

Abstract
Colorectal cancer is a leading cause of cancer‐related deaths world‐wide. Despite the development of new anticancer agents, there will be an estimated 150 000 new cases and 50 000 deaths associated with this disease during the next year. (1) This is due, in part, to the limitations of chemotherapy, resulting from drug resistance and organ system toxicities. To overcome the inherent limitations associated with standard chemotherapy techniques, the development of novel drug targets is of utmost importance in combating this disease. There is accumulating evidence that a small fraction of cancer cells, referred to as cancer stem cells, may play a critical role in the pathogenesis of this disease. In fact, the identification of cancer stem cells can be accomplished based on the expression of surface markers associated with a cancer stem‐like phenotype. This stem‐like phenotype includes indefinite self‐replication, pluripotency, and most importantly, resistance to chemotherapeutics. Therefore, understanding the properties of cancer stem cells may ultimately lead to new therapeutic approaches. Recently, several studies have shown that Notch signaling is critical in maintaining cancer stem cell properties. This review provides a summary of colonic crypt organization and colon carcinogenesis with a focus on stem cells. Moreover, we discuss novel therapeutic strategies that are under development for targeting Notch signaling in cancer stem cells. (Cancer Sci 2011; 102: 1938–1942)
Colonic crypt organization and the stem cell niche
The colon is comprised of histologically distinct layers, including the mucosa, submucosa, muscle layer, and serosa. The innermost layer consists of a mucosa that includes the epithelium, lamina propria, and a thin layer of muscle. The entire surface of the colonic mucosa is comprised of a functional unit referred to as the crypts of Lieberkühn, which contains approximately 2000–3000 cells.( 2 ) The entire colon contains millions of self‐renewing crypts, and it has been estimated that over 6 × 1014 epithelial cells are produced during the lifetime of an individual.
Three terminally differentiated epithelial lineages are present within the crypt: absorptive enterocytes; mucous‐secreting goblet cells; and peptide hormone‐secreting enteroendocrine cells. Each of these epithelial lineages are derived from a pluripotent stem cell located at the base of the crypt. ( 3 ) The colon stem cells show unique properties as they remain in an undifferentiated state. The stem cells have a long half‐life, maintain the ability to self‐renew, and have the potential to generate all three differentiated cells within the colonic crypt compartment. Stem cells usually give rise to two daughter cells by asymmetric division to maintain normal crypt size and homeostasis. ( 4 , 5 ) After division, one cell remains at the bottom of the crypt as a stem cell (self‐renewal) and the other cell commits to a transient amplifying cell for subsequent terminal differentiation (Fig. 1). Stem cells and transient amplifying cells occupy the lower portion of the crypt. It is known that the Wnt, the bone morphogenetic protein, and Notch signaling pathways, ephrin type B receptor tyrosine kinases and their ligands, are involved in the regulation of stem cell behavior, migration, and differentiation. ( 6 ) In particular, Wnt signaling plays an important role in the maintenance of the stem and proliferation cell compartment. ( 7 ) Consistent with these reports, Sato et al. ( 8 ) reported that single stem cells of small intestine can build crypt–villus structures in vitro with Matrigel, but only in the presence of epithelial growth factor, R‐spondin 1 (Wnt agonist), and Noggin (bone morphogenetic protein antagonist). These factors, which regulate stem cell behavior, are provided by the stem cell niche that surrounds the bottom of the crypt, under normal circumstances. ( 9 , 10 ) The pericryptal myofibroblasts are considered a most important component of the stem cell niche. ( 11 ) Furthermore, Sato et al. ( 12 ) showed that the co‐culture of small intestinal single stem cells with Paneth cells improves crypt–villus structure formation in vitro, and Paneth cells are expressing essential signals for stem cell maintenance in culture. These results suggest that Paneth cells, even epithelial cells, represent one component of the stem cell niche, as well as myofibroblasts. However, as there are no Paneth cells in the distal colon, it is unclear which cells are playing the role of Paneth cells in the stem cell niche.
Figure 1.
