

Abstract
The contribution of mutations in amyloid precursor protein (APP) and presenilin (PSEN) to familial Alzheimer's disease (AD) is well established. However, little is known about the molecular mechanisms leading to amyloid β (Aβ) generation in sporadic AD. Increased brain ceramide levels have been associated with sporadic AD, and are a suggested risk factor. Serine palmitoyltransferase (SPT) is the first rate-limiting enzyme in the de novo ceramide synthesis. However, the regulation of SPT is not yet understood. Evidence suggests that it may be posttranscriptionally regulated. Therefore, we investigated the role of miRNAs in the regulation of SPT and amyloid β (Aβ) generation. We show that SPT is upregulated in a subgroup of sporadic AD patient brains. This is further confirmed in mouse model studies of risk factors associated with AD. We identified that the loss of miR-137, -181c, -9, and 29a/b-1 increases SPT and in turn Aβ levels, and provides a mechanism for the elevated risk of AD associated with age, high-saturated-fat diet, and gender. Finally, these results suggest SPT and the respective miRNAs may be potential therapeutic targets for sporadic AD.
Introduction
It is well established that amyloid precursor protein (Aβ) accumulation in familial Alzheimer's disease (AD) is due to mutations in amyloid precursor protein (APP) and presenilin (PSEN) genes (for review, see Cruts and Van Broeckhoven, 1998). However, the mechanisms contributing to Aβ accumulation in sporadic AD are less well understood. Research thus far consistently demonstrates that ceramide, a sphingolipid, is increased in AD patients (Cutler et al., 2004; He et al., 2010) and may contribute to the disease pathogenesis. Membrane ceramides not only are the major component of lipid rafts, but they also contribute to AD pathology by facilitating the mislocation of BACE1 and γ-secretase to lipid rafts, and thereby promoting amyloid β (Aβ) formation (Lee et al., 1998; Vetrivel et al., 2005). Inhibiting de novo ceramide synthesis has been shown to decrease the production of Aβ, while exogenous addition of ceramide increased Aβ production (Puglielli et al., 2003; Patil et al., 2007). Numerous studies suggest a connection between ceramides and Aβ and indicate that increased ceramide levels may be an important risk factor for sporadic AD (Puglielli et al., 2003; Mattson et al., 2005).
SPT is the first rate-limiting enzyme in the de novo ceramide synthesis pathway (Hannun and Obeid, 2008). Activation of SPT elevates ceramide levels (Perry et al., 2000) and inhibition of SPT decreases ceramide levels (Hojjati et al., 2005; Patil et al., 2007) and neuronal cell death by Aβ (Cutler et al., 2004), supporting SPT as an important regulator of ceramide. SPT is a heterodimer composed of serine palmitoyltransferase long chain 1 (SPTLC1) and either serine palmitoyltransferase long chain 2 (SPTLC2) or serine palmitoyltransferase long chain 3 (SPTLC3) (Rotthier et al., 2010). In the brain, SPTLC3 is lowly expressed, while SPTLC1 and SPTLC2 are the major subunits (Hornemann et al., 2006). However, the regulation of these subunits and in turn SPT is not well understood. Cell culture studies demonstrate that SPT activity increases in response to various stimuli (i.e., etoposide or resveratrol), but without concomitant changes in SPTLC1 and SPTLC2 mRNA levels (Perry et al., 2000; Scarlatti et al., 2003), which have led researchers to hypothesize that SPT may be posttranscriptionally regulated.
Gene expression may be posttranscriptionally regulated through miRNAs, endogenous small RNAs of 21–25 nt, that bind to 3′UTR of the target mRNA to cause translational repression or degradation of the mRNAs (He and Hannon, 2004). miRNAs have been associated with neuronal differentiation, synaptic plasticity, and memory formation (Sempere et al., 2004; Mehler and Mattick, 2006; Schratt et al., 2006). From miRNA expression profiles, several miRNAs are differentially expressed in AD patients (Lukiw, 2007; Cogswell et al., 2008; Hébert et al., 2008; Wang et al., 2008), and several have been reported to be specific or enriched in the brain (Sempere et al., 2004). Indeed, a recent study reported altered expressions of several miRNAs in response to Aβ (Schonrock et al., 2010), suggesting the involvement of miRNA in sporadic AD. Therefore, we investigated whether miRNAs mediated the posttranscriptional regulation of SPT with respect to sporadic AD.
Materials and Methods
Patient information.
The AD (n = 7) and control (n = 7) neocortical brain samples were from the University of Kentucky (UK) Alzheimer's disease center tissue bank (ADC) as frozen tissues. The samples have been clinically diagnosed by neurologists, neuropathologists, neuropsychologists, and other staff members in the ADC clinic. Most samples have been obtained in <4 h postmortem interval. All individuals were between the ages of 88–99 years. The reference number, gender, Braak stage, Mini Mental State Examination scores, frontal neuritic plaque numbers, neurofibrillary tangle numbers, and ApoE genotype of the individuals are listed in Table 1. The cause of death of these individuals is multifactorial or unclear with pneumonia being the classical cause of death. The above information was provided by the UK ADC.
Table 1.
Patient information
| Group | Ref # | Age | Sex | Braak stage | PMI | MMSE score | Front NP count | Front NFT count | ApoE allele |
|---|---|---|---|---|---|---|---|---|---|
| Control | 1132 | 95 | F | 0 | 3.50 | 26 | 3 | 0 | 3/4 |
| Control | 1159 | 86 | F | 0 | 3.50 | 28 | 0 | 0 | 3/3 |
| Control | 1165 | 92 | M | 0 | 3.33 | 28 | 10 | 0 | 3/3 |
| Control | 1187 | 85 | F | 0 | 2.50 | 28 | 0 | 0 | 3/3 |
| Control | 5163 | 96 | M | 2 | 7.25 | 27 | 0 | 0 | 2/3 |
| Control | 1206 | 94 | F | 2 | 2.25 | 27 | 0 | 0.4 | 3/3 |
| Control | 1221 | 81 | M | 2 | 2.83 | 29 | 5.6 | 0 | 3/3 |
| AD | 1013 | 80 | M | 5 | 2.50 | 5.8 | 1 | ||
| AD | 1073 | 83 | F | 5 | 4.00 | 4 | 8.6 | 3.4 | 2/4 |
| AD | 1098 | 81 | F | 5 | 2.75 | 15 | 6.2 | 2 | 3/3 |
| AD | 1174 | 96 | M | 5 | 3.50 | 22 | 19.2 | 4 | 2/3 |
| AD | 1194 | 99 | F | 5 | 2.10 | 16 | 15.4 | 3.4 | 3/4 |
| AD | 1201 | 94 | M | 5 | 2.00 | 22 | 6 | 0.2 | 3/3 |
| AD | 1215 | 91 | F | 5 | 3.00 | 13 | 15.2 | 7.2 | 3/3 |
Patient information including reference number (Ref #) age, gender, Braak stage, postmortem interval (PMI), Mini Mental State Examination (MMSE) score, neuritic plaque (NP) count, neurofibrillary tangle (NFT) count, and ApoE genotype. This information was provided by the UK ADC.
Animals.
Wild-type male C57BL/6 mice purchased from The Jackson Laboratory were used in the developmental study. Wild-type mice on a hybrid background, C3H/He (Charles River) × C57BL/6, were used in the diet (all male)- and gender-specific (7 males, 7 females) studies. All procedures conducted were approved by the Institutional Animal Care and Use Committee at Michigan State University.
Primary cell culture.
