

Background: Bacterial glycosylation systems appear to be ideal for glycoengineering purposes.
Results: A Lewis x containing glycoprotein was synthesized using bacterial enzymes from four different species using in vivo and in vitro steps.
Conclusion: Glycosylation systems in bacteria open new avenues for the production of engineered glycoproteins.
Significance: Bacterial glycoengineering is a promising technology for the production of therapeutically valuable glycoproteins with expected high precision and low cost.
Keywords: Glycoconjugate, Glycoprotein, Glycosylation, Glycosyltransferases, Helicobacter pylori, Bacterial Glycoproteins, Fucosyltransferase, Glycoengineering, Oligosaccharyltansferase
Abstract
Glycoproteins constitute a class of compounds of increasing importance for pharmaceutical applications. The manipulation of bacterial protein glycosylation systems from Gram-negative bacteria for the synthesis of recombinant glycoproteins is a promising alternative to the current production methods. Proteins carrying Lewis antigens have been shown to have potential applications for the treatment of diverse autoimmune diseases. In this work, we developed a mixed approach consisting of in vivo and in vitro steps for the synthesis of glycoproteins containing the Lewis x antigen. Using glycosyltransferases from Haemophilus influenzae, we engineered Escherichia coli to assemble a tetrasaccharide on the lipid carrier undecaprenylphosphate. This glycan was transferred in vivo from the lipid to a carrier protein by the Campylobacter jejuni oligosaccharyltransferase PglB. The glycoprotein was then fucosylated in vitro by a truncated fucosyltransferase from Helicobacter pylori. Diverse mass spectrometry techniques were used to confirm the structure of the glycan. The strategy presented here could be adapted in the future for the synthesis of diverse glycoproteins. Our experiments demonstrate that bacterial enzymes can be exploited for the production of glycoproteins carrying glycans present in human cells for potential therapeutic applications.
Introduction
The discovery of general protein glycosylation systems in Gram-negative bacteria inaugurated a new era in the production of recombinant glycoproteins (1, 2). Exploitation of these systems via glycoengineering has the potential to overcome many hurdles of the techniques currently available for the synthesis of glycoconjugates (3, 4). In contrast to protein glycosylation in eukaryotes, bacterial cells tolerate the manipulation of glycan moieties. In higher eukaryotes even minimal alterations in the glycan structure can be lethal, primarily because eukaryotic N-glycosylation is required for protein folding control (5). Although yeasts appear to be a promising system for production of humanized N-glycoproteins, it seems unlikely that these cells can be efficiently used for the production of glycoproteins containing glycans other than the high-mannose type (6). On the other hand, chemical synthesis of conjugates often requires harsh conditions for the attachment of the sugars, which usually affects the properties of the carriers (7). Furthermore, chemical cross-linking is difficult to control and therefore glycoconjugates produced via this method often present reproducibility and homogeneity issues (7). Utilization of living bacteria for the production of glycoproteins combines the specificities of enzymatic reactions for both, the assembly of the glycan and its conjugation to the polypeptide.
Oligosaccharyltransferases (OTases)3 are the key enzymes involved in such bacterial synthesis pathways. They are responsible for the transfer of glycans from the carrier lipid undecaprenyl pyrophosphate (UndPP) onto acceptor proteins (8). Importantly, some of the best characterized OTases display relaxed substrate specificity. Glycans attached to the carrier lipid can be transferred onto acceptor proteins with very limited or no requirements toward the structure of the oligo- or polysaccharide (9, 10). For instance, the presence of an acetamido group at C-2 of the reducing sugar residue and probably a linkage other than 1–4 between the two proximal sugars are the only known substrate requirements of the Campylobacter jejuni OTase PglB (1, 11). Therefore, if the desired glycan structure fulfills these requirements and can be assembled onto UndPP, it will be transferred onto acceptor proteins in the presence of PglB. No structural requirement for the glycan has been found for the Neisseria OTase PglL (9). Interestingly, UndPP serves as a carrier lipid for glycan assembly in many bacterial glycoconjugate pathways (12). One example is the lipopolysaccharide biosynthesis pathway, using UndPP for the assembly of its distal polysaccharide, named O antigen. The O antigen is then conjugated en bloc onto the lipid A-core by the O antigen ligase (13). Indeed, it was demonstrated that UndPP-linked, fully polymerized O antigens can also be transferred onto acceptor proteins by bacterial OTases (9, 10).
OTases recognize specific acceptor sites on acceptor proteins, making enzymatic glycan transfer superior to chemical conjugation in terms of precision and reproducibility. C. jejuni PglB attaches glycans in an N-linkage, recognizing the extended eukaryotic consensus sequence (D/E)XNX(S/T) (14). Any polypeptide engineered to contain this glyco-tag may serve as a C. jejuni PglB glycosylation target in the bacterial periplasm (14). The recognition determinants for other OTases are less clear. Desulfovibrio desulfuricans PglB does not recognize the same sequon as its Campylobacter counterpart (15) and it is unknown how Neisseria PglL recognizes its substrates (16).
Although the best characterized bacterial glycosylation systems originate from the pathogens C. jejuni, Pseudomonas aeruginosa, and Neisseria spp., these glycoprotein assembly machineries were functionally reconstituted in nonpathogenic and fast growing Escherichia coli laboratory strains (2, 17). This has opened up new avenues for the exploitation of these systems for the synthesis of engineered glycoproteins with potential therapeutic applications.