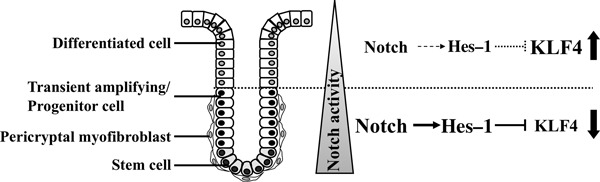
Diagrammatic representation of the colonic crypt compartment and the Notch signaling pathway. Within the epithelium of the normal colonic mucosa, stem cells are located at the base of the crypts. Following asymmetric division, the daughter cells undergo differentiation and migrate upwards to give rise to transient amplifying cells and terminally differentiated cells. Colonic stem cells generate three types of differentiated cells: absorptive columnar cells; hormone‐producing enteroendocrine cells; and mucus‐secreting goblet cells. Pericryptal myofibroblasts surround the crypt base, thought to comprise a stem cell “niche”. These cells regulate epithelial stem cell function by paracrine secretion of growth factors and cytokines. Notch activation is observed within the proliferative zone located within the lower region of the crypt. Krüppel‐like factor 4 (KLF4) is expressed in the differentiated epithelial cells at the top half of the crypt. Hes‐1, the direct transcriptional target of Notch, is a transcriptional repressor of KLF4, therefore, it is generally assumed that KLF4 expression is restricted to differentiated cryptal cells.
Identification of normal colon stem cell markers
During the past decade, considerable research effort has been made to identify the colon stem cell. ( 6 ) Bromodeoxyuridine labeling was initially used to identify stem cells within the colon. This approach was based on the concept that stem cells divide infrequently and retain their DNA label for a longer period of time than the more rapidly dividing progenitor cells. ( 13 ) Recently, however, this method has been replaced by the identification of “stemness” markers that allow for the isolation of stem cells by flow cytometry.
Musashi1 (Msi‐1) was the first colon stem cell marker to be identified. ( 14 ) It was initially reported that Msi‐1 is an RNA‐binding protein that is indispensable for asymmetric cell division of sensory organ precursor cells in Drosophila. ( 14 ) Msi‐1 was also proposed to be required for asymmetric distribution of intrinsic determinants in the developing mammalian nervous system. ( 15 ) In 2003, Msi‐1 positive cells were found in the mouse small intestine ( 16 ) and in the human colon at the base of the crypt compartment, between cell positions 1 and 10. ( 17 )
Fujimoto et al. ( 18 ) reported that the integrin subunit b1 (CD29) was a candidate surface marker for the proliferative zone of the human colonic crypt, which includes stem cells and progenitor cells. Recently, a leucin‐rich repeat‐containing G protein‐coupled receptor 5 (Lgr5) was also identified as a potential intestinal stem cell marker based on cell turnover and lineage tracing analysis. ( 19 ) As reported by the Clevers laboratory, ( 20 ) Lgr5 is an orphan G protein‐coupled receptor of unknown function that is regulated by the Wnt signaling pathway. As mentioned above, single Lgr5‐expressing stem cells can be cultured to form long‐lived, self‐organizing crypt–villus organoids, even in the absence of non‐epithelial niche cells. ( 8 ) More recently, doublecortin and CaM kinase‐like‐1 (DCAMKL‐1), a microtubule‐associated kinase expressed in post‐mitotic neurons, have been proposed as putative colonic stem cell markers. ( 21 ) DCAMKL‐1 was found to be expressed in the same cells as Msi‐1, but its expression was more restricted. Interestingly, DCAMKL‐1 expression is restricted to quiescent cells within columnar cells residing at the base of the crypt, and it is expressed in mitotic cells only after irradiation. ( 21 ) Lgr5 is expressed in rapidly cycling mitotic cells under basal conditions, so DCAMKL‐1 positive cells may be a subset of Lgr5‐expressing stem cells.
