

Abstract
Hashimoto's encephalopathy (H.E.) is probably of autoimmune etiology, and manifests with seizures, stroke-like episodes, cognitive decline, neuropsychiatric symptoms, myoclonus. It is presumed to be autoimmune in origin with high serum titers of antithyroid peroxidase antibodies (anti-TPA). Thyroid function might often be normal. The diagnosis is arrived at by excluding other toxic, metabolic and infectious causes of encephalopathies, supportive clinical profile, elevated thyroid antibodies and optimum steroid response. We present the characteristic phenotypic manifestations, magnetic resonance imaging and electroechography observations and response to immunomodulation with follow-up in three cases of H.E. All the three cases manifested with subacute to chronic progressive encephalopathy, cerebellar dysfunction, seizures, behavioral abnormalities and oculomotor disturbances and had evidence of hypothyroidism, elevated titers of anti-TPA and positive thyroid anti-microsomal antibodies. Atypical and uncommon presentations are known. This report emphasizes that a high index of suspicion is often required in cases with “investigation negative encephalopathy” for early diagnosis of H.E.
Keywords: Antithyroid peroxidase antibodies, Hashimoto's encephalopathy
Introduction
Hashimoto's encephalopathy (H.E.) is an infrequent and intriguing disease of the thyroid gland, of autoimmune origin, and is characterized by seizures, stroke-like episodes, cognitive decline, neuropsychiatric symptoms, myoclonus and high serum titers of antithyroid peroxidase antibodies (anti-TPA) and corticosteroid responsiveness.[1] The therapeutic response to corticosteroids is often rewarding while some cases have also received intravenous immunoglobulin and plasmapheresis. A recent review suggests that there are less than 50 well-diagnosed cases of H.E. in the literature,[2] and reports from India are even rarer.[3]
We discuss the phenotypic manifestations, magnetic resonance imaging (MRI) and EEG observations, and response to immunomodulation in three cases of H.E.
Case Reports
Case 1
Mr. R, a 40-year-old — man, presented with progressive myoclonus for 9 months, cognitive decline and visuospatial disorientation for 8 months and dysarthria for 3 months. One month ago, he developed generalized tonic–clonic seizure, irrelevant speech, gait ataxia and startle response. He consumed alcohol occasionally. His general physical examination and vitals were normal. He scored 11/30 on Mini Mental State Examination (MMSE) and a detailed neuropsychological assessment revealed frontal and parietal lobe impairment. He had stimulus-sensitive multifocal myoclonus, limb rigidity and gait ataxia.
The routine hemogram and serum biochemistry were normal. Chest X-ray and ultrasonography (USG) (abdomen) were normal. MRI (brain) revealed diffuse leucoencephalopathy [Figure 1]. EEG was normal. Serum vasculitis, HIV and VDRL were negative. The cerebrovascular fluid (CSF) revealed two lymphocytes and raised protein (167 mg/dl; range: 25–45 mg/ dl) with normal glucose (68 mg/dl). The CSF was negative for VDRL, oligoclonal bands and antibodies against herpes simplex virus. Thyroid function tests revealed normal T3 (162 ng/dl; range: 70–187) and T4 (8.65 ug/dl; range: 4.5–13) and raised thyroid-stimulating hormone (TSH) (16.1 mU/l; range: 0.3–5.0). The serum anti-TPA level was consistently elevated to 28-times (997 IU/l; reference: <34). A thyroid scan showed diffusely enlarged gland with increased tracer uptake, suggesting hypothyroidism. A diagnosis of H.E. was entertained and he received parenteral monthly pulse methylprednisolone (1 g/day I.V. for 5 days) for the first 6 months and then once in 3 months for 6 months; thyroxine (100 ug/day) and quietapine (150 mg/ day). He started improving from the 1st month onward. The MMSE score was 30/30, with no neurological deficits at 3 months. The serum anti-TPA titer remained elevated (559 IU/l), with normal serum TSH (2.2 micg/dl). At 6 months, he was symptom-free and had normal neuropsychological evaluation. The white matter changes resolved on repeat MRI performed after six pulses of steroids, and this observation of resolution of MRI leucoencephalopathy is rarely documented. After 1½ years, he was asymptomatic, with raised serum anti-TPA (374 IU/l).