Primary astrocytes were isolated and cultured from <24-h-old wild-type Sprague Dawley rat pups and 3-week-old TgCRND8 (Centre for Research in Neurodegenerative Diseases) transgenic mouse pups, containing the APP 695-cDNA with both the Indiana and the Swedish mutations, in a hybrid C3H/He × C57BL/6 background (Chishti et al., 2001) as described previously (Patil et al., 2007). The TgCRND8 mice express the APP transgene at levels fivefold higher than the endogenous APP under the control of the Syrian hamster prion promoter (Chishti et al., 2001).
Protein extraction and Western blot analysis.
Cells, mouse brain cortices, and human brain neocortices (homogenized) were lysed in buffer: 1% (v/v) Triton, 0.1% (w/v) SDS, 0.5% (w/v) deoxycholate, 20 mm Tris, pH 7.4, 150 mm NaCl, 100 mm NaF, 1 mm Na3VO4, 1 mm EDTA, 1 mm EGTA, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor cocktail (all chemicals from Sigma). The lysis was spun at 10,000 rpm for 10 min and then the total protein concentration of the supernatant was measured by Bradford assays and was mixed with reducing loading buffer and heated at 94°C for 5 min. Immunoblot analysis was performed as described previously (Patil et al., 2007). Protein quantifications were conducted by normalizing to GAPDH or β-actin. Western blots were quantified using Quantity One (Bio-Rad) version 4.5.
ELISA.
Protein was extracted from human autopsy brain samples and ELISA was performed for human Aβ42 using KHB3441 (Invitrogen) according to the manufacturer's instruction. The Aβ42 levels were calculated by normalizing to the total protein levels measured by Bradford's assay.
Transfection, plasmid, and luciferase assays.
Primary wild-type and transgenic astrocytes were plated in 12-well plates and transfected for 24–72 h with 100–150 nm Syn-miRNA miScript miRNA mimic or anti-miR-RNA miScript miRNA inhibitor (Qiagen), 500 ng to 2 μg of human SPTLC1 cDNA, or 1.5–2 μg of luciferase vector construct using Lipofectamine RNAi/MAX or Lipofectamine 2000 following the manufacturer's instructions. The SPTLC1 cDNA plasmid and SPTLC1 and SPTLC2 luciferase 3′UTR expression clones, containing the luciferase reporter gene and Renilla tracking gene and driven by the SV40 promoter, were purchased from Genecopoeia. The luciferase assay was conducted with a dual luciferase assay kit (Luc-Pair miR Luciferase Assay Kit) (Genecopoeia). The luciferase expression levels were normalized to Renilla expression levels.
Quantitative RT-PCR.
Total mRNA was extracted using RNeasy Plus Mini Kit (Qiagen) and total RNA was quantified using ND-1000 NanoDrop spectrophotometer. RNA quality control was performed by assessing OD 260/280 ratio. RNA quality control of the control and AD human brain samples was performed using the Agilent Bioanalyzer 2100. In addition, the PCR products were run on agarose gels. Quantitative RT-PCR (qRT-PCR) was conducted using iQSYBR Green Supermix (Bio-Rad) and MyiQ real-time PCR detection system following reverse transcription using iScript cDNA Synthesis Kit according to the manufacturer's instructions. Primers include human SPTLC1: 5′-TGGAAGAGAGCACTGGGTCT-3′ and 5′-GCTACCTCCTTGATGGTGGA-3′; human SPTLC2: 5′-GAGACGCCTGAAAGAGATGG-3′ and 5′-TGGTATGAGCTGCTGACAGG-3′; human GAPDH: 5′-GAGTCAACGGATTTGGTCGT-3′ and 5′-TTGATTTTGGAGGGATCTCG-3′; mouse Sptlc1: 5′-AGTGGTGGGAGAGTCCCTTT-3′ and 5′-CAGTGACCACAACCCTGATG-3′; mouse Sptlc2: 5′-CCTGTCAGCAGCTCATACCA-3′ and 5′-CACACTGTCCTGGGAGGAAT-3′; mouse Gapdh: 5′-AACTTTGGCATTGTGGAAGG-3′ and 5′-ACACATTGGGGGTAGGAACA-3′; rat Sptlc1: 5′-ACCTGGAGCGACTGCTAAAA-3′ and 5′-ATCCCATAGTGCTCGGTGAC-3′; rat Sptlc2: 5′-TTGAGACTCACTGGCCCTCT-3′ and 5′-GGCCAGGAGGAGTCACATAA-3′; rat Gapdh: 5′-AGACAGCCGCATCTTCTTGT-3′ and 5′-CTTGCCGTGGGTAGAGTCAT-3′. Relative human, mouse, and rat SPTLC1 and SPTLC2 expressions were calculated using the comparative CT method normalizing to their corresponding GAPDH expressions.
Total miRNAs were extracted using miRNeasy Mini Kit (Qiagen) and RNeasy MinElute Cleanup Kit (Qiagen) total RNA was quantified using ND-1000 NanoDrop spectrophotometer. RNA quality control was performed by assessing OD 260/280 ratio. In addition the PCR products were run on agarose gels. qRT-PCR was conducted using miScript SYBR Green PCR Kit (Qiagen) and MyiQ real time PCR detection system following reverse transcription using miScript Reverse Transcription Kit (Qiagen) according to the manufacturer's instructions. All miRNA primers were purchased from Qiagen, and the relative expressions were calculated using the comparative CT method using RNU6B as the normalizing control.
Ceramide quantification.
Lipids were extracted from homogenized human brain neocortices and mouse brain cortices according to Bligh and Dyer (1959). Tandem mass spectrometry (MS/MS) was performed using Quattro Premier XE (Waters), Acquity ultra performance liquid chromatography (Waters) (LC-MS/MS), and Mass Lynx 4.1 software. External ceramide standards were purchased from Matreya and Avanti, Polar Lipids. C12:0 (Avanti, Polar Lipids) was used as the internal standard.
Antibodies.
The following antibodies were used: LCB1 (BD Transduction Laboratories), SPTLC1 (Proteintech), SPTLC2 (Abcam), GAPDH (Cell Signaling Technology), β-actin (Sigma), β-amyloid (Cell Signaling Technology), and β-amyloid-4G8 clone (Covance).
Statistical analysis.
Statistical significances were determined by using two-tailed t tests and Spearman correlation (two-tailed t distribution test).
Results
Elevated ceramide and SPT expression in a subgroup of sporadic AD patients
The levels of ceramide and SPT protein expression were measured in the frontal brain cortices of seven sporadic AD patients and seven controls (see Table 1 for information on the patients). Of the vast number of distinct ceramide species (>50 species), d18:1;18:0 and d18:1;16:0 are reported to be the major sphingolipid species in rat neurons (Valsecchi et al., 2007) and human brain (Ladisch et al., 1994). Consistent with previous reports (Cutler et al., 2004; He et al., 2010), ceramide levels, d18:1;16:0 (p = 0.037, Student's t test) and d18:1;18:0 (p = 0.033), were significantly increased in this subgroup of AD patients (Fig. 1A). Several reports have shown that the sphingomyelin levels either increased (Pettegrew et al., 2001; Bandaru et al., 2009) or remained unchanged (Han et al., 2002) in AD brains. In contrast, other researchers have shown that the sphingomyelin levels decreased (Cutler et al., 2004; He et al., 2010) in AD brain. We found that the sphingomyelin d18:1;16:0 levels increased (p = 0.045), while the d18:1;18:0 levels remained unchanged, in the subgroup of AD patient brain cortices studied (Fig. 1B). This suggests that the increased ceramide levels in these patients are from the de novo synthesis pathway. Accordingly, SPTLC1 (p = 0.004) and SPTLC2 (p = 0.007) protein expression were significantly elevated in the autopsy AD brain samples (Fig. 1C,D). However, SPTLC1 and SPTLC2 mRNA levels remained predominantly unchanged in the AD samples as measured by qRT-PCR (Fig. 1E).
