

Abstract
Background
Pediatric dilated cardiomyopathy (DCM) is the leading indication for heart transplantation after age 1 year. Risk factors by etiology at clinical presentation have not been determined separately for death and transplantation in population-based studies. Competing risks analysis may inform patient prioritization for transplantation listing.
Methods and Results
The Pediatric Cardiomyopathy Registry enrolled 1731 children diagnosed with DCM from 1990-2007. Etiologic, demographic and echocardiographic data collected at diagnosis were analyzed with competing risks methods stratified by DCM etiology to identify predictors of death and transplantation. For idiopathic DCM (n=1192), diagnosis after age 6, congestive heart failure (CHF), and lower left ventricular (LV) fractional shortening (FS) z-score were independently associated with both death and transplantation equally. In contrast, increased LV end-diastolic dimension (EDD) z-score was associated only with transplantation while lower height-for-age z-score was associated only with death. For neuromuscular disease (n=139), lower LVFS was associated equally with both endpoints, but increased LVEDD was associated only with transplantation. The risks of death and transplantation were increased equally for older age at diagnosis, CHF, and increased LVEDD among those with myocarditis (n=272) and for CHF and decreased LVFS among those with familial DCM (n=79).
Conclusions
Risk factors for death and transplantation in children varied by DCM etiology. For idiopathic DCM, increased LVEDD was associated with increased transplantation risk but not mortality. Conversely, short stature was significantly related to death but not transplantation. This may present an opportunity to improve the transplantation selection algorithm.
Keywords: cardiomyopathy, pediatrics, cardiac transplantation, heart failure
INTRODUCTION
Pediatric dilated cardiomyopathy (DCM) carries substantial morbidity and mortality and costs US society annually a substantial portion of the estimated $2 billion associated with pediatric heart failure.1-3 DCM is the most common indication for cardiac transplantation among children after the first year of life.1,2 Common unfavorable outcomes of DCM are death, usually from congestive heart failure (CHF) or sudden cardiac death, and cardiac transplantation. Transplantation is commonly combined with death as a poor outcome because it is assumed to be performed in children who would otherwise be at high risk of short-term death. However, transplantation has its own negative consequences: a lifetime of immunosuppressive therapy, potential re-transplantation, associated co-morbidities, and increased risk of premature death.2,4 Two population-based studies, including the Pediatric Cardiomyopathy Registry (PCMR), reported freedom from death and transplant at 1 year to be 72% and 69% and at 5 years to be 63% and 54%.4,5 These survival rates from the time of DCM diagnosis are similar to what they were decades ago.1 Median life expectancy after transplantation is only between 10 and 15 years, although younger children and children receiving more recent transplantations have been observed to have better outcomes.1 Therefore, better identification of children with DCM at high risk for death would avoid unnecessarily exposing them to the risks of potentially poor outcomes following transplantation while ensuring that children who are truly at imminent risk for death receive life-saving surgery. We sought to determine whether the risk factors for death in children with DCM are the same as those used in practice to determine transplantation, or whether other unrecognized risk factors for death are not being weighted sufficiently in the transplantation decision.
Prior analyses from the PCMR5 as well as a systematic review of studies of risk factors for the composite endpoint of death or transplantation in children with DCM at the time of diagnosis have implicated older age, worse left ventricular (LV) fractional shortening (FS) and ejection fraction, and CHF as significant predictors.6 Our current analyses use competing risk methods to separate the effects of these risk factors according to their impact on death and transplantation. The analyses are stratified by the etiology of DCM at the time of diagnosis to simulate the real-world considerations that affect optimal medical management or referral for transplantation.
METHODS
The Pediatric Cardiomyopathy Registry
The Pediatric Cardiomyopathy Registry (PCMR) is a central repository of information on pediatric cardiomyopathy across North American clinical centers.7 Briefly, children (younger than 18 years of age) diagnosed with DCM by a pediatric cardiologist were identified by chart review and enrolled into two cohorts. The retrospective cohort consists of children at 39 tertiary care centers diagnosed between January 1, 1990 and December 31, 1995. The prospective cohort consists of children at 98 pediatric cardiac centers diagnosed after January 1, 1996. The method of data collection was the same (see below). Although the retrospective cohort (N=1214, 72%) compared to the prospective cohort (N=468, 28%) has longer median follow-up time (1.9 versus 1.1 years, P<0.001), the outcomes for the two cohorts are similar.7
Enrollment required echocardiographic evidence of LV dilation and systolic dysfunction, semi-quantitative echocardiographic patterns, diagnostic endomyocardial biopsy results, or other compelling clinical evidence.7 Fourteen criteria excluded secondary cardiomyopathies (i.e., toxic exposures, endocrine disorders, immunologic diseases, etc.). For both the retrospective and prospective cohorts, data were collected from the time of diagnosis, and annually thereafter, through medical record review using standardized case report forms and trained data collectors. Subjects were followed until death, heart transplantation, transfer to a non-participating institution, or until they were lost to follow-up. This report is based on the PCMR database as of January 3, 2008.
All participating PCMR centers obtained institutional review board approval.
This study was limited to children who were recorded as having “pure” DCM at the time of diagnosis (i.e., excluding mixed hypertrophic or restrictive phenotypes). The cases were further classified into 6 etiologic groups at baseline based on the PCMR typology5,8 and on published guidelines:9 inborn errors of metabolism, malformation syndrome, neuromuscular disease (NMD), familial isolated cardiomyopathy (FDCM), myocarditis, and idiopathic DCM.