The flexibility of the bacterial glycosylation pathways prompted us to develop a strategy to exploit these systems for the synthesis of customized glycoproteins. The Lewis x (Lex) antigen is a glycan structure (Galβ1–4[Fucα1–3]GlcNAc-) that is abundantly expressed on a variety of human cells, as well as on some parasitic helminthes and the gastric pathogen Helicobacter pylori (18–20). Glycoconjugates containing Lex have promising applications for the treatment of autoimmune diseases due to their immunosuppressant effect via interaction with the dendritic cell receptor DC-SIGN (21). Experiments in animal models have demonstrated that symptoms associated with autoimmune disorders are ameliorated upon treatment with Lex-containing glycoproteins (22). The “old friend hypothesis” elaborates on this idea, proposing that reduced exposure to immune suppressive signals from microbes due to improved hygiene and generalized use of antibiotics has increased the prevalence of human immune disorders in modern times (23). The use of bacteria for the generation of immunosuppressant and other glycodrugs may be a valuable future technology. In this work, we describe a combined in vivo and in vitro approach for the production of a glycoprotein containing the Lex antigen by exploiting bacterial enzymes.
EXPERIMENTAL PROCEDURES
DNA Manipulations and Plasmid Construction
H. pylori 11639 fucT full-length and truncated versions were excised from pGEM11639fucTfl and pGEM11639fucTtr (24), respectively, using SphI. The DNA ends were then blunted with a Klenow fragment and digested with PstI. The inserts were ligated into pEXT20, which was digested with SmaI and PstI. The resulting plasmids were pIH61, containing the full-length fucT, and pIH62, containing the truncated version.
Bacterial Strains and Growth Conditions
E. coli DH5α or BL21 strains were grown in LB broth supplemented with the appropriate antibiotics to counterselect the eventual loss of plasmids (ampicillin = 100 μg/ml, chloramphenicol = 10 μg/ml in EtOH, trimethoprim = 100 μg/ml).
Production and Detection of Glycoproteins in Living E. coli
E. coli DH5α cells containing Helicobacter influenzae lsg genes on pGEMLOS5 (25), the C. jejuni pglB on pMAF10 or pWA1 containing an inactive pglB for control samples, and the C. jejuni acrA, encoding a periplasmic acceptor protein, on pMH5 (10), were induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside and 0.2% (w/v) arabinose at early log phase and grown overnight at 37 °C with shaking at 200 rpm. Periplasmic contents containing the glycoproteins were extracted using a lysozyme solution as described by Feldman et al. (10). Glycosylation of the acceptor protein AcrA was analyzed after separation of the periplasmic contents on SDS-PAGE by visualization with Coomassie Blue staining or Western blotting using a primary anti-AcrA antibody (Sacri antibody Services, University of Calgary) and a secondary goat anti-rabbit IRDye-680 antibody (Licor biosciences) with an Odyssey Infrared Imager (Licor Biosciences).
Purification of Glycoproteins before Fucosylation
Periplasmic fractions containing glycosylated histidine-tagged AcrA proteins were loaded onto 1-ml HisTrap FF columns (GE Healthcare Life Sciences) in 20 mm Tris-HCl, pH 8.0, containing 0.3 mm NaCl and 10 mm imidazole. The columns were washed with 50 ml of 20 mm Tris-HCl, pH 8.0, containing 20 mm imidazole and 0.3 m NaCl. The proteins were eluted with 20 mm Tris-HCl, pH 8.0, containing 250 mm imidazole and 0.3 m NaCl. Protein concentrations were measured using the Quick Start Bradford dye reagent and a BSA protein standard kit (Bio-Rad).
Expression of H. pylori Fucosyltransferases in E. coli
The H. pylori 11639 fucosyltransferase or its truncated version were either expressed in E. coli BL21 cells containing pGEM11639fucTfl or pGEM11639fucTtr (24) or in E. coli DH5α cells containing pIH61 or pIH62. After induction with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside in early log phase, the cells were incubated for 6 h at 37 °C with shaking at 200 rpm. The cells were washed once with 20 mm HEPES, pH 7.0, buffer and disrupted by sonication in 20 mm HEPES containing a Complete EDTA-free proteinase inhibitor mixture (Roche Applied Science).
Addition of Radioactive Fucose in Vitro
Samples were prepared with 24 μl of E. coli cell extract containing overexpressed fucosyltransferases, 20 μg of purified AcrA, 160 μg of BSA, and 1.2 units of Antarctic phosphatase (New England Biolabs) in 20 mm HEPES, pH 7.0, 50 mm NaCl, 35 mm MgCl2, 20 mm MnCl2, and adjusted to 38 μl with H2O. 2 μl of a mixture of cold (0.2 μm) and hot (80,000 dpm per sample) GDP-fucose was added. The samples were incubated at 37 °C for 3 h. For each sample, 10 μl of Ni-NTA-agarose (Qiagen) was equilibrated with binding buffer (10 mm imidazole, 0.3 m NaCl, 20 mm Tris-HCl, pH 8.0) before the reaction samples were added. Samples were incubated for 1 h at room temperature while shaking, then washed with 20 mm imidazole, 0.3 m NaCl, 20 mm Tris-HCl, pH 8.0. The Ni-NTA-agarose together with the histidine-tagged proteins were then transferred into 10 ml of Ecolite solution and radiation was measured with a scintillation counter.