Figure 1.

SPTLC1 and SPTLC2 are upregulated in sporadic AD brain. A, Ceramide levels, d18:1;16:0 and d18:1;18:0, were significantly increased in the subgroup of AD patients (n = 7). B, Sphingomyelin d18:1;16:0 levels were significantly increased, while d18:1;18:0 levels were unchanged, in the subgroup of AD patients. The frontal brain cortices were analyzed via tandem mass spectrometry and the normalized concentrations are shown as a percentage of the average control (n = 7). The samples were normalized to internal standard (d18:1, 12:0) concentration and to brain total protein concentration. C, Western blot of SPTLC1 (probed with LCB1 antibody), SPTLC2, and Aβ-3 kDa fragment (probed with 4G8 antibody). See Table 1 for neuritic plaque numbers in individual brains. D, SPTLC1 and SPTLC2 protein levels of frontal cortices of control brains (n = 7) versus sporadic AD brains (n = 7) were determined by Western blotting, and the expression levels were quantified by normalizing to GAPDH and are represented as percentages of the control brain average expression. E, qRT-PCR of SPTLC1 and SPTLC2 mRNA from control (n = 7) and sporadic AD patients (n = 7). Relative expressions shown are normalized to GAPDH and control brain average expressions. F, SPTLC1 and SPTLC2 protein quantification of wild-type primary rat astrocytes treated with palmitate for 24 h. Error bars represent SE derived from three independent experiments, normalized to β-actin and represented as a percentage of the control treatment (BSA) expression (*p < 0.05). G, qRT-PCR of Sptlc1 and Sptlc2 mRNA from wild-type primary rat astrocytes treated with palmitate and BSA. Relative expressions shown are normalized to Gapdh and expressions in BSA control treatment. Error bars represent SE derived from six or more independent experiments. The statistical significance between control and AD brains, and between palmitate and BSA treatments, was determined by two-tailed Student's t tests.
Previously, our group found that palmitate (a saturated fatty acid) increased de novo ceramide synthesis in astrocytes through SPT (Patil et al., 2007). Thus, we treated wild-type primary rat astrocytes with palmitate for 24 h and found that the SPTLC1 (p = 0.032) and SPTLC2 (p = 0.015) protein levels (Fig. 1F) increased without a concomitant change in their mRNA levels (Fig. 1G), which is consistent with previous reports (Perry et al., 2000; Scarlatti et al., 2003). Overall, these results support that increased SPTLC1 and SPTLC2 expressions may be posttranscriptionally regulated in a subgroup of sporadic AD patients and in primary astrocytes cultured with palmitate. Thus, we proceeded to further elucidate the potential regulation of SPT by miRNAs.
SPTLC1 and SPTLC2 are miRNA targeted genes
Prediction algorithms miRbase (Griffiths-Jones et al., 2008), Targetscan (Lewis et al., 2005), Pictar (Krek et al., 2005), and miRanda (Betel et al., 2008) were used to select potential miRNAs that bind the human 3′UTR of SPTLC1 or SPTLC2 with strongly conserved (in mammals) target sites. Likely miRNA candidates were filtered according to the following criteria: they must be (1) predicted by at least two algorithms and (2) downregulated in AD patients or enriched in the brain. Of the miRNAs predicted by two or more algorithms to bind the 3′UTR of SPTLC1, miR-15a and miR-181c (Hébert et al., 2008) are reported to be downregulated in sporadic AD patients, while miR-137 and miR-124 (Sempere et al., 2004) are reported to be enriched in the brain. Of the miRNAs predicted by two or more algorithms to bind the 3′UTR of SPTLC2, miR-29a, miR-29b-1, and miR-9 are reported to be downregulated in sporadic AD patients, while miR-9 is also reported to be enriched in the brain.
Two luciferase reporter constructs were generated containing the 3′UTR of human SPTLC1 or SPTLC2. The miRNAs (sense) were cotransfected with the constructs and the luciferase expression was detected in wild-type primary rat astrocytes. While miR-137 (p = 0.000016, Student's t test) and miR-181c (p = 0.0003) significantly decreased the luciferase expression of the construct containing the 3′UTR of SPTLC1, miR-15a and miR-124 did not (Fig. 2A). The luciferase expression of the construct containing the 3′UTR of SPTLC2 decreased significantly upon cotransfection with miR-9 (p = 1.023 × 10−8), miR-29a (p = 1.2 × 10−7), or miR-29b-1 (p = 0.007) (Fig. 2B). These results were confirmed by transfecting primary rat astrocytes with either the sense-miRs or anti-miRs (antisense) of their respective miRNAs following analysis of the endogenous miRNA expression levels in primary rat astrocytes (Fig. 2I). miR-137 and miR-181c significantly suppressed the endogenous SPTLC1 expression and cellular ceramide, levels while anti-miR-137 and anti-miR-181c significantly enhanced the endogenous SPTLC1 (Fig. 2C,E) and cellular ceramide (Fig. 2G) levels upon transient transfection. Similarly, miR-9, miR-29a, and miR-29b-1 significantly suppressed the endogenous SPTLC2 and cellular ceramide levels, while anti-miR-9 and anti-miRs-29a/b-1 significantly enhanced the endogenous SPTLC2 (Fig. 2D,F) and cellular ceramide (Fig. 2H) levels upon transient transfection.
Figure 2.

SPTLC1 and SPTLC2 are miRNA targeted genes. A, Human SPTLC1 3′UTR luciferase and Renilla luciferase constructs were transfected into wild-type rat primary astrocytes with the indicated miRNA oligonucleotides at a final concentration of 100 nm. Normalized (to Renilla) sensor luciferase activity is shown as a percentage of the SPTLC1 3′UTR plasmid. Error bars represent SEs derived from three or more experiments (*p < 0.02). B, Human SPTLC2 3′UTR luciferase and Renilla luciferase constructs were transfected into wild-type rat primary astrocytes with the indicated miRNA oligonucleotides at a final concentration of 100 nm. Normalized (to Renilla) sensor luciferase activity is shown as a percentage of the SPTLC2 3′UTR plasmid. Error bars represent SEs derived from four or more experiments (*p < 0.007). C, D, Representative Western blot analysis of endogenous SPTLC1 (probed with LCB1) and SPTLC2 in wild-type primary astrocytes treated with 100 nm (final concentration) of miRNA (oligonucleotides) or anti-miRs (antisense), with scrambled siRNA as controls. E, F, Quantification of endogenous SPTLC1 and SPTLC2 protein levels by normalizing to β-actin or GAPDH. G, H, Cellular ceramide levels of primary rat astrocytes transfected with miRs or anti-miRs. Cellular ceramide levels were analyzed via tandem mass spectrometry and the normalized concentrations are shown as percentages of the scrambled siRNA. The samples were normalized to internal standard (d18:1, 12:0) concentration and to cellular total protein concentration. Error bars represent SEs derived from three or more experiments conducted with 48–72 h transient transfections astrocytes, neurons, and 6-month-old mouse brain cortical endogenous miRNAs expression levels were quantified by qRT-PCR. Relative expressions shown are normalized to RNU6B. Statistical significance between scrambled siRNA and candidate miRs treated primary astrocytes was determined by two-tailed Student's t tests.