Study Variables
Demographic variables included age, height, weight, and race, with height and weight normalized to z-scores according to CDC standards. Clinical information included etiology as per the PCMR algorithm and familial history of various related diseases and outcomes. Echocardiographic variables, including LVEDD, posterior wall thickness, septal thickness, and mass, were expressed as z-scores conditional on body surface area; LVFS was expressed as a z-score conditional on age.10,11
Statistical Methods
Descriptive data are presented for all children stratified by etiology as percentages or means and standard deviations; skewed continuous data are summarized as medians and quartiles. Children with incomplete baseline data were included in all analyses for which they could contribute data. However, multivariate analyses were limited to those children with complete baseline data for all candidate covariates being considered in the stepwise selection. The two primary outcomes were time-to-death and time-to-transplantation, with censoring occurring when a child was lost to follow-up. Survival and transplantation figures and estimates were calculated using the cumulative incidence of competing risks from the time of DCM diagnosis.12 The Cox proportional hazards regression analysis, modified for competing risks, identified both univariate and multivariate predictors of outcomes for each etiology.12,13 In competing risks analysis, each predictor in the Cox regression model generated two effect estimates and two P-values, one for the impact of the predictor on mortality, and the other for the impact on transplantation. The competing risk model included an interaction term to determine whether the effect of the predictor on mortality differed from its effect on transplantation. If the interaction term was significant, two separate effects were estimated. If the interaction term was not significant, then it was removed and a single effect estimate and P-value were presented reflecting the common impact of the predictor on both outcomes.
Because of the large number of potential predictors being considered, the final multivariable model was developed in a two step process. Initially, we included only those predictors that were significant at P<0.05 in step-wise analyses completed in groups of related variables (demographics, anthropometrics, clinical, and historical). Group winners were then used to build a model in a step-wise selection with significant variables selected at P<0.01. All analyses were conducted using the Statistical Analysis System version 9.1 (SAS, Cary, NC) and a SAS macro for competing risks analysis.13
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
RESULTS
Demographics and Clinical Characteristics
The sample consisted of 1731 children with DCM (Tables 1 and 2). Of these, 6 with malformation syndromes and 43 with inborn errors of metabolism were excluded as there were too few children for analysis of their respective etiology.
Table 1.
Demographic Characteristics of 1682 Children with DCM at the Time of Diagnosis, by Etiology
| Idiopathic DCM | Neuromuscular Disease | Familial Isolated DCM | Myocarditis | |||||
|---|---|---|---|---|---|---|---|---|
| Subjects, n | 1192 | 139 | 79 | 272 | ||||
| Male, n (%) | 593 | (49.8) | 134 | (96.4) | 44 | (55.7) | 134 | (49.3) |
| Age at Diagnosis | ||||||||
| Median, (IQR) | 1.2 | (0–18) | 14.3 | (8–18) | 5.9 | (0–18) | 1.7 | (0–18) |
| Infant (<1 y), n (%) | 558 | (46.8) | 0 | 25 | (31.7) | 83 | (30.5) | |
| 1 to <6 y, n (%) | 249 | (20.8) | 0 | 15 | (19.0) | 99 | (36.7) | |
| 6 to <12 y, n (%) | 167 | (14.0) | 23 | (16.6) | 20 | (25.3) | 33 | (12.4) |
| 12 to <18 y, n (%) | 223 | (18.6) | 116 | (83.5) | 19 | (24.1) | 53 | (19.9) |
| Height-for-Age, n, Mean z-score, SD | 629 | -0.33, 1.6* | 56 | -0.85, 1.7* | 46 | -0.32, 2.1 | 126 | -0.12, 1.5 |
| Weight-for-Age, n, Mean z-score, SD | 725 | -0.26, 1.6* | 77 | 0.15, 1.9 | 51 | 0.03, 2.0 | 157 | -0.29, 1.3* |
| Weight-for-Height, n, Mean z-score, SD | 406 | -0.64, 1.2* | 12 | 1.08, 2.5 | 27 | -0.46, 1.7 | 84 | -0.67, 1.1* |
|
| ||||||||
| Race, n (%) | ||||||||
| White | 644 | (54.0) | 100 | (71.9) | 41 | (51.9) | 144 | (52.9) |
| Black | 251 | (21.1) | 19 | (13.7) | 12 | (15.2) | 67 | (24.6) |
| Hispanic | 197 | (16.5) | 14 | (10.1) | 17 | (21.5) | 43 | (15.8) |
| Other | 77 | (6.5) | 3 | (2.2) | 7 | (8.9) | 13 | (4.8) |
| Missing | 23 | (1.9) | 3 | (2.2) | 2 | (2.5) | 5 | (1.8) |
P<0.01 for two-sided t-test versus z-score=0.
IQR: Interquartile Range, SD: Standard Deviation. Weight-for-height not calculated for children >120 cm.
Table 2.
Clinical Characteristics of 1682 Children with DCM at the Time of Diagnosis, by Etiology
| Idiopathic DCM | Neuromuscular Disease | Familial Isolated DCM | Myocarditis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subjects, n | 1192 | 139 | 79 | 272 | ||||||||
| Congestive Heart Failure, n (%) | 894 | (75.0) | 40 | (28.8) | 44 | (55.7) | 227 | (83.5) | ||||
| LV Echocardiographic, n, mean z-score, SD | ||||||||||||
| End-Diastolic Dimension | 939 | 4.70 | 2.59* | 101 | 1.63 | 1.96* | 60 | 3.42 | 2.49* | 214 | 3.56 | 2.49* |
| End-Diastolic Posterior Wall Thickness | 732 | -0.70 | 2.19* | 84 | -1.70 | 2.20* | 51 | -0.75 | 1.78* | 169 | 0.01 | 2.23 |
| End-Diastolic Septal Thickness | 660 | -1.04 | 1.70* | 78 | -1.40 | 1.57* | 45 | -0.68 | 1.46* | 144 | -0.38 | 1.66* |
| End-Systolic Dimension | 816 | 6.55 | 2.62* | 94 | 3.10 | 2.40* | 54 | 4.86 | 2.82* | 186 | 5.62 | 2.62* |
| Fractional Shortening | 961 | -9.08 | 3.21* | 128 | -5.56 | 3.30* | 64 | -6.99 | 3.75* | 223 | -8.45 | 3.59* |
| Mass | 719 | 2.39 | 2.53* | 83 | -0.19 | 1.85 | 51 | 1.67 | 2.26* | 164 | 2.35 | 2.33* |
| End-Diastolic Posterior Wall Thickness - Dimension ratio, n, median (IQR) | 774 | 0.12 | (.10, .15) | 102 | 0.14 | (.12, .16) | 54 | 0.14 | (.11, .16) | 175 | 0.14 | (.11, .17) |
|
| ||||||||||||
| Familial History at Diagnosis, n with positive history, n, (%) | ||||||||||||
|
| ||||||||||||
| All Categories | 198 | 720 | (27.5) | 36 | 68 | (52.9) | 79 | 79 | (100) | 30 | 118 | (25.4) |
| Cardiomyopathy | 107 | 723 | (14.8) | 12 | 58 | (20.7) | 79 | 79 | (100) | 10 | 118 | (8.5) |
| Sudden Death | 59 | 756 | (7.8) | 3 | 68 | (4.4) | 30 | 56 | (53.6) | 10 | 135 | (7.4) |
| Congenital Heart Disease | 36 | 679 | (5.3) | 1 | 50 | (2.0) | 7 | 43 | (16.3) | 6 | 113 | (5.3) |
| Arrhythmia | 26 | 667 | (3.9) | 3 | 50 | (6.0) | 5 | 43 | (11.6) | 3 | 111 | (2.7) |
| Genetic Syndrome | 32 | 696 | (4.6) | 32 | 75 | (42.7) | 5 | 45 | (11.1) | 8 | 125 | (6.4) |
P<0.01 for two-sided t-test versus z-score=0.