Addition of Cold Fucose in Vitro
40-μl samples were prepared as described for radioactive fucosylation, except using 100 μg of cold GDP-fucose. Samples were incubated overnight at 37 °C, and then analyzed by Western blot and mass spectrometry.
In-gel Protein Digestion and Peptide Purification
After staining with Coomassie Blue, gel pieces containing glycosylated AcrA proteins were treated according to Shevchenko et al. (26) for trypsin digestion. Because AcrA does not contain disulfide bonds, reduction and alkylation steps were not required. Peptides were desalted using C18 Zip-Tips (Millipore).
LC ESI-Q-TOF
Dried desalted peptides were solubilized in 8 μl of 0.1% (v/v) formic acid in H2O. The Q-TOF Premier mass spectrometer (Waters Associates, Milford, MA) equipped with a nanoACQUITY ultraperformance liquid chromatography system (Waters) was operated as described elsewhere (17). Mass spectra were analyzed using MassLynx software (Waters).
LC-MALDI TOF-TOF
In gel trypsin-digested and desalted peptides were dried under vacuum centrifugation followed by resolubilization using 0.1% TFA in H2O and injected onto an Easy nano-LC II system (Proxeon/Thermo Scientific). 10 μl of sample was injected onto a BioSphere C18 (5 μm) 100-μm inner diameter × 20-mm loading/de-salting column, washed with 0.1% TFA for 12 column volumes, and eluted onto a BioSphere C18 (5 μm) 75-μm inner diameter × 100-mm analytical column. The mobile phases used for the elution of analytes from the analytical column were: 0.1% TFA in 98% H2O and 2% acetonitrile as mobile phase A and 0.1% TFA in 95% acetonitrile and 5% H2O as mobile phase B at a flow rate of 300 nl min−1. Elution of the analytes was done by using a linear gradient from 0 B to 5% B over a period of 2 min, 5 B to 50% B over a period of 58 min, 50 B to 100% B over a period of 4 min and held at 100% B over a period of 11 min. The nanoLC separated peptides were directly spotted onto a Bruker Daltonics MTP AnchorChipTM 800/384 target and co-mixed with α-cyano-4-hydroxycinnamic acid matrix running at a flow rate of 1.26 μl min−1 using the Proteineer FcII sample spotting robot (Bruker Daltoinics, Bremen, GmbH). HPLC fractions were collected with an 8-min delay using a spotting frequency of 20 s for a period of 64 min for a total of 192 spots. Mass spectra were obtained in the positive ion mode of ionization using a Bruker Daltonics (Bremen, GmbH) UltrafleXtreme MALDI TOF/TOF mass spectrometer. The FlexAnalysis software provided by the manufacturer was used for analysis of the mass spectra. The MS/MS spectra were obtained manually. The exact m/z used as the precursor m/z for MS/MS was determined first on the MALDI FTICR MS instrument (see below) and the MS/MS spectrum was automatically re-calibrated based upon this m/z.
Pronase E Digestion and Permethylation of Glycan Moieties
Trypsinized peptides were incubated at 37 °C in 10 μl of 0.1 m Tris-HCl, pH 7.5, containing 60 μg of Pronase E. For glycan enrichment, the samples were applied three times onto active charcoal-containing MicroSpin columns (Harvard apparatus), which were previously hydrated for 10 min with H2O and washed three times 80% acetonitrile, 20% H2O containing 0.1% TFA, followed by three washes with H2O) (27). The samples were washed twice with H2O, eluted with 25% acetonitrile, and dried. Permethylation was carried out in MicroTip columns (Harvard Apparatus), which were filled with NaOH beads (until <1 cm from top) and equilibrated with N,N-dimethylformamide (27). The dried glycans were resolubilized with 70% N,N-dimethylformamide, 25% methyliodide, 5% H2O, applied to the MicroTip columns and incubated for 15 min. After centrifugation at 950 × g, 25 μl of methyliodide was added and the flow-through was reapplied for another 15 min. Flow-through and two washes with 100 μl of acetonitrile were pooled. 300 μl of dichloromethane were added and after mixing with 1 ml of 0.5 m NaCl for liquid-liquid extraction. The organic phase was dried after two washes with 0.5 m NaCl and three washes with H2O together with a second extraction with 100 μl of dichloromethane. Permethylated glycans were spotted on a Bruker Daltonics ground steel plate and 1 μl of 2,5-dihydroxybenzoic acid (10 mg/ml in 30% H2O, 70% CH3CN containing 1 mm sodium acetate/ml) was spotted on top and allowed to dry. The permethylated glycans were then analyzed by MALDI-MS on a 9.4T Bruker Daltonics Apex Qe FTICR mass spectrometer (Bruker Daltonics, Billerica, MA) to confirm the exact mass and elemental composition followed by MALDI-MS/MS analysis on a Bruker Daltonics Ultraflextreme mass spectrometer (Bremen, GmbH).