Cancer stem cell theory
Tumors are comprised of a heterogeneous population of cells that vary in their morphology, gene expression patterns, proliferative capacity, and tumorigenic potential. This heterogeneity was originally explained by a stochastic model in which all tumor cells share an equal ability to generate a tumor. ( 22 ) In this model, the morphological heterogeneity of the tumor could be explained by genetic instability of the tumor cells.
However, the cancer stem cell theory has provided a new perspective on tumor heterogeneity. This model proposes that only a small fraction of cells within the tumor, the cancer stem cells, possess cancer‐initiating potential and the ability to sustain tumor growth. Cancer stem cells are considered to have a capacity for self‐renewal and ability to give rise to progeny cells with both tumorigenic and non‐tumorigenic properties. Cancer cells with stem‐like properties were first observed in acute myeloid leukemia ( 23 ) and later found in solid tumors. ( 24 ) Recently, the presence of cancer stem cells within CRC was also reported. ( 25 , 26 , 27 ) These cells were identified by the presence of several markers, including CD133, ( 25 , 26 ) often associated with stem and progenitor populations, as well as CD44 and EpCAM. ( 27 )
The existence of colon cancer stem cells was first reported in 2007. ( 25 , 26 ) O’Brien et al. ( 25 ) isolated CD133+ and CD133− cells from both primary and metastatic human CRCs, and these cells were injected under the renal capsule of NOD/SCID mice. The CD133+ cells gave rise to rapidly growing tumors in this explant model. The tumor cells retained their parental tumor morphology even after serial transfer, whereas CD133− cells did not support tumor growth after transfer. In a related study from another group, CD133+ cells were maintained for more than 1 year in vitro as undifferentiated tumor spheres when maintained in the presence of epidermal growth factor and fibroblast growth factor. ( 26 ) However, the cells will undergo gradual differentiation after withdrawal of these critical growth factors. During the differentiation process, CD133+ cells lose their CD133 expression and their ability to form tumors in immunodeficient mice. ( 26 ) Huang et al. ( 28 ) showed that ALDH1 positive cells isolated from human colon cancer tissue based on its enzymatic activity, formed tumors when transferred into NOD/SCID mice, whereas ALDH1− cells did not form tumors. Furthermore, the population of CD44+/CD133+/ALDH1+ cells increased throughout the crypt compartment and was distributed further up the length of the crypt axis during colon tumor progression. Dalerba et al. ( 27 ) reported that CD44+/CD133+/EpCAMhigh cells isolated from human CRC also establish a tumor that shows an identical morphology in a similar xenograft model. Interestingly, there was no growth of the CD44−/CD133−/EpCAMlow tumor cells. However, in this report, they showed that CD44 expression can also be found in CD133 negative cells. Therefore, the combination of markers with CD44 might be more restrict indication of cancer stem cells than CD133 alone.
Interestingly, as ALDH1 functions as a detoxification enzyme, ( 29 ) its activity could potentially afford protection to cancer stem cells from oxidative insult, allowing for their longevity and enhanced proliferative capacity. In fact, a recent report showed a function for CD44 variant in the regulation of reactive oxygen species defense and tumor growth in vivo. ( 30 ) Thus, biomarkers for colon cancer stem cells are not just a “marker” but will also improve the understanding of the mechanism underlying tumorigenesis and drug resistance.
Cells of origin in colon cancer
Increasing evidence supports the existence of cancer stem cells, but one of the most important questions within the field of cancer biology is the origin of a tumorigenic cell. Presumably, this cell population would sustain the initial mutation, self replicate, and ultimately give rise to the neoplastic foci. Because of the uncertainty in identifying these cells, the terms “cancer‐initiating cell” or “tumor‐initiating cell” are often used interchangeably with “cancer stem cell”. The true definition of a cancer stem cell, however, is based on its capacity for self‐renewal and its ability to give rise to the heterogeneous lineages of cancer cells that comprise a tumor. It is assumed that there are two possible ways for generating the cell‐of‐origin of cancer. First, the accumulation of genetic or epigenetic changes may render a normal tissue stem cell cancerous. The second is that these changes may confer stem‐like abilities on a progenitor or differentiated cell. This might be similar to the reprogramming of differentiated cells to induced pluripotent stem cells by introduction of certain factors. ( 31 )
On the basis of histology, there are two models for the development of colon tumors. In the “top‐down” model, mutant cells appear within the intracryptal zone between the crypt openings. ( 32 ) In fact, those dysplastic cells that show intense staining for β‐catenin are found only at the orifices at the lumenal surface of the crypt. There are two possibilities that the “top‐down” model provides to describe the development of a tumor. Mutant cells at the surface of the mucosa may spread laterally and downward to form new crypts, eventually replacing them. Alternatively, a cell derived from a mutated stem cell may migrate to that area and expand with a second genetic hit that activates its growth potential.