LV: Left Ventricular, IQR: Interquartile Range.
Children with Idiopathic Disease
The largest group had idiopathic DCM (n=1192; 69%) of whom most were white (54%), male (50%), and infants (<1 year, 47%) at time of diagnosis. Growth characteristics were below normal for age. Three-quarters presented with symptoms of CHF. Echocardiographic measurements, recorded in 81%, revealed LV dilation, increased LV mass, and poor LV systolic function. Septal thickness and LV posterior wall thickness z-scores were also below normal. A family history of associated conditions was present in 28%. For 157 (13%), an etiology was subsequently identified at some time after the first month of DCM diagnosis. The most common etiologies discovered were myocarditis (n=86), inborn errors of metabolism (n=31), and FDCM (n=25), and 15 with other causes. However, the initial diagnosis of “idiopathic” was used for all subsequent analyses because the average time between diagnosis and death or transplant was only a few months.
Children with Neuromuscular Disease
Of the 139 children with NMD, 84% had Duchenne (n=117) and 8% had Becker (n=11) muscular dystrophy. Most were male (96%) and white (72%). All presented with cardiomyopathy after age 6 years; 83% presented after age 12 years. Only 39 (28%) had symptoms of CHF at diagnosis. Half had a family history of an associated condition: most commonly a genetic syndrome (32 of 74; 42%) or cardiomyopathy (12 of 58; 20%).
Children with Familial Isolated Cardiomyopathy
Most of the 79 children (72; 91%) with FDCM were believed to have an autosomal dominant inheritance pattern. They were older at diagnosis compared to those with idiopathic DCM or myocarditis (median age, 6 years) and 56% had CHF. This group had the greatest proportion of a family history of sudden deaths (53%).
Children with Myocarditis
Of the 272 children with myocarditis, 112 (41%) met the Dallas pathology criteria,14 with the others classified as “probable cases.” They were similar in baseline characteristics to the idiopathic group, although more presented with CHF (83% versus 75%, P<0.01).
Outcomes
The median follow-up time for the entire sample was 1.3 years (IQR, 0.2 to 4.0 years), and for those who were alive at last follow-up and not transplanted, the median follow-up time was 2.8 years (IQR, 0.9 to 5.5 years). Overall, 665 children (38%) experienced either death or transplantation, with the majority undergoing transplantation (512; 77%). Among patients listed for transplant, the median time from diagnosis to listing for transplantation was 1.4 months; however, 22% who were listed (n=112) did not receive a transplant, and of those, 41% (n=46) died.
Among the 1192 children with idiopathic DCM, 487 had an event, of who 161 died (33%) and 326 received a transplant (67%). Table 3 and Figure 1A show that in this cohort at 1 year after diagnosis, the probability of death was 11%, the probability of transplant was 24%, and the remaining 66% of children were alive and transplant-free. By 5 years, the probability of death had increased to 16% and the probability of transplantation had reached 33%, while the percentage alive and transplant-free had decreased to 51%. The idiopathic DCM group had the largest proportion of deaths among those listed and waiting for transplantation.
Table 3.
Cumulative Event Rates for Death and Cardiac Transplantation
| Time from Diagnosis of DCM (years)
|
||||
|---|---|---|---|---|
| 0.5 | 1 | 5 | ||
| Idiopathic DCM n=1192 | Dead | 0.082 | 0.107 | 0.164 |
| Transplant | 0.183 | 0.237 | 0.325 | |
| Alive | 0.735 | 0.656 | 0.511 | |
| Neuromuscular Disease n=139 | Dead | 0.046 | 0.078 | 0.377 |
| Transplant | 0.031 | 0.055 | 0.072 | |
| Alive | 0.923 | 0.867 | 0.551 | |
| Familial DCM n=79 | Dead | 0.065 | 0.079 | 0.100 |
| Transplant | 0.195 | 0.234 | 0.370 | |
| Alive | 0.740 | 0.687 | 0.530 | |
| Myocarditis n=272 | Dead | 0.087 | 0.087 | 0.087 |
| Transplant | 0.127 | 0.146 | 0.215 | |
| Alive | 0.786 | 0.767 | 0.698 | |
DCM: Dilated Cardiomyopathy.
Figure 1.