Changes in miRNA correlate with SPT expression in AD
The expression levels of miR-137 (p = 0.006, Student's t test), miR-181c (p = 0.006), miR-9 (p = 0.045), miR-29a (p = 0.03), and miR-29b-1 (p = 0.03) (Fig. 3A–C) and miR-15 (p = 0.048) and miR-124 (p = 0.002) (data not shown) were significantly downregulated in the frontal cortices of the subgroup of sporadic AD patient.
Figure 3.
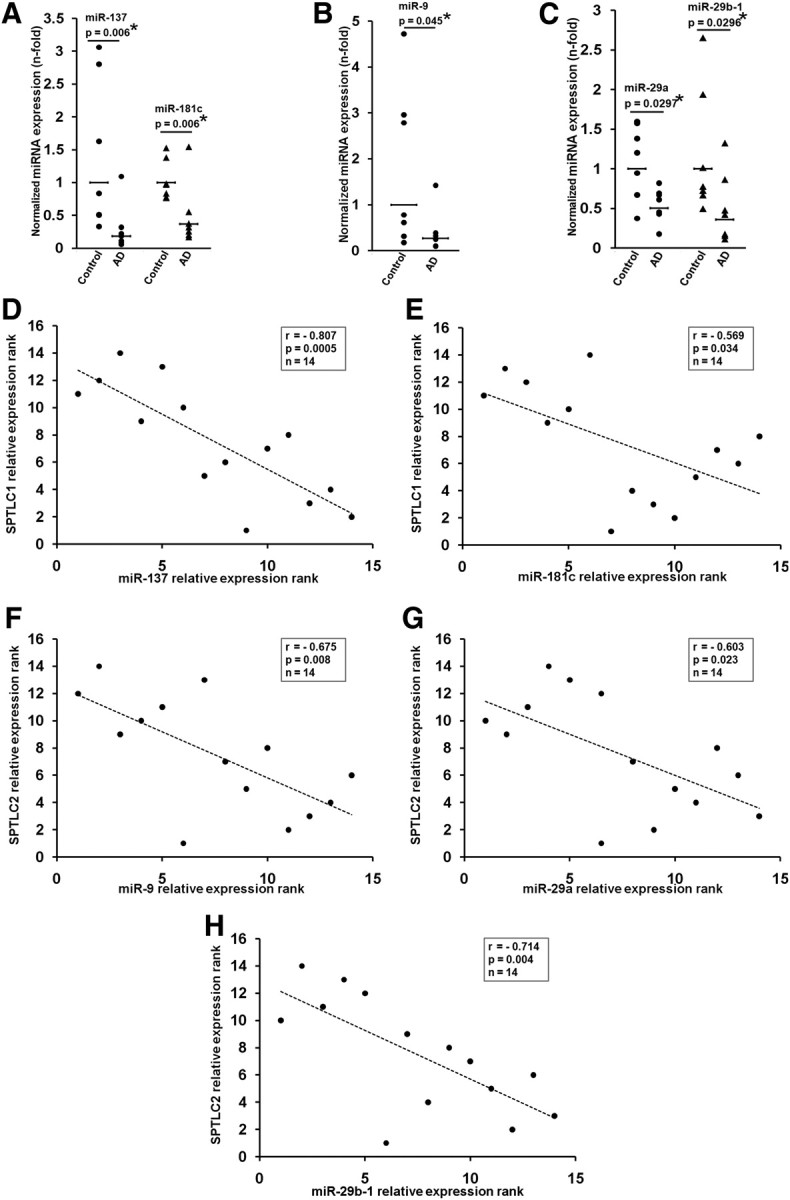
Misregulation of miR-137, -181c, -9, and -29a/b-1 in AD brain. A–C, miR-137 and -181c (A), miR-9 (B), and miR-29a and -29b-1 (C) levels were quantified by qRT-PCR in controls (n = 7) and AD (n = 7) frontal brain cortices. Relative expressions shown are normalized to RNU6B and average control brain expressions. The statistical significance between control and AD brains were determined by two-tailed Student's t tests. D, E, Spearman's correlation test demonstrates significant negative correlation between miR-137, -181c, and SPTLC1 expression in the entire sample set (control and AD) (n = 14). F–H, Spearman's correlation test demonstrates significant negative correlation between miR-9, -29a, -29b-1, and SPTLC2 expression in the entire sample set (control and AD) (n = 14). The significance of the correlation was determined by two-tailed t distribution tests.
Statistically significant negative correlations were observed between SPTLC1 and miR-137 (r = −0.807, p = 0.0005, Spearman's correlation) (Fig. 3D), miR-181c (r = −0.569, p = 0.034) (Fig. 3E), and miR-15a (r = −0.59, p = 0.026) and miR-124 (r = −0.67, p = 0.009) (data not shown) in the control and the subgroup of AD patients. Significant negative correlations were also observed between SPTLC2 and miR-9 (r = −0.675, p = 0.008) (Fig. 3F), miR-29a (r = −0.603, p = 0.023) (Fig. 3G), and miR-29b-1 (r = −0.714, p = 0.004) (Fig. 3H) in the subgroup of AD patients. This negative correlation between the subunits of SPT and their corresponding miRNA expressions, coupled with the transient transfection results, suggests the possibility that changes in miR-137 or miR-181c, and miR-9, miR-29a, or miR-29b-1 contribute, at least in part, to the overall protein expressions of SPTLC1 and SPTLC2, respectively, in AD.
Developmental coregulation of miRNA and SPT in brain
Given that AD is an age-related disorder (Bachman et al., 1992), we assessed the expressions of SPTLC1 (Fig. 4A,B), SPTLC2 (Fig. 4A,C), and their corresponding miRNAs (Fig. 4D–F) with development. The protein, mRNA, and miRNA expressions were evaluated in wild-type mouse brain cortices from postnatal day 0 (P0) up to 18 months. This provided an independent confirmation of the correlation between SPTLC1, SPTLC2, and their respective miRNAs under nonpathological settings. During development, the expression levels of miR-137 and miR-181c (Fig. 4D) and miR-124 (data not shown) increased, while SPTLC1 expression levels decreased, with age (Fig. 4A,B). Consistent with previous reports (Hébert et al., 2008), expression levels of miR-29a and miR-29b-1 were found to increase (Fig. 4E) with development, while the expression levels of SPTLC2 decreased with age (Fig. 4A,C). The Sptlc1 and Sptlc2 mRNA expression levels remained unchanged (stable) over the period analyzed (Fig. 4F), a signature of miRNA regulation. These results suggest that miR-137, miR-181c, miR-29a, and miR-29b-1 are developmentally regulated, with the highest expressions in adult mice. Concomitantly, protein analyses indicate that SPTLC1 and SPTLC2 have lower expression levels in adult mice, thereby further supporting a negative relationship between SPTLC1/2 and their corresponding miRNAs.
Figure 4.

Developmental coregulation of miR-137, -181c, -29a, and 29b-1, SPTLC1, and SPTLC2. A, Western blot analysis of SPTLC1 and SPTLC2 of P0 to 18-month-old mouse brain cortices (n = 3 for each age group). Gapdh was used as the loading control. B, Quantification of SPTLC1 expression from Western blots normalized to GAPDH and the average expressions represented as percentages of average P0 expression. C, Quantification of SPTLC2 expression from Western blots normalized to GAPDH (n = 3 for each age group) and the average expressions represented as percentages of average P0 expression. D, E, Relative expression levels of miR-137 and -181c (D) and miR-9, -29a, and -29b-1 (E) were measured by qRT-PCR in mouse brain cortices with RNU6B used as the normalizing control. F, Relative mRNA expression levels of Sptlc1 and Sptlc2 were measured by qRT-PCR with Gapdh as the normalizing control.