RESULTS
Synthesis of Glycoproteins with Terminal LacNAc Structures in Live Recombinant E. coli
A partial gene cluster from H. influenzae, containing lipooligosaccharide (LOS) synthesis genes (lsgc-f) was previously expressed in E. coli by Phillips and colleagues (25). They analyzed the resulting chimeric lipopolysaccharide and found that the tetrasaccharide Galβ1–4GlcNAcβ1–3Galβ1–3GlcNAc-, derived from the H. influenzae LOS structure, was attached to E. coli lipid A-core. This tetrasaccharide contains a terminal LacNAc structure (Galβ1–4GlcNAc-) and would only require a branching fucose residue attached via an α1–3-linkage to the GlcNAc residue to constitute a Lex antigen structure (Fig. 1). The synthesis of the identified chimeric glycolipid was shown to be dependent on the E. coli enzyme WecA (25). WecA is a glycosyltransferase, initiating the assembly of glycan subunits in the synthesis of the enterobacterial common antigen and the E. coli O antigen, generating UndPP-GlcNAc (13, 28). It can therefore be assumed that the H. influenzae-derived tetrasaccharide is assembled onto UndPP in E. coli. Because this glycan fulfills the substrate requirements of the C. jejuni OTase PglB and it is assembled on the carrier lipid UndPP, it is a potential substrate of PglB and other bacterial OTases for synthesis of glycoproteins.
FIGURE 1.

Strategy for the generation of Lex-glycoproteins. The initiating glycosyltransferase WecA from E. coli was employed for the addition of the first GlcNAc residue onto the lipid carrier UndPP. Glycosyltransferase from the H. influenzae LOS biosynthesis cluster are introduced to the E. coli expression strain for the completion of the precursor glycolipid Galβ1-4GlcNAcβ1–3Galβ1–3GlcNAc-UndPP. The glycan is then conjugated in the same E. coli cells by the C. jejuni oligosaccharyltransferase PglB onto one of its protein acceptors, AcrA. The addition of the fucose residue in an α1–3 linkage onto the exterior GlcNAc residue by H. pylori FucT in vitro completes the synthesis of the Lex-glycoprotein. Squares indicate GlcNAc residues, circles the Gal residues, and triangles the Fuc residues. Sugar residues added by E. coli WecA are dark colored, by the H. influenzae Lsg proteins are light colored, and by the H. pylori FucT are gray colored.
We first tested if PglB was indeed able to generate glycoproteins containing the H. influenzae-derived tetrasaccharide as the glycan moiety. E. coli cells were co-transformed with plasmids containing the required lsg genes, the C. jejuni pglB OTase gene, and the C. jejuni acrA gene encoding an acceptor protein (2). After induction and overnight growth, periplasmic contents were extracted and the production of glycosylated proteins was analyzed by Western blotting (Fig. 2A). In the presence of both, the lsg cluster and pglB, three distinct bands were detected using an anti-AcrA antibody. The fastest migrating band displayed the same electrophoretic mobility as nonglycosylated AcrA. Both slower migrating bands, however, suggested glycosylation and were designated as mono- and diglycosylated AcrA, respectively (Fig. 2A). The histidine-tagged AcrA protein was purified (Fig. 2B) and glycosylation was confirmed by mass spectrometry after digestion with trypsin. The two established AcrA acceptor peptides DFNR (551 Da) and AVFDNNNSTLLPGAFATITSEGFIQK (2755 Da) (the glycosylated asparagines are underlined) were analyzed for the presence of covalently attached glycans (2). The corresponding parental peaks of 1281 and 3485 Da and the fragmentation peaks showing the sequential loss of 162 Da (Hex) and 203 Da (HexNAc) were found in MS and MS/MS spectra, respectively (Fig. 3 shows the MS/MS spectrum for the DFNR glycopeptide). This confirmed that the expected tetrasaccharide, with the sequence Hex-HexNAc-Hex-HexNAc, was transferred onto the AcrA acceptor protein in living E. coli cells. Interestingly, during analysis of the MS spectra, ion peaks compatible with the presence of elongated glycan moieties were occasionally found (supplemental Fig. S1).
FIGURE 2.

Protein glycosylation with the H. influenzae-derived glycan in living E. coli. Glycosylated AcrA acceptor proteins were analyzed by SDS-PAGE and Western blotting. A, periplasmic extracts containing AcrA proteins were analyzed by Western blotting using an anti-AcrA antibody. AcrA from cells containing only the acrA gene (lane 1) or an inactive pglB variant (lane 3) was observed as a single nonglycosylated moiety (−), whereas in the presence of all required functional genes, two additional protein species were produced (lane 2). The monoglycosylated AcrA band is marked with one asterisk, the diglycosylated band with two asterisks. B, glycosylated AcrA containing the H. influenzae-derived glycan was purified by nickel affinity chromatography and analyzed by SDS-PAGE and Coomassie staining.
FIGURE 3.

MS/MS analysis of glycoproteins. Glycosylated AcrA containing the H. influenzae-derived glycan added in vivo was analyzed by LC ESI-Q-TOF. Shown is the fragmentation spectrum of the Galβ1–4GlcNAcβ1–3Galβ1–3GlcNAc-DFNR glycopeptide. Dark squares indicate GlcNAc residues, light circles indicate Gal residues. The parental peak is marked with (P).