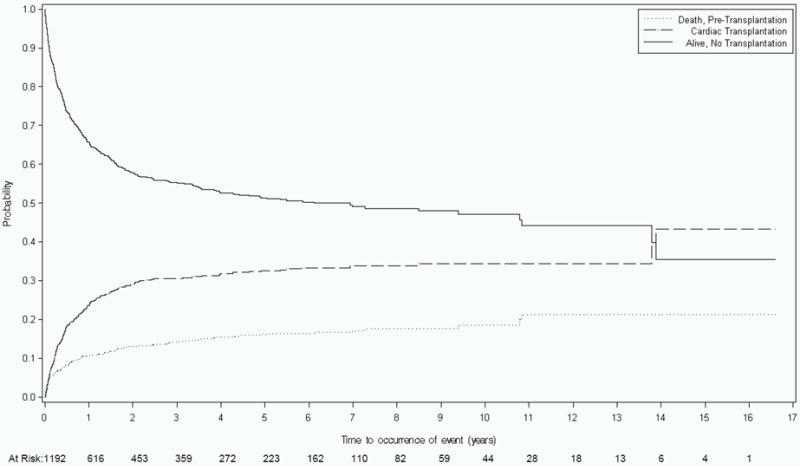
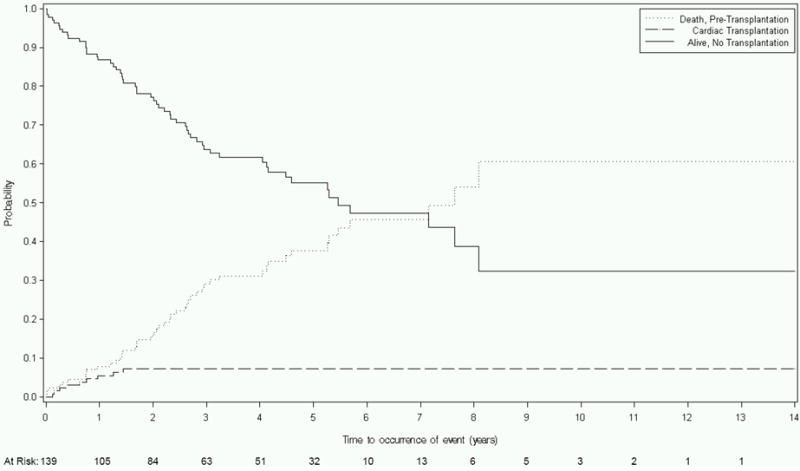
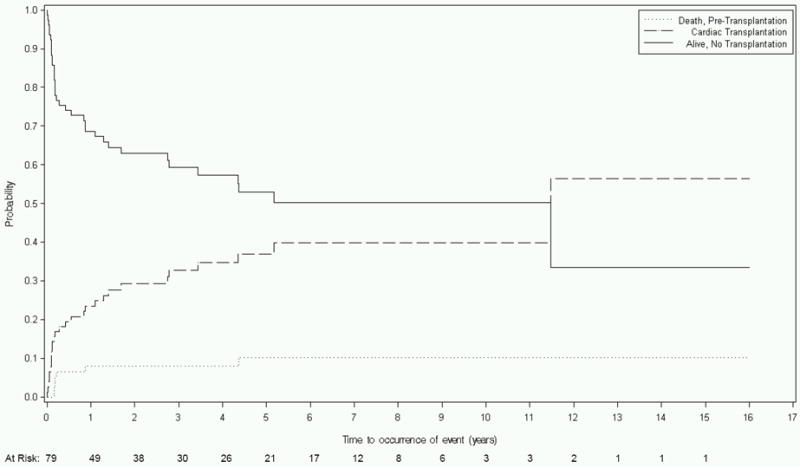
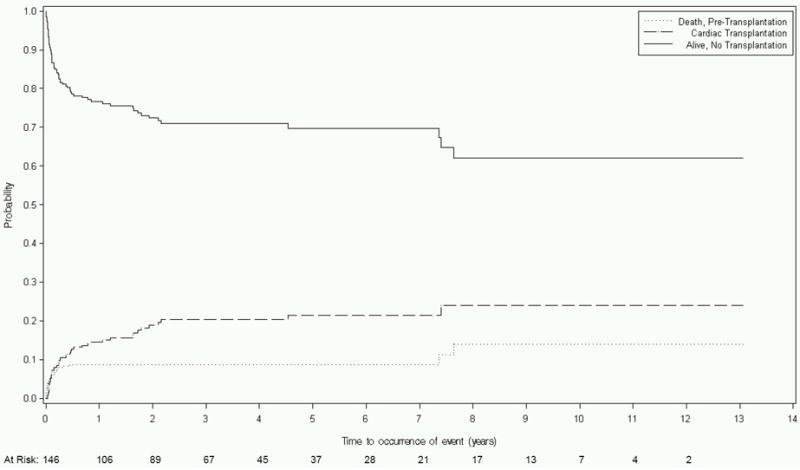
Competing risks estimates of death, cardiac transplant, and survival for children with dilated cardiomyopathy (DCM) caused by: A) Idiopathic DCM (n=1192); B) Neuromuscular Disease (n=139); C) Familial Isolated DCM (n=79); and D) Myocarditis (n=272).
There were 139 children with neuromuscular DCM, of whom 56 had a poor outcome. The outcomes were primarily deaths (47; 84%) while only nine received a heart transplant, of whom, most had Becker muscular dystrophy (n=5), while none had Duchenne muscular dystrophy. At 1 year, the probability of death was 8%, the probability of transplant was 6%, and the remaining 87% of children were alive and transplant-free (Table 3 and Figure 1B). By 5 years, the estimated death and transplant rates were 38% and 7%, respectively, leaving 55% of children alive and transplant-free.
Compared to all other etiologic groups, the 79 children with FDCM had the largest proportion of transplantations relative to the number of poor outcomes. Of 35 children with poor outcomes, 28 (80%) were transplanted, while only 7 died. Moreover, 82% of children listed for cardiac transplantation received a transplant heart. At 1 year, 8% of children had died, 23% had been transplanted, and 69% remained alive and transplant-free (Table 3 and Figure 1C). By 5 years, the respective probabilities of death, transplant, and survival were 10%, 37%, and 53%.