A high-fat diet increases SPT expression with decreased miRNA expression
Increasing evidence in animal models suggest that a high-fat diet aggravates the Aβ burden and thereby the AD pathology (Julien et al., 2010). Indeed, high-fat/high-cholesterol diets have been found to increase plasma ceramide levels in rodents (Shah et al., 2008). Moreover, prior research in our laboratory demonstrated that palmitate, a saturated fatty acid, increases ceramide levels and induces AD-like pathology in primary neuronal cell culture mediated by astrocytes (Patil et al., 2007). Therefore, the expression levels of ceramide, SPTLC1, and SPTLC2 and their corresponding miRNAs were measured in brain cortices of wild-type male mice fed a 60% kcal high-fat diet for a period of 5 months (starting at 4 months of age). While ceramide (Fig. 5A) and SPTLC1 and SPTLC2 (Fig. 5B) expression levels increased in mice fed a high-fat diet, Sptlc1 and Sptlc2 mRNA levels remained unchanged (Fig. 5C), supporting our hypothesis that SPTLC1/2 may be posttranscriptionally regulated by miRNAs. Indeed, miR-137 (p = 0.005, Student's t test) (Fig. 5D), miR-181c (p = 0.026) (Fig. 5D), miR-15a (p = 0.01) (data not shown), and miR-9 (p = 0.0027) (Fig. 5E) expression levels were downregulated in mice fed a high-fat diet. In agreement with our in vivo results, miR-137 (p = 5.6 × 10−5) (Fig. 5F), miR-181c (p = 2.2 × 10−6) (Fig. 5F), and miR-9 (p = 1.9 × 10−5) (Fig. 5G) expression levels were downregulated in wild-type primary rat astrocytes treated with palmitate, whereas SPTLC1/2 protein expression levels were upregulated (Fig. 1F). However, miR-29a and miR-29b-1 expressions did not change with either a high-fat diet (in vivo) or palmitate treatment (in vitro).
Figure 5.

Regulation of miR-137, -181c, and -9, SPTLC1, and SPTLC2 with diet. Male wild-type mice were fed a 60% kcal diet for a period of 5 months starting at 4 months of age. A, Ceramide levels, d18:1;16:0 and d18:1;18:0, were significantly increased in mice fed a high-fat diet (n = 3). The brain cortices were analyzed via tandem mass spectrometry and the normalized concentrations are shown as a percentage of the average control chow diet-fed mice (n = 3). The samples were normalized to internal standard (d18:1, 12:0) concentration and to brain total protein concentration. B, Western blot analysis of SPTLC1 and SPTLC2 in brain cortices of mice fed a control diet (n = 3) or high-fat diet (n = 3). GAPDH was used as the loading control. C, Sptlc1 and Sptlc2 mRNA expression levels were measured by qRT-PCR. Relative expressions shown are normalized to Gapdh and average expression levels in the chow control diet. D, E, The expression levels of miR-137, -181c, -9 (*p < 0.04), -29a, and -29b-1 in high-fat diet-fed mouse cortices were measured by qRT-PCR. Relative expressions shown are normalized to RNU6B and average chow control diet expressions. F, G, The expression levels of miR-137, -181c, -9 (*p < 6.0 × 10−5), -29a, and -29b-1 in palmitate-treated astrocytes were measured by qRT-PCR. Relative expressions shown are normalized to RNU6B and BSA control treatment expressions. Error bars represent SEs derived from six or more experiments. The statistical significance between control chow diet and high-fat diet, and between palmitate and BSA treatments, was determined by two-tailed Student's t tests.
SPT and miRNA are differentially expressed with respect to gender
Evidence suggests that AD pathology may be more prevalent in females than in males (Bachman et al., 1992). Therefore, we evaluated the SPTLC1, SPTLC2, and miRNA expression levels in the brain cortices of female and male wild-type mice (9 months of age). Ceramide species, d18:1;18:0 (p = 0.0042, Student's t test) and d18:1;16:0 (p = 0.0045, Student's t test) (Fig. 6A), and SPTLC1 (p = 0.018) and SPTLC2 (p = 0.014) (Fig. 6B,C) protein expression levels were higher in females as compared to males, while the Sptlc1 and Sptlc2 mRNA levels remained unchanged (Fig. 6D), further indicating that SPTLC1/2 may be posttranscriptionally regulated by miRNAs. Concomitantly, miR-137 (p = 0.011) and miR-181c (p = 0.038) (Fig. 6E), miR-124 (data not shown), and miR-29a (p = 0.031) and miR-29b-1 (p = 0.004) (Fig. 6F), but not miR-9, expression levels are downregulated in female mice, while SPTLC1/2 protein expression levels are increased, further supporting a negative relationship between SPTLC1/2 and their target miRNAs.
Figure 6.

Gender-specific differential regulation of miR-137, -181c, -29a, 29b-1, SPTLC1, and SPTLC2. A, Ceramide species d18:1;18:0 and d18:1;16:0 were measured in 9-month-old male (n = 7) and female (n = 7) wild-type mouse brain cortices by tandem mass spectrometry, and the normalized concentrations are shown as percentages of the average male concentration (n = 7). The samples were normalized to internal standard (d18:1, 12:0) concentration and to brain total protein concentration (*p < 0.005). B, C, SPTLC1 and SPTLC2 protein quantification from Western blots of the brain cortices of females normalized to GAPDH and the average expression levels represented as percentages of the average male expression levels (*p < 0.02). D, Sptlc1 and Sptlc2 mRNA expression levels were measured by qRT-PCR. Relative expressions shown are normalized to Gapdh and average male expressions. E, F, The expression levels of miR-137 and -181c (*p < 0.04) (E) and miR-29a/b-1 (*p < 0.04) and -9 (F) were measured by qRT-PCR. Relative expressions shown are normalized to RNU6B and average male expressions. The statistical significance between male and female expressions was determined by two-tailed Student's t tests.
miRNA modulates SPT and Aβ
A casual relationship between miR-29a/b-1, BACE1 activity, and Aβ has been established by Hébert et al. (2008). Therefore, we assessed whether a relationship exists between miR-137/181c and Aβ, mediated by SPTLC1. Statistically significant positive correlations were observed between SPTLC1 (Western blot) and Aβ(42) protein levels (from ELISA) (r = 0.76, p = 0.002, Spearman's correlation) (Fig. 7A), and between SPTLC2 (Western blot) and Aβ(42) protein levels (r = 0.67, p = 0.007) (Fig. 7B) in the control and the subgroup of AD patients. Additionally, statistically significant negative correlations were observed between Aβ(42) and miR-137 (r = −0.75, p = 0.003), miR-181c (r = −0.57, p = 0.037), miR-9 (r = −0.7, p = 0.007), miR-29a (r = −0.64, p = 0.01), and miR-29b-1 (r = −0.569, p = 0.037) in the control and the subgroup of AD patients. Furthermore, we performed gain- and loss-of-function experiments in primary astrocytes derived from transgenic mice expressing the human APP Swedish mutation. In these cells, overexpressing miR-137 or miR-181c downregulated the endogenous expression levels of SPTLC1 (p = 0.001) and Aβ (p = 0.01) (Fig. 7D,F). The functional affects were reversed upon transfection with the complementary anti-miRs-137 and -181c (Fig. 7C,E). Thus, the loss of the suppressing activity of miR-137 and miR-181c led to increased Aβ production in cell culture. Additionally, transient overexpression of SPTLC1 (p = 0.033) restored/increased Aβ expression levels in cells cotransfected with miR-137/-181c (p = 0.005) (Fig. 7D,F). To assess the direct role of miR-137, miR-181c, and thus SPTLC1 on Aβ expression, “target protectors” were designed against the targeted site on SPTLC1 for miR-137 and miR-181c. Primary astrocytes expressing the human APP Swedish mutation were transiently transfected with miR-137 or miR-181c along with their respective “target protectors” (Fig. 7G–J). Both SPTLC1 and Aβ expression levels decreased significantly upon transfection with miR-137 (Fig. 7G,I) or miR-181c (Fig. 7H,J) along with a negative target protector. SPTLC1 and Aβ expression levels remained unchanged upon transfection with miR-137 (Fig. 7G,I) or miR-181c (Fig. 7H,J) along with their respective target protectors. Additionally, the transfection of anti-miR-137 (Fig. 7G,I) or anti-miR-181c (Fig. 7H,J) significantly increased Aβ and SPTLC1 expression levels.