The other, perhaps more intuitive model, is the “bottom‐up” model. This model is based on findings that the more frequent lesion in familial adenomatous polyposis is the unicryptal or monocryptal adenoma, where the dysplastic epithelial cells occupy an entire single crypt. ( 33 ) It proposes that the malignant transformation process takes place among the stem cell population at the crypt base, then the transformed stem cell migrates to the apex of the crypt where it expands. Recently, technological progress to identify the specific set of genes expressed in stem cells of the intestine has allowed the monitoring of the cell of origin of adenomas. Barker et al. and Zhu et al. ( 34 , 35 )made use of a Cre‐inducible activation of the Wnt pathway, with Cre being expressed from either the endogenous Lgr5 or the CD133 locus of these stem cell marker genes. Both Lgr5‐ and CD133‐induced activation of the Wnt pathway leads to efficient tumor formation in the small intestine of transgenic mice, respectively. Importantly, they also show that tumor formation does not occur when the activation of the Wnt pathway is induced in progenitor or differentiated cells. A novel intestinal stem cell marker is Bmi1. ( 36 ) Bmi1 is expressed in discrete cells located near the bottom of crypts in the small intestine, predominantly four cells above the base of the crypt. This cell population is shown to be susceptible to tumorigenesis by deregulated Wnt signaling using an Bmi1‐Cre‐ER knock‐in system. ( 36 ) Based on the apparently more rapid entry of Lgr5 positive cells into the proliferative state, relative to Bmi1 positive cells, it is considered that these two markers label different states of intestinal stem cells: a more quiescent state (Bmi1), and a state more prone to enter proliferation (Lgr5). However, it is still unclear in colon as these results suggest that only stem cells can be a cell of origin in cancer of the small intestine. These reports also suggest that the stemness as observed in “stem cells” as well as the “cancer stem cells” plays an important role in the tumorigenesis.
In cancer stem cells, the stem‐like properties include indefinite self‐renewal, puluripotency and, importantly, resistance to chemotherapeutic agents. Therefore, it is entirely plausible that cancer stem cells, regardless of their cellular origin, may escape standard chemotherapy, ultimately causing disease recurrence, even after apparently complete remission. Thus, the selective targeting of cancer stem cells with novel therapeutic agents has become a rapidly emerging field of molecular oncology. Thus, the Notch pathway has become one of the most intensively studied putative therapeutic targets in cancer stem cells.
Notch signaling in normal intestine
Notch signaling plays an important role in the determination of cell fate. In recent years, this signaling pathway has been shown to play a critical role in regulating the balance between cell proliferation, differentiation, and apoptosis of cells in various tissues. ( 37 , 38 ) In mammals, four Notch genes are expressed, each of which encode a single‐pass trans‐membrane receptor (Notch1–4). The interaction between Notch receptors and their ligands (Jagged 1 and 2, and Delta‐like 1, 3, and 4) results in proteolytic cleavage of Notch by the γ‐secretase and other proteases, which releases NICD from the plasma membrane and initiates its subsequent translocation into the nucleus. After nuclear translocation, NICD binds to, and forms a complex with, one of three transcriptional regulators, referred to as CSL (a collective name of CBP or RBP‐Jκ in vertebrates, Su (H) in Drosophila, and Lag‐1 in Caenorhabditis elegans), MAML‐1, and p300/CBP. ( 39 , 40 ) These complexes activate the transcription of the HES‐1, ‐5, ‐7, HEY‐1, ‐2, and HEYL genes encoding basic helix–loop–helix/orange domain transcriptional repressors. ( 39 , 40 ) Signal transduction from typical Notch ligands to the CSL–NICD–MAML‐1 cascade is also known as the “canonical” Notch signaling pathway. It is reported that NICD also interacts with p50 or c‐Rel in the nucleus to enhance nuclear factor (NF)‐κB activity, ( 41 ) which is referred to as the “non‐canonical” Notch signaling pathway. Notch signaling to the NF‐κB–NICD complex augments transcriptional activation of NF‐κB target genes, such as IFN‐γ.