In Vitro Fucosylation of an Engineered Bacterial Glycoprotein
Knowing that the H. influenzae-derived tetrasaccharide can be transferred onto protein acceptors in living E. coli cells, the next step was to fucosylate the terminal LacNAc structure to produce a glycoconjugate containing the Lex antigen (Fig. 1). FucT from H. pylori is one of the most extensively characterized fucosyltransferases (24). It was shown that the fucosyltransferase activity of FucT from H. pylori 11639 readily modified LacNAc structures, whereas the similar type I disaccharide Galβ1–3GlcNAc- was not a suitable acceptor (29). Therefore, it was expected that this fucosyltransferase would modify the H. influenzae-derived tetrasaccharide at the terminal LacNAc structure to generate a Lex-epitope, and leaving the internal GlcNAc residue unchanged. A truncated version of this enzyme (FucT116391–441) was previously demonstrated to have enhanced expression levels and improved solubility in E. coli (24). This recombinant enzyme seemed therefore ideal for this glycoengineering purpose. Due to the fact that E. coli cells do not synthesize GDP-fucose in normal lab growth conditions, and that stable coexpression of the OTase, the protein carrier, the Lsg and FucT glycosyltransferases, and the enzymes required for the synthesis of GDP-fucose is challenging and would require extensive genetic engineering of the E. coli cells, we decided to test the fucosylation in vitro.
Purified glycosylated AcrA containing the tetrasaccharide generated by the H. influenzae glycosyltransferases in E. coli as described previously was incubated with radioactively labeled GDP-fucose and crude extracts of E. coli cells expressing either the full-length or truncated H. pylori 11639 FucT (FucTFL or FucTTR). The glycoproteins were then purified once more using Ni-NTA-agarose. The rate of radioactive decays as a measure for the incorporated fucose residues was determined using a scintillation counter. Elevated counts in samples containing FucTFL, and especially FucTTR, suggested that fucose was transferred onto the glycoproteins in vitro (Fig. 4). Based on these results, we predicted that glycoproteins containing terminal Lex-epitopes were produced via this combined in vivo and in vitro glycoengineering approach.
FIGURE 4.
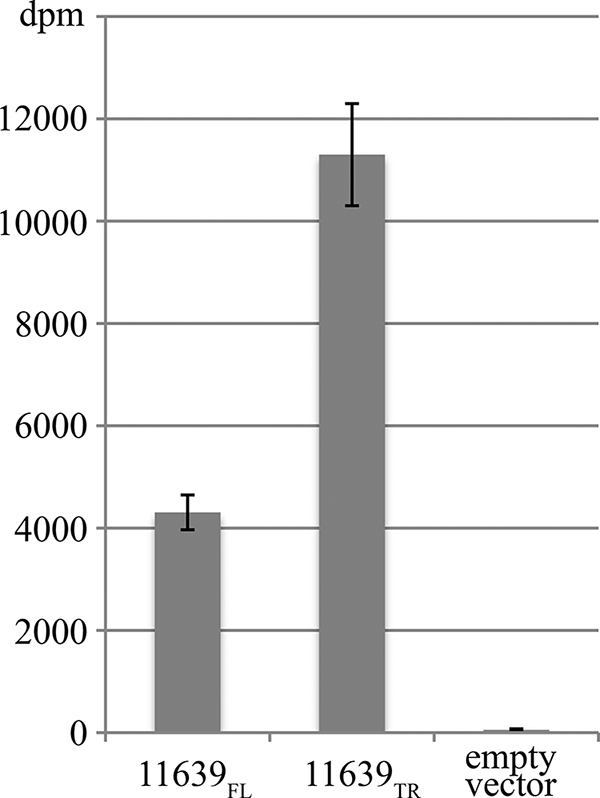
In vitro activity of the H. pylori FucT on the in vivo generated precursor glycoprotein. Fucosylation was determined after in vitro incubation of purified AcrA glycoproteins containing the H. influenzae-derived tetrasaccharide with extracts of E. coli cells expressing the H. pylori fucosyltransferases and radiolabeled GDP-fucose. The fucosyltransferases used were H. pylori 11639 FucT full-length (11639FL) and truncated (11639TR) versions or as negative control the empty vector pEXT20. The counts were collected from experiments conducted in triplicates.
To further characterize the fucosylated glycoproteins, the in vitro assay was carried out using FucTTR and unlabeled GDP-fucose. The resulting fucosylated glycoproteins were then purified, digested with trypsin, and the peptides were analyzed by mass spectrometry. Addition of a fucose residue increases the molecular mass of a molecule by 146.1 Da, leading to expected parental peaks at 1427 and 3631 Da for the fucosylated AcrA-glycopeptides. Indeed, these signals were detected with MALDI-MS and the peak corresponding to the nonfucosylated peptide, m/z = 3485 Da, decreased close to background intensity, suggesting nearly complete fucosylation of the acceptor glycoprotein (data not shown). MS/MS spectra of the 1427- and 3631-Da fragments contained a sequence of expected peaks confirming the successful incorporation of fucose residues (Fig. 5).
FIGURE 5.

MS/MS analysis of Lex-glycoproteins. Glycoproteins were analyzed by MS after in vitro fucosylation. The LC-MALDI TOF-TOF MS/MS fragmentation spectrum of the (M + H)+ of the DFNR glycopeptide is shown in A, the MS/MS spectrum of the (M + Na)+ peak of the larger AcrA glycopeptide is shown in B, with its corresponding MS spectrum shown under supplemental Fig. S3. The mass difference between adjacent peaks in the glycan region of the MS/MS accurately corresponds to successive glycan loss although the absolute m/z shifts slightly higher than the expected mass as m/z increases. Note the MS spectrum (supplemental Fig. S3) accurately shows that the expected m/z was observed within 0.05 Da. Dark squares indicate GlcNAc residues, light circles indicate Gal residues, and gray triangles indicate Fuc residues. Asterisks mark unexpected peaks that misleadingly suggested fucosylation at the internal GlcNAc residue.