The 272 children with myocarditis had the best outcomes of any group with transplant-free survival rates of 77% at 1 year and 70% at 5 years. During the course of follow-up, 70 poor outcomes occurred, including 24 deaths and 46 transplantations. These resulted in a 9% death rate and a 15% transplant rate at 1 year, and a 9% death rate and a 22% transplant rate at 5 years (Table 3 and Figure 1D).
Competing Risks Analysis: Risk Factors
The results of multivariate modeling of risk factors for death or transplantation for each etiology are shown in Table 4. For children with idiopathic DCM, older age, depressed LVFS, and the presence of CHF independently predicted both death and transplantation. Lower height-for-age was associated with an increased risk of death, but not of transplantation. In contrast, higher LVEDD was associated with an increased risk of transplantation, but a decreased risk of death. In fact, among the children who have not been transplanted, an elevated LVEDD is associated with decreased, not increased, mortality.
Table 4.
Results of Multivariate Modeling of Competing Risks of Death and Heart Transplantation by Etiology
| Group | Factor at Diagnosis | Hazard Ratio (95% CI) P-value | ||
|---|---|---|---|---|
| Combined (Death or Transplant) | Death | Transplant | ||
| Idiopathic DCM n=536 | Age at Diagnosis | |||
| 1 to <6 vs. Infants (<1 year) | 1.20 (0.76, 1.98) 0.5 | |||
| 6 to <12 vs. Infants (<1 year) | 4.22 (2.83, 6.61) <.001 | |||
| 12 to <18 vs. Infants (<1 year) | 3.52 (2.37, 5.23) <.001 | |||
| CHF (present vs. absent) | 3.48 (2.24, 5.41) <.001 | |||
| LVEDD z-score (per 1 SD increase) | 0.87 (0.77, 0.98) 0.019 | 1.11 (1.02, 1.21) 0.02 | ||
| LVFS z-score (per 1 SD increase) | 0.86 (0.8, 0.91) <.001 | |||
| Height-for-Age z-score (per 1 SD increase) | 0.77 (0.67, 0.9) <.001 | 0.99 (0.89, 1.1) 0.69 | ||
|
| ||||
| Neuromuscular Disease* n=98 | LVEDD z-score (per 1 SD increase) | 1.23 (0.96, 1.59) 0.1 | 3.45 (1.74, 6.84) <.001 | |
| LVFS z-score (per 1 SD increase) | 0.85 (0.74, 0.97) | |||
|
| ||||
| Myocarditis n=212 | Age (per 1 year increase) | 1.10 (1.05, 1.16) <.001 | ||
| CHF (present vs. absent) | 5.51 (1.32, 23) 0.02 | |||
| LVEDD z-score (per 1 SD increase) | 1.20 (1.06, 1.36) 0.005 | |||
|
| ||||
| Familial Isolated DCM n=64 | CHF (present vs. absent) | 2.22 (0.45, 11) 0.33 | ||
| LVFS z-score (per 1 SD increase) | 0.85 (0.68, 1.07) 0.17 | |||
Adjusted for subgroup (Duchenne, Becker or Other).
LV: Left Ventricular, CHF: Congestive Heart Failure, FS: Fractional Shortening, EDD: End-Diastolic Dimension, SD: Standard Deviation, DCM: Dilated Cardiomyopathy.
For children with neuromuscular DCM, the final competing risks multivariate model included LVFS and LVEDD, for which an interaction with outcome was present for the latter. As was seen for the children with idiopathic DCM, depressed LVFS was significantly and equally associated with both death and transplantation. LVEDD also showed a similar result with elevated LVEDD strongly associated with increased transplantation but only weakly and non-significantly associated with death.
In the myocarditis group, the multivariate model showed that increased age, CHF, and increased LVEDD at presentation were associated similarly with both death and transplantation. For FDCM, although several factors were associated with increased risk of either death or transplantation in univariate analyses (CHF, increased LVEDD, worse LVFS, LV end-diastolic septal thickness, or weight-for-height), a multivariate model with independently significant factors could not be fitted. However, the best model is presented (Table 4) after excluding those with significant collinearity or missing data.
DISCUSSION
Compared to the initial PCMR report on the incidence of pediatric DCM and its outcomes,5 this study assessed the differential effects of demographic and clinical factors determined at the time of DCM diagnosis on the subsequent risk of death or transplantation for each etiology. Using a competing risks analytical model, we identified novel differential risk factors for death and transplantation. Identifying these differences may improve the initial assessment, treatment, and survival of children with DCM.
Previous studies differ on the effect of age at diagnosis on mortality in children with DCM.6 In idiopathic DCM, we found that age at diagnosis of 6 years or older was associated with a 3-4 times increase in the risk of death or transplantation compared to infants. The effect of age in children with myocarditis also showed an association with older age and death or transplantation, however, it was best explained as a direct linear association between the outcomes and age at diagnosis. At presentation, in idiopathic DCM, older children may have more deleterious disease, perhaps because myocardial damage has had more time to occur or, conversely, in myocarditis, the younger heart has more reserve.15 The cumulative, long-term effects of this smoldering disease may diminish the reserve in a maturing heart, ultimately making recovery from structural and functional problems less likely, as opposed to a specific insult in those with myocarditis, whose age effect reflects reserve. The fact that age was not predictive of death or transplantation in the NMD group is not surprising because children are generally diagnosed with DCM at a much later age, usually during adolescence.
Congestive heart failure present when DCM is diagnosed is the single strongest predictor of death or transplantation for idiopathic DCM and myocarditis, but not in patients with NMD. In our population, only 29% of children with NMD had CHF at the time of DCM diagnosis, compared with greater than 50% for all other etiologies, which is not surprising as a large proportion of this population is prospectively screened by echocardiography and DCM is captured prior to overt symptomatic CHF. The mortality rate was much greater than the transplantation rate for this group, likely indicative of the fact that in children with NMD, the presence of respiratory compromise makes them less suitable for transplantation than compared to children with other etiologies of DCM.16 For those with idiopathic DCM and myocarditis, the severity of symptoms is associated with death and the decision to perform a cardiac transplantation reflects the practice patterns of pediatric cardiologists utilizing appropriate management.