In the colon, abundant expression of Notch 1 and Jagged 1 was observed in the proliferative zone located within the middle‐third of the crypt, ( 42 ) whereas Notch 2 expression was absent. Jagged 2 appeared to be more uniformly expressed across the entire crypt. Notch signaling is essential for regulating the differentiation, and maintaining, stem and progenitor cells. The role of Notch signaling in the control of differentiation and proliferation was recently confirmed using transgenic mice. Depletion of Hes‐1, the most abundant and direct target gene of Notch signaling, was associated with a significant increase in the secretory lineage of intestinal epithelial cells. ( 43 ) Conditional gut‐specific inactivation of CSL results in the complete loss of proliferating crypt progenitor cells followed by their conversion into post‐mitotic goblet cells. ( 44 ) In reciprocal gain‐of‐function studies, expression of NICD in the intestine inhibits differentiation of crypt progenitor cells, resulting in a large increase in undifferentiated transient amplifying cells. ( 45 ) These studies indicate an essential role for Notch signaling as a gatekeeper of the intestinal progenitor compartment.
Notch signaling in CRC
Under normal circumstances, Notch signaling clearly plays an important role in the maintenance of the colon crypt compartment. More recently, inappropriate activation of Notch signaling has been associated with the pathogenesis of colon cancers. Significant upregulation of Notch1 ( 46 ) and Hes1 ( 47 ) has been detected in colon adenocarcinomas, but not in normal differentiated epithelial cells. It was also reported that the activation of Notch signaling is essential for the development of adenomas in Apc Min/+ mice ( 44 ) and self‐renewal of tumor‐initiating cells. ( 48 ) Importantly, Hes1 is known to suppress the expression of Krüppel‐like factor 4, a transcriptional repressor. ( 49 ) A zinc finger‐containing transcription factor, KLF4 is highly expressed in terminally differentiated epithelial cells of the intestine (Fig. 1). ( 50 , 51 ) It is also reported that overexpression of KLF4 inhibits colon cancer cell proliferation ( 52 , 53 ) and haploinsufficiency of KLF4 promotes the development of intestinal adenomas in Apc Min/+ mice. ( 54 ) Expression of KLF4 is reduced in colorectal neoplasia relative to normal mucosa, including both adenomas and carcinomas. ( 55 ) We have also recently examined KLF4 expression in colon tumors and adjacent normal mucosa of AOM‐treated mice. Consistent with these earlier findings in the Apc Min/+ model, we have observed that KLF4 expression is restricted to the most differentiated epithelial cells residing within the upper half of normal colonic crypts that maintain normal membrane β‐catenin staining. In the AOM‐induced tumor, β‐catenin exists within the cytoplasm, and KLF4 expression seems to be suppressed (Fig. 2). Thus, Notch signaling does not always function as an oncogenic factor, but may also play an important role in colon carcinogenesis.
Figure 2.
