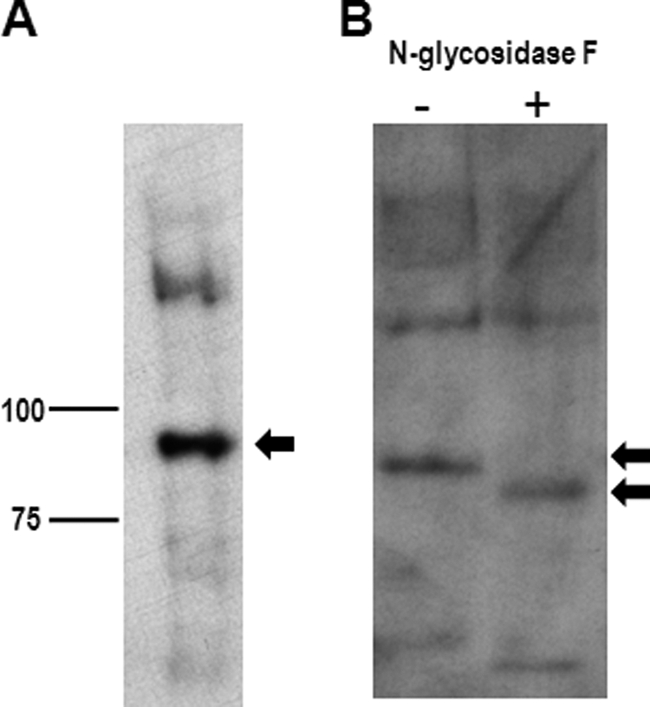
CILP-2 is cartilage extracellular matrix glycoprotein. A, immunoblot analysis of CILP-2 in 2-week-old cartilage guanidine HCl extract reveals the N-terminal fragment at ∼90 kDa. B, cartilage protein extract was subjected to N-glycosidase treatment. Products of the reaction were reduced and resolved on a 7.5% SDS-PAGE gel. The proteins were blotted to a PVDF membrane and probed with CILP-2 antiserum. The relative shift in mobility with N-glycosidase treatment indicates that CILP-2 contains N-linked oligosaccharides.
In addition, the relative mobility of CILP-2 changed with treatment of N-glycosidase F (Fig. 5B), from ∼90 to ∼80 kDa, confirming that CILP-2 contains N-linked oligosaccharides. This finding is consistent with bioinformatic data indicating putative N-glycosylation sites (supplemental Fig. S3), and is similar to CILP-1, in which up to 10% of CILP-1 content can be attributed to N-linked oligosaccharides (6).
CILP-2 Is Co-localized with Collagen VI ECM Suprastructures
In human articular cartilage, CILP-2 protein was detected in the inter-territorial matrix, in the intermediate to deep zone (Fig. 6B), and in particular in the pericellular matrix. To investigate the suprastructural organization of CILP-2 in human cartilage, CILP-2 was visualized in homogenates of human cartilage by immunogold electron microscopy using the polyclonal CILP-2 antibody generated in this study. Immunogold labeling of articular cartilage confirmed the occurrence and the distribution of CILP-2 in this tissue. CILP-2 was located in the amorphous extrafibriller material (Fig. 6C) and in close association with the heterotypic collagen containing II fibrils, visible as large cross-banded fibrils (Fig. 6D). Because collagen VI expression overlaps CILP-2 expression, and is also localized to the pericellular matrix of chondrocytes (supplemental Fig. S4), we used immunogold electron microscopy to study localization of CILP-2 (18-nm particles) and collagen VI (12-nm particles) in human cartilage samples. This revealed that a number of CILP-2 particles were located in close proximity to collagen VI particles (Fig. 6, C and D) suggesting that in human articular cartilage CILP-2 may associate with collagen VI into larger multimeric suprastructures.
FIGURE 6.