Figure 7.

Modulation of SPTLC1 and Aβ by miR-137 and -181c. A, B, Spearman's correlation test demonstrates significant positive correlation between SPTLC1 protein (Western blot) and Aβ(42) protein (ELISA) expressions (A) and between SPTLC2 protein (Western blot) and Aβ(42) protein (ELISA) expressions (B) in the brain cortices of the subgroup of sporadic AD samples (control and AD) n = 14). The significance of the correlation was determined by two-tailed t distribution tests. C, E, Western blot analysis of SPTLC1 (probed with LCB1 antibody) and cellular Aβ (3 kDa-probed with Aβ antibody) levels in primary astrocytes expressing APP Swedish mutation following transfection with miRs alone or in combination with anti-miRs. D, F, Western blot analysis of SPTLC1 (probed with LCB1 antibody) and cellular Aβ (3 kDa-probed with Aβ antibody) levels in primary astrocytes expressing APP Swedish mutation following transfection with miRNA alone or in combination with human SPTLC1 cDNA. G–J, miR-137 along with either negative or positive “target protectors” or anti-miR-137 (G, I) and miR-181c along with either negative or positive “target protectors” or anti-miR-181c (H, J) were transfected into primary astrocytes expressing APP Swedish mutation. β-Actin or GAPDH was used as the loading controls. Error bars represent SEs derived from three or more experiments conducted with 48 h transfections (final concentration of 100 nm). Statistical significance between scrambled siRNA- and candidate miR-treated primary astrocytes was determined by two-tailed Student's t tests.
Discussion
We found that a subgroup of sporadic AD patients exhibited increased levels of ceramides (this study; Cutler et al., 2004; He et al., 2010), suggesting that ceramide may be a potential target for the treatment of AD. Increased ceramide levels have been associated with increased neutral SMase (N-SMase) levels in AD where Aβ induced N-SMase production (Jana and Pahan, 2010). In this study, we observed that the Aβ levels increased with overexpression of SPTLC1. Therefore, ceramide rise through the de novo synthesis pathway upregulates Aβ levels, and the Aβ in turn may induce N-SMase activity to reinforce the production of ceramide, and thereby propagate a continual cycle of ceramide-Aβ generation.
We identified that a subgroup of sporadic AD patients exhibit increased levels of ceramides with concomitant increase in SPTLC1 and SPTLC2 protein expression levels in their brain cortices. This coupled with our animal and cell culture studies suggests that SPT may be a novel target for the treatment of AD. Further, SPTLC1/2 mRNA levels in these AD patient samples did not differ significantly from the levels in the control samples. This in combination with the luciferase assays and primary cell culture data suggests that SPTLC1/2 may be posttranscriptionally regulated through miRNAs. Along these lines, we found negative correlations/relationships between the expression levels of miR-137/-181c and SPTLC1, and between miR-9/-29a/b-1 and SPTLC2 protein expressions, in sporadic AD brains, and developing diet- and gender-specific mouse brains.
Apart from changes in miRNA expressions, binding sites for the transcription factor NFκB have been identified in the promoter region of Sptlc2 (Chang et al., 2011). However, this regulation may be tissue and stimuli specific as the tested experimental conditions did not impact brain SPT activity (Memon et al., 2001). Further, a significant increase in SPTLC2 protein levels was observed in human glioma tissue with only a slight increase in SPTLC2 mRNA (An et al., 2009). Interestingly, miR-29b is downregulated in glioblastomas (Cortez et al., 2010), suggesting that miR-29b could be involved in elevating the SPTLC2 protein levels. In this present study, we observed that SPTLC1 and SPTLC2 mRNA expression levels remained unchanged in the brain cortices of AD patients, as well as in primary astrocytes treated with palmitate and in mice fed a high-fat diet. This was also observed with development and in both genders of mice. Further, correlation analyses coupled with the transfection studies in cells suggest that changes in the miRNA levels, miR-137, -181c, -9, and 29a/b-1, could contribute to altered SPTLC1 and SPTLC2 expression levels in this subgroup of sporadic AD patient samples.
Of the miRNAs identified to regulate SPT expression, increased expression levels of miR-137 has been shown to induce neurogenesis in hippocampus (Szulwach et al., 2010) while miR-9 is involved in neurogenesis and differentiation (Coolen and Bally-Cuif, 2009; Gao, 2010). In addition, Hébert et al. (2008) and Cogswell et al. (2008) observed downregulated miR-9 levels in AD patient brains, whereas Lukiw (2007) detected an upregulation in AD. In contrast, miR-29a/b-1 was observed to be consistently downregulated by Hébert et al. (2008), Wang et al. (2008), and Shioya et al. (2010) in AD brains. Similarly, Hébert et al. (2008) also detected downregulated miR-181c expression levels in these patients. Consistent with these reports, we found that miR-181c, -9, and 29a/b-1 levels are downregulated in the frontal cortex of the subgroup of AD patients in this study. In addition, we observed that miR-137 was also downregulated in the frontal cortex of these seven AD patients. In support of this, chromosomes 1p13.3–q31.1 region, which includes the map location of miR-137, chromosome 1p21, has been linked to late-onset AD (Butler et al., 2009). The map location of miR-181c, chromosome 19p13.13, has also been linked to late-onset AD (Butler et al., 2009). We observed that the suppression of SPTLC1 by miR-137 and miR-181c reduced Aβ expression levels in a target-specific manner, while overexpression of SPTLC1 and inhibition of miR-137 and miR-181c increased Aβ expression levels. This, coupled with the fact that overexpression of BACE1 did not increase Aβ levels even though it increased β-CTF levels (Hébert et al., 2008), leaves open a possible role of ceramide, mediated by SPT, in transporting BACE1 and γ-secretase to the lipid rafts for amyloidogenic processing of APP. Inactive BACE1 and γ-secretase resides outside of the lipid rafts under nonpathological settings, allowing nonamyloidogenic processing of APP, while under disease state the ceramides facilitate the trafficking of these pathogenic secretases to lipid rafts, where they become active to produce Aβ (Cordy et al., 2003; Vetrivel et al., 2005; Ebina et al., 2009). Ceramide also increases Aβ production by stabilizing BACE1 (Puglielli et al., 2003; Costantini et al., 2007; Patil et al., 2007) through increased acetylation (Ko and Puglielli, 2009). miR-9 and miR-29a/b previously have been identified as potential suppressors of BACE1 and thus associated with sporadic AD (Hébert et al., 2008). Given that SPT is also regulated by miR-9 and miR-29a/b-1, it further strengthens the contribution of SPT to the etiology of sporadic AD.