Further analysis of the fragmentation spectra of the DFNR-glycopeptide showed unexpected peaks at m/z = 900 and 1062 Da (marked with asterisks in Fig. 5A). Depending on the mass spectrometer used, these peaks were either prominent using ESI-Q-TOF (supplemental Fig. S2) or close to the background level in the MALDI TOF-TOF spectra (Fig. 5A). Both peaks matched with fragments still containing the DFNR peptide and a dHex (fucose) residue in the remaining glycan moiety. However, not more than one HexNAc residue appeared to be present in either glycopeptide fragment. This suggested that the external GlcNAc residue, which was the predicted acceptor site for the fucose residue, was not required for fucosylation, and that the fucose was linked to another residue. Assuming that fucose was attached to the internal GlcNAc residue, the 900-Da fragment could correspond to Fucα1–4GlcNAc-DFNR and the 1062-Da fragment to Galβ1–3[Fucα1–4]GlcNAc-DFNR. These MS data indicated therefore the presence of the unexpected glycan structure Galβ1–4GlcNAcβ1–3Galβ1–3[Fucα1–4]GlcNAc- (LacNAc-Lea). Although the specificity of H. pylori 11639 FucT was previously determined to be restricted to type II acceptors and α1–3 fucosyltransferase activity (29), it was a possibility that in the applied in vitro conditions, and offering the enzyme an unusual acceptor with an internal type I and an external type II structure, the enzyme specificity was altered. An alternative explanation was that these peaks were MS artifacts. It has been reported that fucose residues can rearrange during MS analysis, which can lead to misinterpretations concerning the sugar sequence of glycan moieties (30).
To conclusively resolve the structure of the fucosylated glycan moieties, the trypsinized peptides were Pronase E digested, leaving only the asparagine-linked glycans (31). These were then permethylated and subjected to MS/MS analysis (27). Permethylation overcomes the problem of fucose moieties re-arranging during MS/MS analysis. As free hydroxyl and amino groups are modified with methyl groups, adding a mass change of 14 Da at each permethylation site, this technique allows the determination of the number of linkages in which a sugar residue is involved. Analysis by MALDI-MS on the 9.4T FTICR MS was completed to determine the elemental composition of the permethylated glycan. The expected m/z for the sodium-cationized permethylated glycan was 1252.6045 and the observed m/z was 1252.6046 corresponding to an expected elemental composition of (C55H95N3O27+Na)+. The obtained MS/MS spectrum from the MALDI TOF/TOF instrument contained two major peaks at 615 and 660 Da (Fig. 6). Both masses corresponded to the expected fragments of the permethylated asparagine-glycan moiety containing a terminal Lex-epitope. Consistent with what was observed by Liu et al. (31) the permethylated modified Asn shows a 143-Da loss in MS/MS observed as the mass difference between the precursor m/z 1252 and the first fragment ion at m/z 1109 (exact m/z by FTICR MS was 1109.5469 observed, 1109.5463 expected (C49H86N2O24+Na)+ (31). The peak at m/z 615 is consistent with a sodium- cationized y2 ion containing the modified permethylated Asn plus HexNAc and hexose (by MALDI FTICR the exact m/z 615.2736, observed; and 615.2737, expected (C26H44N2O13+Na)+). Similarly, the peak at m/z 660.35 corresponded to a sodium-cationized b2 ion (the remaining part of the permethylated molecule) consisting of a branched HexNAc (230 Da) and terminal Hex (219 Da) and terminal dHex (189 Da) (by MALDI FTICR the exact m/z 660.3202, observed; 660.3202, expected (C29H51NO14+Na)+). This showed conclusively that fucose was located on the HexNAc residue remote from the Asn and not on the HexNAc moiety attached to the Asn. Therefore, a glycoprotein containing a Lex-epitope was successfully produced with combined in vivo and in vitro techniques, using selected bacterial enzymes.
FIGURE 6.

MALDI TOF-TOF analysis of permethylated glycans to verify the Lex structure. Shown is the MALDI TOF-TOF fragmentation spectrum of the permethylated Asn-glycan, confirming the position of the fucose residue at the exterior GlcNAc residue (the Lex structure). Peaks at 615.3 and 660.3 Da can only be derived from fragmentation of the expected Lex-containing glycan. The peak at 472.2 Da may correspond to a Gal-GlcNAc fragment, which may be the internal or external disaccharide fragments, as they cannot be distinguished by the mass. The inset shows the correct glycan (containing the Lex-epitope) in comparison to the alternative glycan containing the Fuc residue at the internal GlcNAc residue (Lea-epitope). GlcNAc residues are indicated by dark squares, Gal residues by light colored circles, and Fuc residues by gray triangles.
DISCUSSION
The synthesis of defined glycoconjugates is an important but challenging task for the pharmaceutical industry. In the future, the exploitation of glycoconjugate biosynthesis pathways functionally expressed in nonpathogenic bacteria may become a valuable technology for the production of such compounds. In this work, we have produced a glycoprotein containing an epitope commonly found in human tissues, using a combined approach consisting of in vivo and in vitro glycosylation steps. The strategy used is presented in Fig. 1.