Localization of CILP-2 in human articular cartilage extracts. A and B, in human articular cartilage, CILP-2 protein was detected in the inter-territorial matrix, in the intermediate to deep zone (B). Control sections were probed with CILP-2 preimmune sera (A). Scale bar, 50 μm. C and D, representative micrographs confirming CILP-2 distribution in human articular cartilage visualized by electron microscopy. Immunogold particles (CILP-2, 18-nm particles black arrowheads; collagen VI, 12-nm particles, white arrowheads) were localized within collagen VI containing suprastructures (C), and also as part of collagen VI containing suprastructures in close association with the heterotypic collagen II containing cartilage fibrils (D). Scale bar, 100 nm.
Cilp2 Expression Is Down-regulated during Experimental Osteoarthritis
Cilp1 and Cilp2 mRNA expression was determined in articular cartilage from mice where OA is induced by surgical DMM. In this model at 2 weeks postsurgery early focal degeneration in the medial tibial plateau is evident, exemplified by loss of aggrecan in the noncalcified articular cartilage. By 6 weeks aggrecan loss had progressed and cartilage fibrillation is evident (16). Cartilage was microdissected from the region of focal lesion in DMM joints, and from the same region in sham-operated contralateral joints that do not develop OA. Microarray expression profiling showed that at 2 weeks after surgery (early OA) Cilp1 expression was increased 3.6-fold (n = 4, p = 0.026) and Cilp2 expression decreased 1.7-fold (n = 4, p = 0.023). At 6 weeks Cilp2 expression decreased 1.8-fold (n = 4, p = 0.012). The change in Cilp1 and Cilp2 expression measured using qRT-PCR showed some variability between the individual animals (Fig. 7). Nevertheless, the mean fold-change had a similar temporal pattern to that observed in microarray analysis. Although there was an increase in Cilp1 at 2 weeks (mean change 5.2-fold, n = 4), Cilp2 expression was slightly down-regulated (mean change −1.3-fold, n = 4). However, at 6 weeks postsurgery, Cilp2 expression was significantly down-regulated compared with the sham joint (mean change −3.0-fold, n = 4). These data, showing an increase in Cilp1 and decrease in Cilp2 expression in surgically induced OA in mice, were further confirmed in an independent expression profiling experiment on pooled microdissected cartilage lesion tissues from 1, 2, and 6 postsurgery DMM and sham-operated mouse joints (data not shown). Immunohistochemical analysis of the medial tibial plateau cartilage demonstrated reduced CILP-2 protein at 4 and 8 weeks postoperative DMM (Fig. 8, B, C, E, and F) compared with control cartilage (Fig. 8, A and D) consistent with the loss of aggrecan in the developing OA cartilage lesion (Fig. 8, H and I).
FIGURE 7.

Cilp1 and Cilp2 mRNA expression in a mouse model of osteoarthritis. qPCR of Cilp1 and Cilp2 expression in sham and DMM joints at 2 and 6 weeks postsurgery. The results of four mice at each time point are shown (average of three technical replicates) and the data from sham and DMM cartilage of each mouse are shown in corresponding colors. The results are shown as the ΔCT between Cilp1 and Cilp2 and the geometric mean expression of two housekeeping genes, Atp5b and Rpl10 (HKGs). The median result at each time point is indicated by a horizontal line.
FIGURE 8.

CILP-2 protein is reduced in during cartilage degeneneration in a mouse model of osteoarthritis. Immunohistochemistry for CILP-2 in control (A and D) and DMM joints at 4 weeks postsurgery (B and E) and 6 weeks postsurgery (C and F) show loss of CILP-2 protein from the chondrocyte territorial and inter-territorial matrix in the deep articular cartilage zone during cartilage degeneration, visualized by loss of toluidine staining (H and I). Scale bar, 100 μm.
Cilp1 and Cilp2 Are Also Expressed in Specific Noncartilaginous Tissues
The expression of Cilp1 and Cilp2 was analyzed by RT-PCR on various mouse tissues (Fig. 9). Cilp1 and Cilp2 were expressed in muscle and heart tissue. In addition, Cilp2 was expressed in the brain.
FIGURE 9.