Of the miRNAs identified to regulate SPTLC1 expression, miR-137 was shown to be negatively regulated in adult neural stem cells, epigenetically and transcriptionally by MeCP2 and Sox2 through direct binding to the 5′ regulatory region (Szulwach et al., 2010). Research conducted to treat Rett syndrome, a disease caused by mutations in MeCP2, indicates that a high-fat diet may increase MeCP2 levels (Haas et al., 1986; Liebhaber et al., 2003), providing a potential explanation for the reduced miR-137 levels in mice fed a high-fat diet. The other miRNA identified to regulate SPTLC1, miR-181c, is positively regulated by Akt1 at the transcriptional level (Androulidaki et al., 2009). Akt activity is reduced in response to high-fat diet (Tremblay et al., 2001), providing a possible mechanism for our observations that miR-181c expression levels are reduced in mice fed a high-fat diet. Of the miRNAs identified to regulate SPTLC2 expression, miR-9 is negatively regulated by RE1-silencing transcription factor (REST) but positively regulated by cAMP-response element binding protein (CREB) (Laneve et al., 2010). High fat has been shown to suppresses CREB protein expression in the liver (Inoue et al., 2005), providing a possible explanation for the reduced miR-9 expression levels observed in high-fat diet-fed mice.
Many studies suggest that dysregulation of miRNA expression is aging-associated and contributes to AD (Niwa et al., 2008). Additionally, increased DNA methylation have led to downregulated expressions of miR-137 (Langevin et al., 2010) and miR-29 family (Koturbash et al., 2011) in females, indicative of differential expression of miRNA in a gender-specific manner and further supporting our observations in the mice study. Furthermore, maternal high-fat diet has been shown to influence differential expression of mouse hepatic miRNAs in offspring, including downregulation of miR-29a (Zhang et al., 2009). We observed a reduction of miR-137, -181, and -29a/b-1 expressions in females compared to males and a downregulation of miR-137, -181c, and -9 expression levels with high dietary fat intake. This raises an intriguing possibility that women consuming high-fat diets may be at higher risk for SPT dysregulation and thus AD. Therefore, our results lend support to epidemiological factors such as age, gender, and diet epigenetically regulating miRNAs and contributing to loss-of-function of miR-137, -181c, -9, and 29a/b-1, resulting in reduced suppression of SPT expression, and thereby increasing the ceramide levels and Aβ generation seen in sporadic AD.
Footnotes
This work was supported in part by the National Institute of Health (R01GM079688, R01GM089866, and R21RR024439), the National Science Foundation (CBET 0941055 and CBET-1049127), and the Michigan State University (MSU) Foundation. We thank Dr. Peter Nelson and the UK ADC NIA P30-AG0-28383 for providing the human autopsy brain samples. We thank Dr. Daniel Jones and Lijun Chen at the MSU mass spectrometry facility for their support. We thank Aditi Upadhye for her assistance.
References
- An JH, Lee SY, Jeon JY, Cho KG, Kim SU, Lee MA. Identification of gliotropic factors that induce human stem cell migration to malignant tumor. J Proteome Res. 2009;8:2873–2881. doi: 10.1021/pr900020q. [DOI] [PubMed] [Google Scholar]
- Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, Margioris AN, Tsichlis PN, Tsatsanis C. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 2009;31:220–231. doi: 10.1016/j.immuni.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman DL, Wolf PA, Linn R, Knoefel JE, Cobb J, Belanger A, D'Agostino RB, White LR. Prevalence of dementia and probable senile dementia of the Alzheimer type in the Framingham Study. Neurology. 1992;42:115–119. doi: 10.1212/wnl.42.1.115. [DOI] [PubMed] [Google Scholar]
- Bandaru VV, Troncoso J, Wheeler D, Pletnikova O, Wang J, Conant K, Haughey NJ. ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimer's but not normal brain. Neurobiol Aging. 2009;30:591–599. doi: 10.1016/j.neurobiolaging.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Butler AW, Ng MY, Hamshere ML, Forabosco P, Wroe R, Al-Chalabi A, Lewis CM, Powell JF. Meta-analysis of linkage studies for Alzheimer's disease—a web resource. Neurobiol Aging. 2009;30:1037–1047. doi: 10.1016/j.neurobiolaging.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Chang ZQ, Lee SY, Kim HJ, Kim JR, Kim SJ, Hong IK, Oh BC, Choi CS, Goldberg IJ, Park TS. Endotoxin activates de novo sphingolipid biosynthesis via nuclear factor kappa B-mediated upregulation of Sptlc2. Prostaglandins Other Lipid Mediat. 2011;94:44–52. doi: 10.1016/j.prostaglandins.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, Kelnar K, Kemppainen J, Brown D, Chen C, Prinjha RK, Richardson JC, Saunders AM, Roses AD, Richards CA. Identification of miRNA changes in Alzheimer's disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis. 2008;14:27–41. doi: 10.3233/jad-2008-14103. [DOI] [PubMed] [Google Scholar]
- Coolen M, Bally-Cuif L. MicroRNAs in brain development and physiology. Curr Opin Neurobiol. 2009;19:461–470. doi: 10.1016/j.conb.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ. Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2003;100:11735–11740. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, Nicoloso MS, Shimizu M, Rossi S, Gopisetty G, Molina JR, Carlotti C, Jr, Tirapelli D, Neder L, Brassesco MS, Scrideli CA, Tone LG, Georgescu MM, Zhang W, Puduvalli V, Calin GA. miR-29b and miR-125a regulate podoplanin and suppress invasion in glioblastoma. Genes Chromosomes Cancer. 2010;49:981–990. doi: 10.1002/gcc.20808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini C, Ko MH, Jonas MC, Puglielli L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. The Biochemical journal. 2007;407:383–395. doi: 10.1042/BJ20070040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Van Broeckhoven C. Molecular genetics of Alzheimer's disease. Ann Med. 1998;30:560–565. doi: 10.3109/07853899809002605. [DOI] [PubMed] [Google Scholar]
- Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebina M, Futai E, Tanabe C, Sasagawa N, Kiso Y, Ishiura S. Inhibition by KMI-574 leads to dislocalization of BACE1 from lipid rafts. J Neurosci Res. 2009;87:360–368. doi: 10.1002/jnr.21858. [DOI] [PubMed] [Google Scholar]
- Gao FB. Context-dependent functions of specific microRNAs in neuronal development. Neural Dev. 2010;5:25. doi: 10.1186/1749-8104-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas RH, Rice MA, Trauner DA, Merritt TA. Therapeutic effects of a ketogenic diet in Rett syndrome. Am J Med Genet Suppl. 1986;1:225–246. doi: 10.1002/ajmg.1320250525. [DOI] [PubMed] [Google Scholar]
- Han X, Holtzman DM, McKeel DW, Jr, Kelley J, Morris JC. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer's disease: potential role in disease pathogenesis. J Neurochem. 2002;82:809–818. doi: 10.1046/j.1471-4159.2002.00997.x. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- He X, Huang Y, Li B, Gong CX, Schuchman EH. Deregulation of sphingolipid metabolism in Alzheimer's disease. Neurobiol Aging. 2010;31:398–408. doi: 10.1016/j.neurobiolaging.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojjati MR, Li Z, Zhou H, Tang S, Huan C, Ooi E, Lu S, Jiang XC. Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoE-deficient mice. J Biol Chem. 2005;280:10284–10289. doi: 10.1074/jbc.M412348200. [DOI] [PubMed] [Google Scholar]