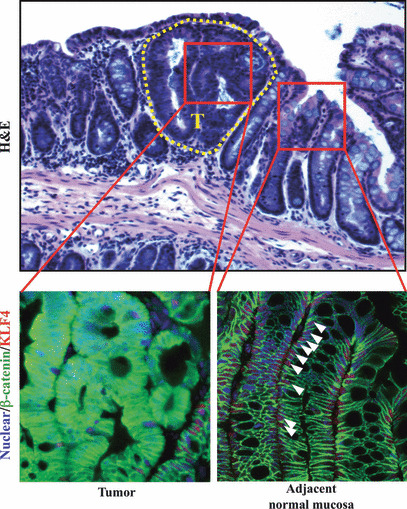
Krüppel‐like factor 4 (KLF4) and β‐catenin expression in azoxymethane‐induced tumor and normal adjacent mucosa. Double staining for β‐catenin (green) and KLF4 (red) using mouse colonic tissue is indicated. In the normal adjacent mucosa, β‐catenin is located at the cell membrane. KLF4 is expressed within the epithelial cells at the top half of the crypt (white arrowheads). In azoxymethane‐induced tumors, β‐catenin is translocated into cytosol and KLF4 expression is decreased. T, tumor area (delineated by dotted line).
Conclusion
This review has summarized our current understanding of the colonic crypt structure and organization and provided an overview of colon carcinogenesis from the point of view of the cancer stem cell. The studies summarized strongly suggest that the stem‐like properties of cancer stem cells could provide an excellent druggable target for novel colon cancer therapies. For example, one γ‐secretase inhibitor molecule, dibenzazepine, which inhibits the cleavage of Notch, showed growth suppressive effects on small intestinal tumors in Apc Min/+ mice. ( 44 ) Before Notch‐based therapies can be more broadly developed, however, additional studies must first focus on key issues such as target cell specificity. This is of foremost importance because of the overlapping features of normal stem cells and cancer stem cells. Although this is obviously a challenging issue that must be resolved, it is not insurmountable, and we believe that these novel therapeutic approaches could ultimately provide treatment strategies that may overcome resistance to chemotherapeutic agents.
Disclosure Statement
The authors have no conflicts of interest.
Abbreviations
- ALDH1
aldehyde dehydrogenase 1
- AOM
azoxymethane
- Bmi1
B‐cell‐specific Moloney murine leukemia virus integration site 1
- CRC
colorectal cancer
- DCAMKL‐1
doublecortin and CaM kinase‐like‐1
- KLF4
Krüppel‐like factor 4
- Msi‐1
Musashi1
- NICD
Notch intracellular domain
Acknowledgments
This work supported by National Institutes of Health Grant CA125691 (D.W. Rosenberg).
References
- 1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59: 225–49. [DOI] [PubMed] [Google Scholar]
- 2. Booth C, Potten CS. Gut instincts: thoughts on intestinal epithelial stem cells. J Clin Invest 2000; 105: 1493–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marshman E, Booth C, Potten CS. The intestinal epithelial stem cell. Bioessays 2002; 24: 91–8. [DOI] [PubMed] [Google Scholar]
- 4. Barker N, van de Wetering M, Clevers H. The intestinal stem cell. Genes Dev 2008; 22: 1856–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brittan M, Wright NA. Gastrointestinal stem cells. J Pathol 2002; 197: 492–509. [DOI] [PubMed] [Google Scholar]
- 6. van der Flier LG, Clevers H. Stem cells, self‐renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol 2009; 71: 241–60. [DOI] [PubMed] [Google Scholar]
- 7. Korinek V, Barker N, Moerer P et al. Depletion of epithelial stem‐cell compartments in the small intestine of mice lacking Tcf‐4. Nat Genet 1998; 19: 379–83. [DOI] [PubMed] [Google Scholar]
- 8. Sato T, Vries RG, Snippert HJ et al. Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 2009; 459: 262–5. [DOI] [PubMed] [Google Scholar]
- 9. Scoville DH, Sato T, He XC, Li L. Current view: intestinal stem cells and signaling. Gastroenterology 2008; 134: 849–64. [DOI] [PubMed] [Google Scholar]
- 10. Yeung TM, Chia LA, Kosinski CM, Kuo CJ. Regulation of self‐renewal and differentiation by the intestinal stem cell niche. Cell Mol Life Sci 2011; 68: 2513–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yen TH, Wright NA. The gastrointestinal tract stem cell niche. Stem Cell Rev 2006; 2: 203–12. [DOI] [PubMed] [Google Scholar]