Cilp1 and Cilp2 are expressed in noncartilaginous tissues. RT-PCR analysis of various mouse tissues demonstrates that Cilp1 is also expressed in heart and skeletal muscle as well as cartilage. Cilp2 is also expressed in heart, skeletal muscle, and brain in addition to cartilage. Gapdh is shown as a loading control. The control sample contains water, and -RT indicates cDNA synthesized without the presence of reverse transcriptase enzyme.
DISCUSSION
Although investigating the differential transcriptome of permanent articular cartilage and transient growth plate cartilage, we identified a novel ECM molecule, CILP-2 that is expressed uniquely by articular chondrocytes. We characterized the mRNA and protein distribution of CILP-2 in developing cartilage, compared it to its isoform CILP-1. We report that CILP-2 is structurally similar to CILP-1, specifically localized to the intermediate to deep zone of mature articular cartilage where it is deposited in the inter-territorial matrix and is also expressed throughout the meniscal cartilage. Our data also suggests that CILP-2 may be part of collagen VI containing suprastructures in human articular cartilage. We also demonstrated distinct differences in the regulation of CILP-1 and CILP-2 expression in articular chondrocytes during the development of post-traumatic OA.
CILP-2 protein and mRNA is restricted to articular cartilage and the meniscus, and was not expressed throughout the cartilage anlage of the developing limb. The absence of Cilp2 mRNA and protein in growth plate cartilage suggests that CILP-2 is a component of permanent cartilage, rather than transient cartilage that will undergo ossification, and further defining the specialized composition of the extracellular matrix synthesized by articular chondrocytes. Although CILP-1 and CILP-2 are structurally similar and may have redundant functions, we uncovered some differences in their mRNA and protein expression, suggesting possible functional differences. mRNA expression of Cilp1 and Cilp2 is broadly similar until 2 weeks postnatal, where Cilp2 is expressed throughout the intermediate zone of articular cartilage and widespread in the meniscus, but Cilp1 is restricted to the surface of articular and meniscal cartilage. However, by 8 weeks postnatal, both Cilp1 and Cilp2 are expressed throughout the intermediate zone of articular cartilage.
The lack of similarity between CILP-2 and other proteins (apart from CILP-1), and the absence of knock-out or transgenic mouse models of CILP-1, makes it difficult to speculate on the function and role of CILP-2 in articular cartilage. Initial studies reported that a fragment of CILP-1 (C-terminal) was a homologue to porcine nucleotide pyrophosphohydrolase (NTPPH) and was possibly linked to NPP activity (10). However, neither the CILP-1 or CILP-2 genes have the domains required for NPP activity. Testing recombinant CILP-1 and CILP-2 for NPP activity confirmed that CILP-1 and CILP-2 did not exhibit intrinsic NPP activity (5).
The presence of a TSP-1 domain in CILP-2 raises the possibility that it may be able to anchor to other matrix constituents. The functions of the three type 1 repeated sequence motifs that are present in TSP-1 have been extensively studied. They reportedly function as (a) attachment sites for many cell types, (b) inhibitors of angiogenesis, (c) protein-binding sites, and (d) glycosaminoglycan (GAG)-binding sites (reviewed by Refs. 29 and 30)). The ability to bind GAGs is important to the function of many extracellular and membrane proteins and TSP-1 repeat sequence motifs have been reported to mediate GAG binding in several proteins. When the TSP-1 domain is deleted in ADAMTS-4, a cartilage enzyme, it is unable to bind to the glycosaminoglycans of the aggrecan molecule suggesting that the TSP-1 domain is critical for substrate recognition and cleavage (31). The W(S/G)XW has been implicated in the interaction of thrombospondin with the sulfated GAG chains of heparin, heparin sulfate, and chondroitin sulfate (32, 33), whereas the CSVTCG motif is able to bind to CD36, which is the thrombospondin receptor (34). Additionally, the W(S/G)XW motif is a well defined consensus sequence within the TSP-1 domain that is able to bind to TGF-β1 (35, 36). Recently, the CILP-1 protein has been shown to bind to and inhibit TGF-β1 (8) and CILP-1 mRNA expression has been shown to be induced by TGF-β1 and dependent upon signaling via TGF-β receptors (9). These motifs are conserved in CILP-2, thus CILP-2 may have the potential to recognize and bind to other matrix constituents. We present data in support of this hypothesis. We show that CILP-2 is located in the human cartilage matrix in close proximity to collagen VI containing suprastructures. This data suggests a potential interaction between CILP-2 and collagen VI, or that CILP-2 may be associated with collagen VI containing microfibrils via intermediate molecules. The latter is observed with matrilin-1, where biglycan/matrilin-1 or decorin/matrilin-1 complexes act as a linkage between collagen VI microfibrils (37).