- Hornemann T, Richard S, Rütti MF, Wei Y, von Eckardstein A. Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase. J Biol Chem. 2006;281:37275–37281. doi: 10.1074/jbc.M608066200. [DOI] [PubMed] [Google Scholar]
- Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, Suzuki Y, Saito H, Kohgo Y, Okumura T. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun. 2005;336:215–222. doi: 10.1016/j.bbrc.2005.08.070. [DOI] [PubMed] [Google Scholar]
- Jana A, Pahan K. Fibrillar amyloid-beta-activated human astroglia kill primary human neurons via neutral sphingomyelinase: implications for Alzheimer's disease. J Neurosci. 2010;30:12676–12689. doi: 10.1523/JNEUROSCI.1243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, Calon F. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging. 2010;31:1516–1531. doi: 10.1016/j.neurobiolaging.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Ko MH, Puglielli L. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J Biol Chem. 2009;284:2482–2492. doi: 10.1074/jbc.M804901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koturbash I, Zemp F, Kolb B, Kovalchuk O. Sex-specific radiation-induced microRNAome responses in the hippocampus, cerebellum and frontal cortex in a mouse model. Mutat Res. 2011;722:114–118. doi: 10.1016/j.mrgentox.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Ladisch S, Li R, Olson E. Ceramide structure predicts tumor ganglioside immunosuppressive activity. Proc Natl Acad Sci U S A. 1994;91:1974–1978. doi: 10.1073/pnas.91.5.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laneve P, Gioia U, Andriotto A, Moretti F, Bozzoni I, Caffarelli E. A minicircuitry involving REST and CREB controls miR-9-2 expression during human neuronal differentiation. Nucleic Acids Res. 2010;38:6895–6905. doi: 10.1093/nar/gkq604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin SM, Stone RA, Bunker CH, Grandis JR, Sobol RW, Taioli E. MicroRNA-137 promoter methylation in oral rinses from patients with squamous cell carcinoma of the head and neck is associated with gender and body mass index. Carcinogenesis. 2010;31:864–870. doi: 10.1093/carcin/bgq051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT, Jr, Kosik KS. A detergent-insoluble membrane compartment contains A beta in vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Liebhaber GM, Riemann E, Baumeister FA. Ketogenic diet in Rett syndrome. J Child Neurol. 2003;18:74–75. doi: 10.1177/08830738030180011801. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer's disease hippocampus. Neuroreport. 2007;18:297–300. doi: 10.1097/WNR.0b013e3280148e8b. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cutler RG, Jo DG. Alzheimer peptides perturb lipid-regulating enzymes. Nat Cell Biol. 2005;7:1045–1047. doi: 10.1038/ncb1105-1045. [DOI] [PubMed] [Google Scholar]
- Mehler MF, Mattick JS. Non-coding RNAs in the nervous system. J Physiol. 2006;575:333–341. doi: 10.1113/jphysiol.2006.113191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memon RA, Holleran WM, Uchida Y, Moser AH, Grunfeld C, Feingold KR. Regulation of sphingolipid and glycosphingolipid metabolism in extrahepatic tissues by endotoxin. J Lipid Res. 2001;42:452–459. [PubMed] [Google Scholar]
- Niwa R, Zhou F, Li C, Slack FJ. The expression of the Alzheimer's amyloid precursor protein-like gene is regulated by developmental timing microRNAs and their targets in Caenorhabditis elegans. Dev Biol. 2008;315:418–425. doi: 10.1016/j.ydbio.2007.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S, Melrose J, Chan C. Involvement of astroglial ceramide in palmitic acid-induced Alzheimer-like changes in primary neurons. Eur J Neurosci. 2007;26:2131–2141. doi: 10.1111/j.1460-9568.2007.05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DK, Carton J, Shah AK, Meredith F, Uhlinger DJ, Hannun YA. Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J Biol Chem. 2000;275:9078–9084. doi: 10.1074/jbc.275.12.9078. [DOI] [PubMed] [Google Scholar]
- Pettegrew JW, Panchalingam K, Hamilton RL, McClure RJ. Brain membrane phospholipid alterations in Alzheimer's disease. Neurochem Res. 2001;26:771–782. doi: 10.1023/a:1011603916962. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Ellis BC, Saunders AJ, Kovacs DM. Ceramide stabilizes beta-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid beta-peptide biogenesis. J Biol Chem. 2003;278:19777–19783. doi: 10.1074/jbc.M300466200. [DOI] [PubMed] [Google Scholar]
- Rotthier A, Auer-Grumbach M, Janssens K, Baets J, Penno A, Almeida-Souza L, Van Hoof K, Jacobs A, De Vriendt E, Schlotter-Weigel B, Löscher W, Vondráček P, Seeman P, De Jonghe P, Van Dijck P, Jordanova A, Hornemann T, Timmerman V. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am J Hum Genet. 2010;87:513–522. doi: 10.1016/j.ajhg.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlatti F, Sala G, Somenzi G, Signorelli P, Sacchi N, Ghidoni R. Resveratrol induces growth inhibition and apoptosis in metastatic breast cancer cells via de novo ceramide signaling. FASEB J. 2003;17:2339–2341. doi: 10.1096/fj.03-0292fje. [DOI] [PubMed] [Google Scholar]
- Schonrock N, Ke YD, Humphreys D, Staufenbiel M, Ittner LM, Preiss T, Götz J. Neuronal microRNA deregulation in response to Alzheimer's disease amyloid-beta. PloS one. 2010;5:e11070. doi: 10.1371/journal.pone.0011070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439:283–289. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome biology. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah C, Yang G, Lee I, Bielawski J, Hannun YA, Samad F. Protection from high fat diet-induced increase in ceramide in mice lacking plasminogen activator inhibitor 1. J Biol Chem. 2008;283:13538–13548. doi: 10.1074/jbc.M709950200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioya M, Obayashi S, Tabunoki H, Arima K, Saito Y, Ishida T, Satoh J. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol Appl Neurobiol. 2010;36:320–330. doi: 10.1111/j.1365-2990.2010.01076.x. [DOI] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Smrt RD, Li Y, Luo Y, Lin L, Santistevan NJ, Li W, Zhao X, Jin P. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J Cell Biol. 2010;189:127–141. doi: 10.1083/jcb.200908151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay F, Lavigne C, Jacques H, Marette A. Defective insulin-induced GLUT4 translocation in skeletal muscle of high fat-fed rats is associated with alterations in both Akt/protein kinase B and atypical protein kinase C (zeta/lambda) activities. Diabetes. 2001;50:1901–1910. doi: 10.2337/diabetes.50.8.1901. [DOI] [PubMed] [Google Scholar]
- Valsecchi M, Mauri L, Casellato R, Prioni S, Loberto N, Prinetti A, Chigorno V, Sonnino S. Ceramide and sphingomyelin species of fibroblasts and neurons in culture. J Lipid Res. 2007;48:417–424. doi: 10.1194/jlr.M600344-JLR200. [DOI] [PubMed] [Google Scholar]
- Vetrivel KS, Cheng H, Kim SH, Chen Y, Barnes NY, Parent AT, Sisodia SS, Thinakaran G. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J Biol Chem. 2005;280:25892–25900. doi: 10.1074/jbc.M503570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer's disease and may accelerate disease progression through regulation of β-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang F, Didelot X, Bruce KD, Cagampang FR, Vatish M, Hanson M, Lehnert H, Ceriello A, Byrne CD. Maternal high fat diet during pregnancy and lactation alters hepatic expression of insulin like growth factor-2 and key microRNAs in the adult offspring. BMC Genomics. 2009;10:478. doi: 10.1186/1471-2164-10-478. [DOI] [PMC free article] [PubMed] [Google Scholar]