- 12. Sato T, van Es JH, Snippert HJ et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011; 469: 415–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lipkin M, Bell B, Sherlock P. Cell proliferation kinetics in the gastrointestinal tract of man. I. Cell renewal in colon and rectum. J Clin Invest 1963; 42: 767–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakamura M, Okano H, Blendy JA, Montell C. Musashi, a neural RNA‐binding protein required for Drosophila adult external sensory organ development. Neuron 1994; 13: 67–81. [DOI] [PubMed] [Google Scholar]
- 15. Okabe M, Sawamoto K, Imai T, Sakakibara S, Yoshikawa S, Okano H. Intrinsic and extrinsic determinants regulating cell fate decision in developing nervous system. Dev Neurosci 1997; 19: 9–16. [DOI] [PubMed] [Google Scholar]
- 16. Potten CS, Booth C, Tudor GL et al. Identification of a putative intestinal stem cell and early lineage marker; musashi‐1. Differentiation 2003; 71: 28–41. [DOI] [PubMed] [Google Scholar]
- 17. Nishimura S, Wakabayashi N, Toyoda K, Kashima K, Mitsufuji S. Expression of Musashi‐1 in human normal colon crypt cells: a possible stem cell marker of human colon epithelium. Dig Dis Sci 2003; 48: 1523–9. [DOI] [PubMed] [Google Scholar]
- 18. Fujimoto K, Beauchamp RD, Whitehead RH. Identification and isolation of candidate human colonic clonogenic cells based on cell surface integrin expression. Gastroenterology 2002; 123: 1941–8. [DOI] [PubMed] [Google Scholar]
- 19. Barker N, van Es JH, Kuipers J et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007; 449: 1003–7. [DOI] [PubMed] [Google Scholar]
- 20. Van der Flier LG, Sabates‐Bellver J, Oving I et al. The intestinal Wnt/TCF signature. Gastroenterology 2007; 132: 628–32. [DOI] [PubMed] [Google Scholar]
- 21. May R, Riehl TE, Hunt C, Sureban SM, Anant S, Houchen CW. Identification of a novel putative gastrointestinal stem cell and adenoma stem cell marker, doublecortin and CaM kinase‐like‐1, following radiation injury and in adenomatous polyposis coli/multiple intestinal neoplasia mice. Stem Cells 2008; 26: 630–7. [DOI] [PubMed] [Google Scholar]
- 22. Little MP, Wright EG. A stochastic carcinogenesis model incorporating genomic instability fitted to colon cancer data. Math Biosci 2003; 183: 111–34. [DOI] [PubMed] [Google Scholar]
- 23. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3: 730–7. [DOI] [PubMed] [Google Scholar]
- 24. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100: 3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007; 445: 106–10. [DOI] [PubMed] [Google Scholar]
- 26. Ricci‐Vitiani L, Lombardi DG, Pilozzi E et al. Identification and expansion of human colon‐cancer‐initiating cells. Nature 2007; 445: 111–15. [DOI] [PubMed] [Google Scholar]
- 27. Dalerba P, Dylla SJ, Park IK et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA 2007; 104: 10158–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang EH, Hynes MJ, Zhang T et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res 2009; 69: 3382–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Riveros‐Rosas H, Julian‐Sanchez A, Pinã E. Enzymology of ethanol and acetaldehyde metabolism in mammals. Arch Med Res 1997; 28: 453–71. [PubMed] [Google Scholar]
- 30. Ishimoto T, Nagano O, Yae T et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(‐) and thereby promotes tumor growth. Cancer Cell 2011; 19: 387–400. [DOI] [PubMed] [Google Scholar]
- 31. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126: 663–76. [DOI] [PubMed] [Google Scholar]
- 32. Shih IM, Wang TL, Traverso G et al. Top‐down morphogenesis of colorectal tumors. Proc Natl Acad Sci USA 2001; 98: 2640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Preston SL, Wong WM, Chan AO et al. Bottom‐up histogenesis of colorectal adenomas: origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res 2003; 63: 3819–25. [PubMed] [Google Scholar]