Our studies show for the first time that Cilp2 gene expression is dysregulated during cartilage degeneration in a mouse model of osteoarthritis reduced severalfold compared with sham operated joints (−1.8-fold by microarray; −3-fold by qPCR) at 6 weeks postsurgery. At this stage cartilage erosion is apparent and the altered expression may result as a consequence of cartilage degradation. This would be consistent with greater effects on Cilp2 with OA progression, that is, a greater effect at 6 weeks than at 2 weeks. However, it is also possible that reduced Cilp2 expression may result from the biomechanical changes in this joint destabilization model. Furthermore, we show that at the protein level CILP-2 is significantly reduced in the deep cartilage underlying the OA lesion. This reduction in CILP-2 may be a direct result of the reduced Cilp2 transcription, or may also involve CILP-2 degradation. Whereas CILP1 has been linked to a number of cartilage degenerative diseases (8, 13, 14) this is the first connection between Cilp2 and cartilage disease. The down-regulation of Cilp2 is in stark contrast to the up-regulation of Cilp1 in OA, demonstrated in our studies and previously reported (11) in human OA cartilage. This differential response suggests that Cilp1 and Cilp2 may play distinct roles in disease pathology.
Expression of Cilp2 at the surface of articular cartilage and throughout the meniscus, and the reduced CILP-2 in the mouse OA cartilage therefore suggests that CILP-2 may be useful to explore as a biomarker for the detection of cartilage damage in joint diseases. Recently, the C-terminal polypeptide of CILP-2 has been shown to be susceptible to specific metalloprotease proteolysis in diseased cartilage (38). Four CILP-2 peptides were identified as part of the 20 most abundant peptide fragments released from cartilage by metalloproteases. These data are consistent with our finding of reduced CILP-2 protein in mouse OA cartilage and suggest that CILP-2 peptides could be potential biomarkers of arthritis or of metalloprotease activity in articular cartilage (38).
Our data also demonstrates that CILP-1 and CILP-2 are not exclusively produced by chondrocytes. We detected Cilp1 and Cilp2 in heart and skeletal muscle. A number of matrix molecules are components of both tissues, including collagen types I, III, V, and VI (39–43), tenascin-C, and decorin (6, 21, 28), thus CILP-2 may contribute to the structural organization and matrix architecture within these connective tissues.
Acknowledgments
We thank Dr. Katrina Bell (MCRI, Bioinformatics) for assistance with microarray data analysis and Professor Bruce Caterson (Cardiff) and Dr. Pilar Lorenzo (Lund) for their suggestions and for critically reading the manuscript.
This work was supported in part by National Health and Medical Research Council of Australia Grant 607399), the Murdoch Childrens Research Institute, the Victorian Government's Operational Infrastructure Support Program, and Sonderforschungsbereich Grant SFB 492, TP A2.

The on-line version of this article (available at http://www.jbc.org) contains Tables S1 and S2 and Figs. S1–S4.
- ECM
- extracellular matrix
- CILP-2
- cartilage intermediate layer protein 2
- qPCR
- quantitative PCR
- TSP-1
- thrombospondin type 1
- DMM
- medial meniscal destabilization
- OA
- osteoarthritis
- P14
- postnatal 14.