The absence of LVFS as a predictor of death or transplantation for children with DCM due to myocarditis may be explained by the temporal process of the disease and time-dependent effects of circulating pro-inflammatory cytokines (e.g., TNF and IL-6).17 Cytokine release is hypothesized to adversely affect LV systolic function. The decrease in LVFS would be evident early in the course of most children with myocarditis, but this is not necessarily true for children with NMD and idiopathic DCM. As demonstrated, children with preexistent cardiac structural damage, i.e. the idiopathic and NMD groups, who have a decrease in LVFS at the time of diagnosis, appear to have worse outcomes. In these groups, the decreased LVFS and its continued deterioration could possibly relate to a chronic inflammatory response, but the outcome appears to be different than in isolated myocarditis, which represents an acute process that usually resolves on its own. The hypothesized role of chronic cytokine release is supported by, non-randomized studies of anti-inflammatory agents, such as intravenous immunoglobulin or steroids, that have reported some benefit in small case series of children with DCM,18 as well as in a larger Italian cohort with 13 years of follow-up.19 We have seen similar findings in children with HIV and LV dysfunction treated with immunoglobulins.20 Additionally, equivocal findings in trials of adults and a Cochrane review21 support the argument that transient (as opposed to chronic) inflammation may explain the particular constellation of predictors uncovered by this analysis.
Nevertheless some children with myocarditis do quite poorly. Other theories have been put forth regarding myocarditis pathology to explain why these children do not fully recover, including: the cleavage of myocardial structural proteins, like dystrophin, by viral proteases; and the persistence of Coxsackievirus B in the myocardium, leading to a low-grade, non-cytolytic, chronic infection in the heart. Either theory may explain worse outcomes with longer follow-up time although with structural damage as its mechanism.22
Higher LVEDD z-scores predicted a significantly greater risk of either death or cardiac transplantation among children with myocarditis; a greater risk of transplantation, but a greater likelihood of survival, in children with idiopathic DCM; and a greater risk of transplantation, but not death, in children with NMD. Although LV dilation is expected by definition in all children with DCM, greater LV dilation appeared to be a significant factor in the decision to perform transplantation for those with idiopathic DCM and NMD. For example, in children with idiopathic DCM, the median LVEDD z-score was 5.7 among children who were listed for transplantation but only 4.3 in children who were not listed (P<0.001). However, the role of elevated LVEDD in mortality risk is less clear. It is possible that the children with the highest levels of LVEDD were appropriately triaged to transplantation, but it is impossible to examine this empirically since we cannot know what would have happened to these children if the transplant had not occurred. We do know, however, that among 69 children who were listed but not transplanted, the median LVEDD z-score was 5.8 in children who were still alive at last follow-up while it was only 5.2 in those who died (and 5.7 in the children who were transplanted). There is at least the possibility that while elevated LVEDD is bad, there are other risk factors which, when present, are even worse.
Another explanation may be that comparatively greater LVEDD is a surrogate for children with worse symptoms and reflects the listing practices based on that criterion. In comparison, an associated study evaluating echocardiograms performed within 6 months of listing has linked greater LVEDD to increased mortality both during the post-listing period and within 6 months post-transplantation in children with DCM. There was a stronger effect noted in those younger than 5 years of age at the time of diagnosis.23 Once the decision is made to list for cardiac transplantation, the effect of severe LV dilation appears to be more detrimental to survival than in the pre-listed population. LV dilation should be monitored throughout the triage process for transplantation listing because it affects the success of both medical and surgical management.
The association of height-for-age with death, but not transplantation, corroborates the findings from another PCMR study looking at the association between growth parameters and mortality in children with idiopathic DCM.24 Impaired height-for-age may be an important indicator of the severity of heart failure and does not currently appear to be used as a strong indication for transplantation listing. Based on the current study, using short stature as a transplantation indication could improve survival by an estimated 50% for children whose height-for-age is below 2 standard deviations (HR 1.5; 95% CI, 1.03 to 2.18). Growth failure, determined using either height or weight, is a criterion for Status 1B listing by the United Network for Organ Sharing and Organ Procurement and Transplantation Network.25 Height as a proxy for overall growth and health status appears to be a simple criterion that does not appear to factor into the transplantation listing process as frequently as it ought to be. However, once such a deficit is noted, whether active interventions to prevent or reduce growth retardation would ultimately affect survival and defer transplantation is unknown. Although several studies have investigated growth hormone supplementation in cardiac disease for its hypothesized direct effects on the myocardium, it is unclear whether positive long-term effects would be seen in children with DCM and, if they did, whether that benefit would be reflected in height-for-age z-scores.26-28
This analysis was possible largely because of the strength of the PCMR, whose observational data on children with DCM spans almost 15 years across many clinical sites. This has allowed our analysis to propose and test potentially predictive algorithms of clinical outcomes, as was done in a prior PCMR analysis on establishing of an etiology through clinical and laboratory testing.29
Limitations
Several limitations should be considered when evaluating this analysis. Children in the PCMR may have more severe disease because all were identified as part of a clinical interaction with a health care provider, and not as part of asymptomatic screening. Although the PCMR has the largest reported population of children with DCM, the power to detect statistically significant differences in risk factors for death versus transplantation is limited by the small sample size for certain etiologic subgroups. For example, data from the Registry likely underestimates the true proportion of familial cases because not all families were thoroughly evaluated for the presence of cardiac disease.
Lastly, the relatively short follow-up period limits the ability to extrapolate our results. However, given that the majority of events occur within the first year after diagnosis of DCM, our overall conclusions should be clinically applicable. Continued follow-up is warranted to establish more accurate predictors of long-term events.