Figure 1.
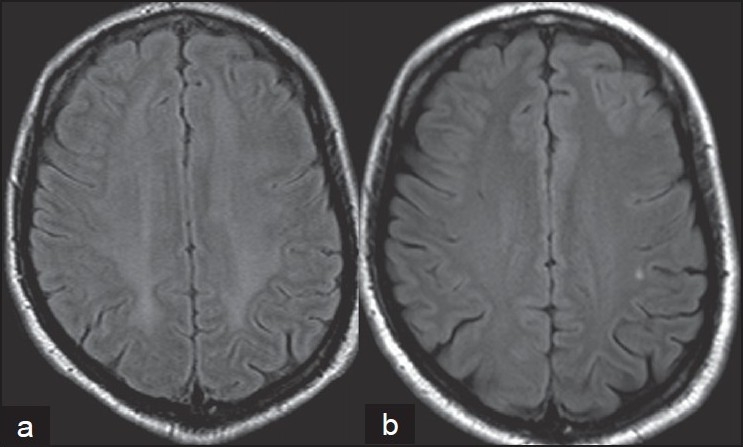
Magnetic resonance imaging (MRI) axial of the brain (FLAIR sequence) of case 1 showed (a) a symmetrical hyperintensity in the cerebral subcortical white matter with sparing of U fibers and (b) a repeat MRI carried out after 6 months revealed resolution of the lesion
Case 2
This 17-year-old girl had fever without any systemic complaints for 1 week, which improved with “over the counter” medications 3 weeks ago. About 2 weeks prior to presentation to us, she developed an acute onset of altered sensorium. She improved significantly and spontaneously over the next 3 days in sensorium, could speak, identify relatives and responded to daily needs. But, she complained of diplopia on looking to either side, with gait ataxia.
At admission, there was thyromegaly, medial deviation of both the eyes in primary position with full range of extraocular movements and ataxia. MRI (brain) was normal. The CSF study showed 12 lymphocytes/cu.mm and raised protein (52 mg/dl). The CSF study was negative for oligoclonal bands and VDRL. Serum HIV, vasculitis and VDRL were negative. Chest X-ray and USG abdomen were normal. The thyroid profile revealed low T3 (52 ng/dl) and T4 (2 ng/dl) and raised TSH (70 mU/l). The thyroid scan confirmed hypothyroidism. The thyroid antimicrosomal antibody level was positive and there was a high titer of anti-TPA (>600 IU/ml; normal: ≤34).
She was suspected to have H.E. and received monthly pulse methylprednisolone (1 g/day I.V. for 5 days) and oral thyroxine (100 ug/day). There was improvement in the situation within 2 months. After six pulses of steroids, she was symptom-free and had attained an euthyroid state. The serum anti-TPA was 316.6 IU/ml and the antimicrosomal antibody levels were positive. After 1 year, she resumed her university studies.
Case 3
This 46-year-old gentleman manifested with gait unsteadiness for 2 months and slurred speech for 20 days. He was recently detected to have hypertension. He had truncal obesity, mild spastic dysarthria, brisk reflexes, ataxic gait and reduced arm swing. Routine hemogram, basic biochemical investigations, chest X-ray and USG abdomen, MRI (brain) and EEG were normal. The CSF was acellular, with normal protein/glucose. Oligoclonal bands and VDRL were negative in the CSF. Serum vasculitis workup, HIV and VDRL were nonreactive. The thyroid profile suggested sub-clinical hypothyroidism: Low T3 (2.03 ng/ dl), T4 (88.52 ug/dl) and raised TSH (14.22 mU/l). The serum anti-TPA was markedly elevated to >600 IU/ml (normal: ≤34 IU/ml).