REFERENCES
- 1. Alman B. A. (2008) Clin. Genet. 73, 24–30 [DOI] [PubMed] [Google Scholar]
- 2. Bateman J. F., Boot-Handford R. P., Lamande S. R. (2009) Nat. Rev. Genet. 10, 173–183 [DOI] [PubMed] [Google Scholar]
- 3. Mundlos S., Olsen B. R. (2002) in Connective Tissue and Its Heritable Disorders. Molecular, Genetic, and Medical Aspects (Royce P. M., Steinmann B. eds) Second Ed., pp. 993–1023, Wiley-Liss, Inc., New York [Google Scholar]
- 4. Dehne T., Karlsson C., Ringe J., Sittinger M., Lindahl A. (2009) Arthritis Res. Ther. 11, R133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson K., Farley D., Hu S. I., Terkeltaub R. (2003) Arthritis Rheum. 48, 1302–1314 [DOI] [PubMed] [Google Scholar]
- 6. Lorenzo P., Bayliss M. T., Heinegård D. (1998) J. Biol. Chem. 273, 23463–23468 [DOI] [PubMed] [Google Scholar]
- 7. Lorenzo P., Neame P., Sommarin Y., Heinegård D. (1998) J. Biol. Chem. 273, 23469–23475 [DOI] [PubMed] [Google Scholar]
- 8. Seki S., Kawaguchi Y., Chiba K., Mikami Y., Kizawa H., Oya T., Mio F., Mori M., Miyamoto Y., Masuda I., Tsunoda T., Kamata M., Kubo T., Toyama Y., Kimura T., Nakamura Y., Ikegawa S. (2005) Nat. Genet. 37, 607–612 [DOI] [PubMed] [Google Scholar]
- 9. Mori M., Nakajima M., Mikami Y., Seki S., Takigawa M., Kubo T., Ikegawa S. (2006) Biochem. Biophys. Res. Commun. 341, 121–127 [DOI] [PubMed] [Google Scholar]
- 10. Masuda I., Iyama K. I., Halligan B. D., Barbieri J. T., Haas A. L., McCarty D. J., Ryan L. M. (2001) J. Bone Miner. Res. 16, 868–875 [DOI] [PubMed] [Google Scholar]
- 11. Lorenzo P., Bayliss M. T., Heinegård D. (2004) Matrix Biol. 23, 381–391 [DOI] [PubMed] [Google Scholar]
- 12. Tsuruha J., Masuko-Hongo K., Kato T., Sakata M., Nakamura H., Nishioka K. (2001) Arthritis Rheum. 44, 838–845 [DOI] [PubMed] [Google Scholar]
- 13. Valdes A. M., Hart D. J., Jones K. A., Surdulescu G., Swarbrick P., Doyle D. V., Schafer A. J., Spector T. D. (2004) Arthritis Rheum. 50, 2497–2507 [DOI] [PubMed] [Google Scholar]
- 14. Valdes A. M., Van Oene M., Hart D. J., Surdulescu G. L., Loughlin J., Doherty M., Spector T. D. (2006) Arthritis Rheum. 54, 533–539 [DOI] [PubMed] [Google Scholar]
- 15. Belluoccio D., Bernardo B. C., Rowley L., Bateman J. F. (2008) Biochim. Biophys. Acta 1779, 330–340 [DOI] [PubMed] [Google Scholar]
- 16. Glasson S. S., Blanchet T. J., Morris E. A. (2007) Osteoarthritis Cartilage 15, 1061–1069 [DOI] [PubMed] [Google Scholar]
- 17. Smyth G. K., Speed T. (2003) Methods 31, 265–273 [DOI] [PubMed] [Google Scholar]
- 18. Smyth G. K. (2005) in Bioinformatics and Computational Biology Solutions using R and Bioconductor (Genetleman R., Carey V. J., Huber W., Irizarry R. A., Dudoit S. eds) pp. 397–420, Springer Science and Business Media, Inc., New York [Google Scholar]
- 19. Wettenhall J. M., Smyth G. K. (2004) Bioinformatics 20, 3705–3706 [DOI] [PubMed] [Google Scholar]
- 20. Smyth G. K. (2004) Stat. Appl. Genet. Mol. Biol. 3, Article 3 [DOI] [PubMed] [Google Scholar]
- 21. Bock H. C., Michaeli P., Bode C., Schultz W., Kresse H., Herken R., Miosge N. (2001) Osteoarthritis Cartilage 9, 654–663 [DOI] [PubMed] [Google Scholar]