Fucosylation of free oligosaccharides with LacNAc-epitopes for the assembly of the Lex antigen was previously accomplished in live E. coli cells using H. pylori fucosyltransferases (32). The design of the glycoengineering approach followed here, however, imposes that the glycans must be built onto the UndPP carrier to allow en bloc transfer to acceptor proteins. This requires the participation of a distinct class of initiating glycosyltransferases, capable of adding a sugar residue onto the lipid carrier (12). The substrate for the next glycosyltransferases is therefore fixed to the inner membrane. It is possible that due to sterical hindrance the fixed glycans might not be able to reach the active sites of some glycosyltransferases, which prevents the use of some enzymes for this second step. Based on previous work (25), it was correctly predicted that the set of H. influenzae glycosyltransferases selected for the in vivo glycosylation step was able to generate the required glycolipid anchored in the inner membrane. In the presence of the C. jejuni OTase PglB, the tetrasaccharide was transferred onto the AcrA acceptor protein. Some side products were generated, having slightly elongated glycans with an additional HexNAc residue or a Hex-HexNAc disaccharide. This was surprising and indicated a partial reduction in acceptor specificity of the responsible glycosyltransferases in the heterologous expression system.
Because of its terminal LacNAc structure, the tetrasaccharide assembled with the H. influenzae glycosyltransferases may be modified in various ways to generate humanized antigens. Addition of sialic acid, galactose, or GalNAc residues could result in epitopes mimicking gangliosides or blood group antigens. The goal of this work was the synthesis of a Lex-containing glycoprotein by fucosylation of the LacNAc acceptor. The fucose incorporation was successfully accomplished by in vitro fucosylation of the in vivo assembled glycoprotein. Although MS/MS peaks at 900 and 1062 Da misleadingly indicated an unexpected site of fucosylation (LacNAc-Lea-), the analysis of the permethylated product confirmed that the expected Lex-epitope was indeed produced. Curiously, fucose rearrangement in the gas phase was prominent using Q-TOF MS (supplemental Fig. S2), but only minor using MALDI-MS (Fig. 5A). This can be explained considering that the compounds spend several milliseconds in the collision cell of a Q-TOF but only microseconds of interaction time in a MALDI TOF/TOF.
The presented work demonstrates a step toward a novel technology for the production of glycoproteins. Our work was preceded by an analogous, although conceptually different approach for the synthesis of homogenous N-glycoproteins. The strategy, also consisting of an initial in vivo glycosylation step, and several modification steps in vitro, was recently reported by Schwarz et al. (33). In that work, AcrA was glycosylated in vivo, the glycan was trimmed by glycanases in vitro, and subsequently subjected to an enzymatic transglycosylation reaction to obtain N-glycoproteins carrying oligosaccharides of the high mannose type. Unlike the strategy presented by Schwarz et al. (33), our approach can be adapted to generate glycoproteins carrying human glycan epitopes of diverse nature. This includes glycans unlikely to be synthesized in engineered yeast or mammalian cells such as those present on glycolipids. The ultimate goal of the glycoengineering approach is to achieve complete synthesis in vivo, followed by simple purification of the customized glycoconjugates. Using an optimized set of glycosyltransferases, the all-enzymatic in vivo synthesis is expected to assure the highest product quality and low production costs. With this approach an almost limitless combination of designed glycan and protein acceptor structures appears possible. One limitation is that bacterial strains will have to be engineered to produce the activated sugar donors not available in E. coli under standard growth conditions, for example, GDP-fucose and CMP-sialic acid. However, strains producing these activated sugars have been constructed (34). A main focus of future work should be the thorough analysis and optimization of metabolic pathways and the expression and control of activity of glycosyltransferases originating from various species, in E. coli. These efforts are currently ongoing in our laboratory.
Acknowledgments
We thank M. Veronica Ielmini and M. Florencia Haurat for critical reading of the manuscript.
This work was supported in part by grants from Alberta Innovates Health Solutions and the Alberta Innovates Centre for Carbohydrate Science (AICCS) (to M. F. F.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- OTase
- oligosaccharyltransferases
- UndPP
- undecaprenyl pyrophosphate
- Ni-NTA
- nickel-nitrilotriacetic acid
- FTICR
- Fourier-transform ion cyclotron resonance.