With a possibility of H.E., he received intravenous methyprednisolone (1 g × 5 days) and showed marked improvement in gait and speech in 2 weeks. After four pulses of parenteral methylprednisolone, he had resumed work and the serum anti-TPA was normal.
Discussion
All the three cases had manifested with subacute to chronic encephalopathy, ataxia, seizures, behavioral and oculomotor disturbances and hypothyroidism, elevated serum anti-TPA and thyroid antimicrosomal antibodies. Following high-dose steroids, all exhibited marked and sustained improvement at the 4- to 18-months follow-up. The clinical spectrum of H.E. is variable and includes myoclonus, seizures, dementia and disturbed consciousness.[1] Florid neuropsychiatric manifestations as the initial symptom have been reported.[4] Our case 1 had rapid evolution of encephalopathy that led to suspicion of Creutzfeldt-Jakob disease, limbic encephalitis, neurosyphilis, central nervous system vasculitis and HIV encephalopathy. However, all these disorders could be excluded by investigations.[5] Case 2 had presented with rapidly resolving altered sensorium at the onset, which is less often observed. The third case had manifested with subacute cerebellar syndrome. There are only few reports of such presentation in cases with H.E.[6]
Hashimoto's thyroiditis is an autoimmune disease affecting mainly middle-aged women, and a third of them are hypothyroid. In almost all cases, antibodies against thyroglobulin and/or thyroid peroxidase are detectable.[1,7] Our cases had raised serum levels of anti-TPA that remained high in two cases even after steroid therapy and clinical improvement. It is reported that even in the general population, elevated anti-TPA might be detected.[8]
The EEG findings in H.E. are nonspecific and include slowing of background, reduced amplitude of waveforms, rarely triphasic waves and photomyogenic response.[1,9] In our cases, the EEG was normal. The MRI of the brain is often normal (cases 2 and 3) and reveal reversible, diffuse, high intensity in the white matter on diffusion- and T2-weighted images, mimicking leukoencephalopathy (case 1).[10] Most cases have elevated CSF protein, the cause of which is not fully understood.[11]
The precise pathogenesis of H.E. is uncertain. Autoimmune vasculitis, possibly with deposition of immune complexes, reversible leukoencephalopathy with surrounding edema or the presence of a shared antigen in the thyroid gland and in the brain have been postulated.[1,5] Similar manifestations like H.E. have been reported with lithium-induced silent thyroiditis.[11]
Long-term maintenance steroids may be required for remission. In steroid-resistant cases, intravenous immunoglobulin or plasma-exchange could be effective.[1–3] In addition, thyroid hormonal replacement may be required in cases with hypothyroidsm. Our cases had shown a rapid improvement with steroids, which was maintained on follow-up. All were on thyroid hormone replacement.
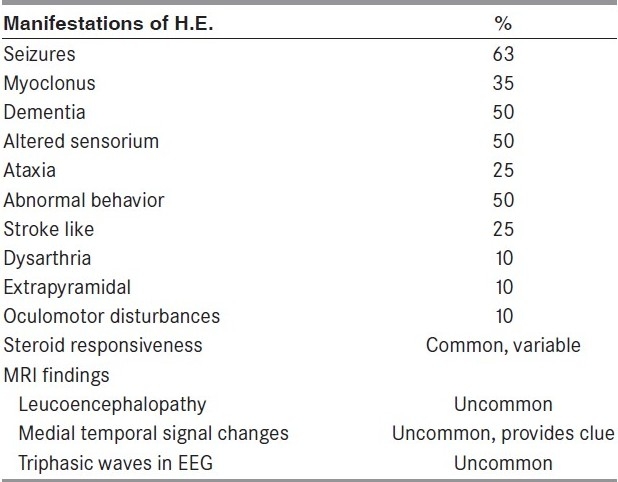
Diagnosis of H.E. requires a high index of suspicion and should be considered more often in cases with “investigation negative encephalopathy” in view of the potential therapeutic options [Table 1].[12] Thyroid antibody assessment should be carried out more often in cases manifesting with myoclonus, seizures, dementia and disturbed consciousness, with or without triphasic waves in EEG and leucoencephalopathy/medial temporal signal alterations in MRI.