- 22. Cameron T. L., Belluoccio D., Farlie P. G., Brachvogel B., Bateman J. F. (2009) BMC Dev. Biol. 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pfaffl M. W. (2001) Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Belluoccio D., Wilson R., Thornton D. J., Wallis T. P., Gorman J. J., Bateman J. F. (2006) Proteomics 6, 6549–6553 [DOI] [PubMed] [Google Scholar]
- 25. Allen J. M., Bateman J. F., Hansen U., Wilson R., Bruckner P., Owens R. T., Sasaki T., Timpl R., Fitzgerald J. (2006) J. Biol. Chem. 281, 7341–7349 [DOI] [PubMed] [Google Scholar]
- 26. Kassner A., Hansen U., Miosge N., Reinhardt D. P., Aigner T., Bruckner-Tuderman L., Bruckner P., Grässel S. (2003) Matrix Biol. 22, 131–143 [DOI] [PubMed] [Google Scholar]
- 27. Hauser N., Paulsson M., Heinegârd D., Mörgelin M. (1996) J. Biol. Chem. 271, 32247–32252 [DOI] [PubMed] [Google Scholar]
- 28. Pacifici M. (1995) Matrix Biol. 14, 689–698 [DOI] [PubMed] [Google Scholar]
- 29. Chen H., Herndon M. E., Lawler J. (2000) Matrix Biol. 19, 597–614 [DOI] [PubMed] [Google Scholar]
- 30. Tan K., Duquette M., Liu J. H., Dong Y., Zhang R., Joachimiak A., Lawler J., Wang J. H. (2002) J. Cell Biol. 159, 373–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tortorella M., Pratta M., Liu R. Q., Abbaszade I., Ross H., Burn T., Arner E. (2000) J. Biol. Chem. 275, 25791–25797 [DOI] [PubMed] [Google Scholar]
- 32. Gantt S. M., Clavijo P., Bai X., Esko J. D., Sinnis P. (1997) J. Biol. Chem. 272, 19205–19213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guo N. H., Krutzsch H. C., Nègre E., Zabrenetzky V. S., Roberts D. D. (1992) J. Biol. Chem. 267, 19349–19355 [PubMed] [Google Scholar]
- 34. Magnetto S., Bruno-Bossio G., Voland C., Lecerf J., Lawler J., Delmas P., Silverstein R., Clezardin P. (1998) Cell Biochem. Funct. 16, 211–221 [DOI] [PubMed] [Google Scholar]
- 35. Young G. D., Murphy-Ullrich J. E. (2004) J. Biol. Chem. 279, 47633–47642 [DOI] [PubMed] [Google Scholar]
- 36. Murphy-Ullrich J. E., Poczatek M. (2000) Cytokine Growth Factor Rev. 11, 59–69 [DOI] [PubMed] [Google Scholar]
- 37. Wiberg C., Klatt A. R., Wagener R., Paulsson M., Bateman J. F., Heinegård D., Mörgelin M. (2003) J. Biol. Chem. 278, 37698–37704 [DOI] [PubMed] [Google Scholar]
- 38. Zhen E. Y., Brittain I. J., Laska D. A., Mitchell P. G., Sumer E. U., Karsdal M. A., Duffin K. L. (2008) Arthritis Rheum. 58, 2420–2431 [DOI] [PubMed] [Google Scholar]
- 39. Eyre D. (2002) Arthritis Res. 4, 30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Light N., Champion A. E. (1984) Biochem. J. 219, 1017–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Listrat A., Lethias C., Hocquette J. F., Renand G., Ménissier F., Geay Y., Picard B. (2000) Histochem. J. 32, 349–356 [DOI] [PubMed] [Google Scholar]
- 42. Listrat A., Picard B., Geay Y. (1999) Tissue Cell 31, 17–27 [DOI] [PubMed] [Google Scholar]
- 43. Passerieux E., Rossignol R., Chopard A., Carnino A., Marini J. F., Letellier T., Delage J. P. (2006) J. Struct. Biol. 154, 206–216 [DOI] [PubMed] [Google Scholar]