Conclusions
Using competing risk analysis, we found that at the time of DCM diagnosis, the presence of CHF, echocardiographic evidence of more severe disease, and increased age at diagnosis predicted worse outcomes, with certain key differences between etiologies. For children with idiopathic disease, diagnosis after age 6 years, CHF, decreased LVFS, increased LVEDD, and decreased height-for-age predicted worse outcomes. Increased LV dilation was predictive of use of cardiac transplantation, but not of mortality in idiopathic DCM, calling into question the importance of this factor when deciding to list a child for transplantation. Our analysis showed that children with idiopathic DCM and lower height-for-age z-scores might be dying prior to consideration for listing or receipt of potentially beneficial heart transplantation suggesting that impaired height should be an important consideration in the decision to list a child for transplantation. Whether height, as a product of nutrition, growth, or disease can be modified earlier in the disease course to decrease the risk of death may be a new area of research. For children with neuromuscular disease, decreased LVFS predicted death or transplantation, while increased LVEDD predicted transplant, but not death. For children with myocarditis, older age at diagnosis, CHF, and increased LVEDD predicted death or transplantation. These results suggest that the etiology of DCM modifies the importance of certain predictive factors. The ability to predict failed medical management may improve if the etiology of cardiomyopathy can be established earlier. Additionally, analyzing registry data in this manner allows for the postulation of predictive algorithms of clinical outcomes.
Prior studies of outcomes for children with dilated cardiomyopathy (DCM) have used a composite endpoint of death and cardiac transplantation and may have missed identifying predictive factors differentially affecting outcomes, which affect the transplantation listing process. Using competing risk analysis, this study found that at the time of DCM diagnosis, the presence of congestive heart failure (CHF), echocardiographic evidence of more severe disease, and increased age at diagnosis predicted worse outcomes, with certain key differences between etiologic groups. For children with idiopathic disease, diagnosis after age 6 years, CHF, decreased LVFS, increased LVEDD, and decreased height-for-age predicted worse outcomes. Increased LV dilation was predictive of use of transplantation, but not of mortality in idiopathic DCM, calling into question the importance of this factor in the decision to list for transplantation. Routine height measurement could highlight those with idiopathic DCM and lower height-for-age z-scores who might require more urgent consideration for transplantation listing. For children with neuromuscular disease, decreased LVFS predicted death or transplantation, while increased LVEDD predicted transplant, but not death. For children with myocarditis, older age at diagnosis, CHF, and increased LVEDD predicted death or transplantation. These results suggest that the etiology of DCM modifies the importance of particular predictive factors. The ability to predict failed medical management may improve if the etiology of the cardiomyopathy can be established earlier. Additionally, analyses of registry data in this manner allow for the postulation of predictive algorithms of clinical outcomes.
Acknowledgments
We thank the participating clinical centers for patient recruitment.
Funding Sources This study was supported by National Heart Lung and Blood Institute (NHLBI) F31HL094100 (J.A. Alvarez). The PCMR is supported by NHLBI R01HL53392 (S.E. Lipshultz).
Footnotes
Disclosures None.
References
- 1.Kirk R, Edwards LB, Aurora P, Taylor DO, Christie J, Dobbels F, Kucheryavaya AY, Rahmel AO, Hertz MI. Registry of the International Society for Heart and Lung Transplantation: Eleventh Official Pediatric Heart Transplantation Report--2008. J Heart Lung Transplant. 2008;27:970–977. doi: 10.1016/j.healun.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 2.Bublik N, Alvarez JA, Lipshultz SE. Pediatric cardiomyopathy as a chronic disease: A perspective on comprehensive care programs. Prog Pediatr Cardiol. 2008;25:103–111. doi: 10.1016/j.ppedcard.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossano JW, Kim JJ, Decker JA, Price JF, Zafar F, Graves DE, Morales DL, Heinle JS, Bozkurt B, Denfield SW, Dreyer WJ, Jefferies JL. Increasing prevalence and hospital charges in pediatric heart failure related hospitalizations in the United States: a population-based study. Circulation. 2010;122:A13740. [Google Scholar]
- 4.Daubeney PE, Nugent AW, Chondros P, Carlin JB, Colan SD, Cheung M, Davis AM, Chow CW, Weintraub RG. Clinical features and outcomes of childhood dilated cardiomyopathy: results from a national population-based study. Circulation. 2006;114:2671–2678. doi: 10.1161/CIRCULATIONAHA.106.635128. [DOI] [PubMed] [Google Scholar]
- 5.Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, Canter C, Wilkinson JD, Lipshultz SE. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 6.Alvarez JA, Wilkinson JD, Lipshultz SE. Outcome predictors for pediatric dilated cardiomyopathy: A systematic review. Prog Pediatr Cardiol. 2007;23:25–32. doi: 10.1016/j.ppedcard.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grenier MA, Osganian SK, Cox GF, Towbin JA, Colan SD, Lurie PR, Sleeper LA, Orav EJ, Lipshultz SE. Design and implementation of the North American Pediatric Cardiomyopathy Registry. Am Heart J. 2000;139:S86–95. doi: 10.1067/mhj.2000.103933. [DOI] [PubMed] [Google Scholar]