Table 1.
Description of the clinical and imaging observations in Hashimoto's encephalopathy[12]
Footnotes
Source of Support: Nil
Conflict of Interest: Nil
References
- 1.Mocellin R, Walterfang M, Velakoulis D. Hashimoto's Encephalopathy: Epidemiology, pathogenesis and management. CNS Drugs. 2007;21:799–811. doi: 10.2165/00023210-200721100-00002. [DOI] [PubMed] [Google Scholar]
- 2.Marshall GA, Doyle JJ. Long term treatment of Hashimoto's encephalopathy. J Neuropsychiatry Clin Neurosci. 2006;18:14–20. doi: 10.1176/jnp.18.1.14. [DOI] [PubMed] [Google Scholar]
- 3.Nagpal T, Pande S. Hashimoto's encephalopathy: Response to plasma exchange. Neurol India. 2004;52:245–7. [PubMed] [Google Scholar]
- 4.Wilcox RA, To T, Koukourou A, Frasca J. Hashimoto's Encephalopathy Masquerading as acute psychosis. J Clin Neurosci. 2008;15:1301–4. doi: 10.1016/j.jocn.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 5.Ferraci F, Bertiato G, Moretto G. Hashimoto's Encephalopathy: Epidemiological data and pathogenetic considerations. J Neurol Sci. 2004;15:165–8. doi: 10.1016/j.jns.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 6.Nakagawa H, Yoneda M, Fujii A, Kinomoto K, Kuriyama M. Hashimoto's Encephalopathy presenting as progressive cerebellar ataxia. J Neurol Neurosurg Psychiatry. 2007;8:196–7. doi: 10.1136/jnnp.2006.093005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tamagno G, Federspil G, Murialdo G. Clinical and diagnostic aspects of encephalopathy associated with autoimmune thyroid disease. Intern Emerg Med. 2006;1:15–23. doi: 10.1007/BF02934715. [DOI] [PubMed] [Google Scholar]
- 8.Yoneda M, Fujii A, Ito A, Yokohama H, Nakagawa H, Kuriyama M. High Prevalence of Serum Auto antibodies against the amino terminal of amino enolase in Hashimoto's Encephalopathy. J Neuroimmunology. 2007;185:195–200. doi: 10.1016/j.jneuroim.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez AJ, Jicha GA, Steeves TD, Benarroch EE, Westmoreland BF. EEG changes in a case with steroid-responsive encephalopathy associated with antibodies to Thyroperoxidase (SREAT, Hashimoto's encephalopathy) J Clin Neurophysiol. 2006;23:371–3. doi: 10.1097/01.wnp.0000214542.21735.49. [DOI] [PubMed] [Google Scholar]
- 10.Okamoto K, Mori C, Kamogawa K, Tominaga K, Okuda B. Case of Hashimoto's encephalopathy with diffuse white matter lesions on diffusion-weighted MRI. Rinsho Shinkeigaku. 2007;47:112–5. [PubMed] [Google Scholar]
- 11.Nagamine M, Yoshino A, Ishii M, Ogawa T, Kurauchi S, Yoshida T, et al. Lithium-induced Hashimoto's encephalopathy: A case report. Bipolar disord. 2008;10:846–8. doi: 10.1111/j.1399-5618.2008.00605.x. [DOI] [PubMed] [Google Scholar]
- 12.Seipelt M, Zerr I, Nau R, Mollenhauer B, Kropp S, Steinhoff BJ, et al. Hashimoto's encephalitis as a differential diagnosis of Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 1999;66:172–6. doi: 10.1136/jnnp.66.2.172. [DOI] [PMC free article] [PubMed] [Google Scholar]