REFERENCES
- 1. Wacker M., Feldman M. F., Callewaert N., Kowarik M., Clarke B. R., Pohl N. L., Hernandez M., Vines E. D., Valvano M. A., Whitfield C., Aebi M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 7088–7093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wacker M., Linton D., Hitchen P. G., Nita-Lazar M., Haslam S. M., North S. J., Panico M., Morris H. R., Dell A., Wren B. W., Aebi M. (2002) Science 298, 1790–1793 [DOI] [PubMed] [Google Scholar]
- 3. Langdon R. H., Cuccui J., Wren B. W. (2009) Future Microbiol. 4, 401–412 [DOI] [PubMed] [Google Scholar]
- 4. Feldman M. (2009) in Microbial Glycobiology (Moran A. ed) pp. 904–914, Elsevier Science Publishing, Amsterdam, The Netherlands [Google Scholar]
- 5. Helenius A., Aebi M. (2001) Science 291, 2364–2369 [DOI] [PubMed] [Google Scholar]
- 6. Hamilton S. R., Davidson R. C., Sethuraman N., Nett J. H., Jiang Y., Rios S., Bobrowicz P., Stadheim T. A., Li H., Choi B. K., Hopkins D., Wischnewski H., Roser J., Mitchell T., Strawbridge R. R., Hoopes J., Wildt S., Gerngross T. U. (2006) Science 313, 1441–1443 [DOI] [PubMed] [Google Scholar]
- 7. Jones C. (2005) An. Acad. Bras. Cienc. 77, 293–324 [DOI] [PubMed] [Google Scholar]
- 8. Nothaft H., Szymanski C. M. (2010) Nat. Rev. Microbiol. 8, 765–778 [DOI] [PubMed] [Google Scholar]
- 9. Faridmoayer A., Fentabil M. A., Haurat M. F., Yi W., Woodward R., Wang P. G., Feldman M. F. (2008) J. Biol. Chem. 283, 34596–34604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feldman M. F., Wacker M., Hernandez M., Hitchen P. G., Marolda C. L., Kowarik M., Morris H. R., Dell A., Valvano M. A., Aebi M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 3016–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen M. M., Glover K. J., Imperiali B. (2007) Biochemistry 46, 5579–5585 [DOI] [PubMed] [Google Scholar]
- 12. Hug I., Feldman M. F. (2011) Glycobiology 21, 138–151 [DOI] [PubMed] [Google Scholar]
- 13. Raetz C. R., Whitfield C. (2002) Annu. Rev. Biochem. 71, 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kowarik M., Young N. M., Numao S., Schulz B. L., Hug I., Callewaert N., Mills D. C., Watson D. C., Hernandez M., Kelly J. F., Wacker M., Aebi M. (2006) EMBO J. 25, 1957–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ielmini M. V., Feldman M. F. (2011) Glycobiology 21, 734–742 [DOI] [PubMed] [Google Scholar]
- 16. Vik A., Aas F. E., Anonsen J. H., Bilsborough S., Schneider A., Egge-Jacobsen W., Koomey M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 4447–4452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Faridmoayer A., Fentabil M. A., Mills D. C., Klassen J. S., Feldman M. F. (2007) J. Bacteriol. 189, 8088–8098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chan N. W., Stangier K., Sherburne R., Taylor D. E., Zhang Y., Dovichi N. J., Palcic M. M. (1995) Glycobiology 5, 683–688 [DOI] [PubMed] [Google Scholar]
- 19. Oriol R., Le Pendu J., Mollicone R. (1986) Vox Sang. 51, 161–171 [DOI] [PubMed] [Google Scholar]
- 20. Srivatsan J., Smith D. F., Cummings R. D. (1992) J. Biol. Chem. 267, 20196–20203 [PubMed] [Google Scholar]
- 21. van Die I., van Vliet S. J., Nyame A. K., Cummings R. D., Bank C. M., Appelmelk B., Geijtenbeek T. B., van Kooyk Y. (2003) Glycobiology 13, 471–478 [DOI] [PubMed] [Google Scholar]
- 22. Atochina O., Harn D. (2006) Exp. Dermatol. 15, 461–468 [DOI] [PubMed] [Google Scholar]
- 23. Rook G. A. (2009) Immunology 126, 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma B., Audette G. F., Lin S., Palcic M. M., Hazes B., Taylor D. E. (2006) J. Biol. Chem. 281, 6385–6394 [DOI] [PubMed] [Google Scholar]
- 25. Phillips N. J., Miller T. J., Engstrom J. J., Melaugh W., McLaughlin R., Apicella M. A., Gibson B. W. (2000) J. Biol. Chem. 275, 4747–4758 [DOI] [PubMed] [Google Scholar]
- 26. Shevchenko A., Wilm M., Vorm O., Mann M. (1996) Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 27. Alley W. R., Jr., Madera M., Mechref Y., Novotny M. V. (2010) Anal. Chem. 82, 5095–5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meier-Dieter U., Barr K., Starman R., Hatch L., Rick P. D. (1992) J. Biol. Chem. 267, 746–753 [PubMed] [Google Scholar]
- 29. Ma B., Wang G., Palcic M. M., Hazes B., Taylor D. E. (2003) J. Biol. Chem. 278, 21893–21900 [DOI] [PubMed] [Google Scholar]
- 30. Harvey D. J., Mattu T. S., Wormald M. R., Royle L., Dwek R. A., Rudd P. M. (2002) Anal. Chem. 74, 734–740 [DOI] [PubMed] [Google Scholar]
- 31. Liu X., McNally D. J., Nothaft H., Szymanski C. M., Brisson J. R., Li J. (2006) Anal. Chem. 78, 6081–6087 [DOI] [PubMed] [Google Scholar]
- 32. Dumon C., Bosso C., Utille J. P., Heyraud A., Samain E. (2006) ChemBioChem 7, 359–365 [DOI] [PubMed] [Google Scholar]
- 33. Schwarz F., Huang W., Li C., Schulz B. L., Lizak C., Palumbo A., Numao S., Neri D., Aebi M., Wang L. X. (2010) Nat. Chem. Biol. 6, 264–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Antoine T., Priem B., Heyraud A., Greffe L., Gilbert M., Wakarchuk W. W., Lam J. S., Samain E. (2003) ChemBioChem 4, 406–412 [DOI] [PubMed] [Google Scholar]