- 8.Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, Lurie PR, McCoy KL, McDonald MA, Messere JE, Colan SD. The incidence of pediatric cardiomyopathy in two regions of the United States. N Eng J Med. 2003;348:1647–1655. doi: 10.1056/NEJMoa021715. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz ML, Cox GF, Lin AE, Korson MS, Perez-Atayde A, Lacro RV, Lipshultz SE. Clinical approach to genetic cardiomyopathy in children. Circulation. 1996;94:2021–2038. doi: 10.1161/01.cir.94.8.2021. [DOI] [PubMed] [Google Scholar]
- 10.Colan SD, Parness IA, Spevak PJ, Sanders SP. Developmental modulation of myocardial mechanics: age- and growth-related alterations in afterload and contractility. J Am Coll Cardiol. 1992;19:619–629. doi: 10.1016/s0735-1097(10)80282-7. [DOI] [PubMed] [Google Scholar]
- 11.Sluysmans T, Colan SD. Theoretical and empirical derivation of cardiovascular allometric relationships in children. J Appl Physiol. 2005;99:445–457. doi: 10.1152/japplphysiol.01144.2004. [DOI] [PubMed] [Google Scholar]
- 12.Lunn M, McNeil D. Applying Cox regression to competing risks. Biometrics. 1995;51:524–532. [PubMed] [Google Scholar]
- 13.Tai BC, Machin D, White I, Gebski V. Competing risks analysis of patients with osteosarcoma: a comparison of four different approaches. Stat Med. 2001;20:661–684. doi: 10.1002/sim.711. [DOI] [PubMed] [Google Scholar]
- 14.Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio JJ, Jr, Olsen EG, Schoen FJ. Myocarditis. A histopathologic definition and classification. Am J Cardiovasc Pathol. 1987;1:3–14. [PubMed] [Google Scholar]
- 15.Burch M, Siddiqi SA, Celermajer DS, Scott C, Bull C, Deanfield JE. Dilated cardiomyopathy in children: determinants of outcome. Brit Heart J. 1994;72:246–250. doi: 10.1136/hrt.72.3.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Connuck DM, Sleeper LA, Colan SD, Cox GF, Towbin JA, Lowe AM, Wilkinson JD, Orav EJ, Cuniberti L, Salbert BA, Lipshultz SE. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: a comparative study from the Pediatric Cardiomyopathy Registry. Am Heart J. 2008;155:998–1005. doi: 10.1016/j.ahj.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freeman GL, Colston JT, Zabalgoitia M, Chandrasekar B. Contractile depression and expression of proinflammatory cytokines and iNOS in viral myocarditis. Am J Physiol Heart Circ Physiol. 1998;274:H249–258. doi: 10.1152/ajpheart.1998.274.1.H249. [DOI] [PubMed] [Google Scholar]
- 18.Drucker NA, Colan SD, Lewis AB, Beiser AS, Wessel DL, Takahashi M, Baker AL, Perez-Atayde AR, Newburger JW. Gamma-globulin treatment of acute myocarditis in the pediatric population. Circulation. 1994;89:252–257. doi: 10.1161/01.cir.89.1.252. [DOI] [PubMed] [Google Scholar]
- 19.Gagliardi MG, Bevilacqua M, Bassano C, Leonardi B, Boldrini R, Camassei FD, Fierabracci A, Ugazio AG, Bottazzo GF. Long term follow up of children with myocarditis treated by immunosuppression and of children with dilated cardiomyopathy. Heart. 2004;90:1167–1171. doi: 10.1136/hrt.2003.026641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipshultz SE, Orav EJ, Sanders SP, Colan SD. Immunoglobulins and left ventricular structure and function in pediatric HIV infection. Circulation. 1995;92:2220–2225. doi: 10.1161/01.cir.92.8.2220. [DOI] [PubMed] [Google Scholar]
- 21.Robinson J, Hartling L, Vandermeer B, Crumley E, Klassen TP. Intravenous immunoglobulin for presumed viral myocarditis in children and adults. Cochrane Database Syst Rev. 2005:CD004370. doi: 10.1002/14651858.CD004370.pub2. [DOI] [PubMed] [Google Scholar]
- 22.Cooper LT., Jr Myocarditis. N Engl J Med. 2009;360:1526–1538. doi: 10.1056/NEJMra0800028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh TP, Sleeper LA, Lipshultz S, Cinar A, Canter C, Webber SA, Bernstein D, Pahl E, Alvarez JA, Wilkinson JD, Towbin JA, Colan SD. Association of left ventricular dilation at listing for heart transplant with postlisting and early posttransplant mortality in children with dilated cardiomyopathy. Circ Heart Fail. 2009;2:591–598. doi: 10.1161/CIRCHEARTFAILURE.108.839001. [DOI] [PubMed] [Google Scholar]
- 24.Miller TL, Orav EJ, Wilkinson JD, Sleeper LA, Towbin JA, Colan SD, Hsu DT, Canter CE, Webber SA, Lipshultz S Pediatric Cardiomyopathy Registry Study Group. Nutritional status is associated with cardiac outcomes and mortality in children with idiopathic dilated cardiomyopathy. Circulation. 2009;120:S861. [Google Scholar]
- 25.Organ Procurement and Transplantation Network. Organ Distribution: Allocation of Thoracic Organs. [August 8, 2010]; http://optn.transplant.hrsa.gov/PoliciesandBylaws2/policies/pdfs/policy_9.pdf. Updated November 17, 2009.
- 26.McElhinney DB, Colan SD, Moran AM, Wypij D, Lin M, Majzoub JA, Crawford EC, Bartlett JM, McGrath EA, Newburger JW. Recombinant human growth hormone treatment for dilated cardiomyopathy in children. Pediatrics. 2004;114:e452–e458. doi: 10.1542/peds.2004-0072. [DOI] [PubMed] [Google Scholar]
- 27.Parissis JT, Adamopoulos S, Karatzas D, Paraskevaidis J, Livanis E, Kremastinos D. Growth hormone-induced reduction of soluble apoptosis mediators is associated with reverse cardiac remodeling and improvement of exercise capacity in patients with idiopathic dilated cardiomyopathy. Eur J Cardiovasc Prev Rehabil. 2005;12:164–168. doi: 10.1097/01.hjr.0000159320.70090.3d. [DOI] [PubMed] [Google Scholar]
- 28.Lipshultz SE, Vlach SA, Lipsitz SR, Sallan SE, Schwartz ML, Colan SD. Cardiac changes associated with growth hormone therapy among children treated with anthracyclines. Pediatrics. 2005;115:1613–1622. doi: 10.1542/peds.2004-1004. [DOI] [PubMed] [Google Scholar]
- 29.Cox GF, Sleeper LA, Lowe AM, Towbin JA, Colan SD, Orav EJ, Lurie PR, Messere JE, Wilkinson JD, Lipshultz SE. Factors associated with establishing a causal diagnosis for children with cardiomyopathy. Pediatrics. 2006;118:1519–1531. doi: 10.1542/peds.2006-0163. [DOI] [PubMed] [Google Scholar]