- 34. Barker N, Ridgway RA, van Es JH et al. Crypt stem cells as the cells‐of‐origin of intestinal cancer. Nature 2009; 457: 608–11. [DOI] [PubMed] [Google Scholar]
- 35. Zhu L, Gibson P, Currle DS et al. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 2009; 457: 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet 2008; 40: 915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Artavanis‐Tsakonas S, Rand M, Lake R. Notch signaling: cell fate control and signal integration in development. Science 1999; 284: 770–6. [DOI] [PubMed] [Google Scholar]
- 38. Baron M. An overview of the Notch signalling pathway. Semin Cell Dev Biol 2003; 14: 113–19. [DOI] [PubMed] [Google Scholar]
- 39. Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol 2003; 194: 237–55. [DOI] [PubMed] [Google Scholar]
- 40. Katoh M. Notch signaling in gastrointestinal tract (review). Int J Oncol 2007; 30: 247–51. [PubMed] [Google Scholar]
- 41. Shin HM, Minter LM, Cho OH et al. Notch1 augments NF‐kappaB activity by facilitating its nuclear retention. EMBO J 2006; 25: 129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sander GR, Powell BC. Expression of notch receptors and ligands in the adult gut. J Histochem Cytochem 2004; 52: 509–16. [DOI] [PubMed] [Google Scholar]
- 43. Jensen J, Pedersen EE, Galante P et al. Control of endodermal endocrine development by Hes‐1. Nat Genet 2000; 24: 36–44. [DOI] [PubMed] [Google Scholar]
- 44. van Es JH, van Gijn ME, Riccio O et al. Notch/gamma‐secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005; 435: 959–63. [DOI] [PubMed] [Google Scholar]
- 45. Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis‐Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature 2005; 435: 964–8. [DOI] [PubMed] [Google Scholar]
- 46. Zagouras P, Stifani S, Blaumueller CM, Carcangiu ML, Artavanis‐Tsakonas S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc Natl Acad Sci USA 1995; 92: 6414–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reedijk M, Odorcic S, Zhang H et al. Activation of Notch signaling in human colon adenocarcinoma. Int J Oncol 2008; 33: 1223–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sikandar SS, Pate KT, Anderson S et al. NOTCH signaling is required for formation and self‐renewal of tumor‐initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res 2010; 70: 1469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ghaleb AM, Aggarwal G, Bialkowska AB, Nandan MO, Yang VW. Notch inhibits expression of the Krüppel‐like factor 4 tumor suppressor in the intestinal epithelium. Mol Cancer Res 2008; 6: 1920–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shields JM, Christy RJ, Yang VW. Identification and characterization of a gene encoding a gut‐enriched KrA?ppel‐like factor expressed during growth arrest. J Biol Chem 1996; 271: 20009–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dang DT, Pevsner J, Yang VW. The biology of the mammalian KrA?ppel‐like family of transcription factors. Int J Biochem Cell Biol 2000; 32: 1103–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen X, Johns DC, Geiman DE et al. KrA?ppel‐like factor 4 (gut‐enriched KrA?ppel‐like factor) inhibits cell proliferation by blocking G1/S progression of the cell cycle. J Biol Chem 2001; 276: 30423–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yoon HS, Chen X, Yang VW. Kruppel‐like factor 4 mediates p53‐dependent G1/S cell cycle arrest in response to DNA damage. J Biol Chem 2003; 278: 2101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ghaleb AM, McConnell BB, Nandan MO, Katz JP, Kaestner KH, Yang VW. Haploinsufficiency of Krüppel‐like factor 4 promotes adenomatous polyposis coli dependent intestinal tumorigenesis. Cancer Res 2007; 67: 7147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of Krüppel‐like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene 2004; 23: 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]