

Abstract
Oxidative stress has been implicated in the pathogenesis of methylmercury (MeHg) neurotoxicity. Studies of mature neurons suggest that the mitochondrion may be a major source of MeHg-induced reactive oxygen species and a critical mediator of MeHg-induced neuronal death, likely by activation of apoptotic pathways. It is unclear, however, whether the mitochondria of developing and mature neurons are equally susceptible to MeHg. Murine embryonal carcinoma (EC) cells, which differentiate into neurons following exposure to retinoic acid, were used to compare the differentiation-dependent effects of MeHg on ROS production and mitochondrial depolarization. EC cells and their neuronal derivatives were pre-incubated with the ROS indicator 2’,7’-dichlorofluoroscin diacetate or tetramethylrhodamine methyl ester, an indicator of mitochondrial membrane potential, with or without cyclosporin A (CsA), an inhibitor of mitochondrial permeability transition pore opening, and examined by laser scanning confocal microscopy in the presence of 1.5 μM MeHg. To examine consequences of mitochondrial perturbation, immunohistochemical localization of cytochrome c (cyt c) was determined after incubation of cells in MeHg for 4 hours. MeHg treatment induced earlier and significantly higher levels of ROS production and more extensive mitochondrial depolarization in neurons than in undifferentiated EC cells. CsA completely inhibited mitochondrial depolarization by MeHg in EC cells but only delayed this response in the neurons. In contrast, CsA significantly inhibited MeHg-induced neuronal ROS production. Cyt c release was also more extensive in neurons, with less protection afforded by CsA. These data indicate that neuronal differentiation state influences mitochondrial transition pore dynamics and MeHg-stimulated production of ROS.
Keywords: Methylmercury, Neurotoxicity, Mitochondrial Permeability Transition, Reactive Oxygen Species, Apoptosis
1. Introduction
The mechanisms underlying the age dependency of MeHg neurotoxicity remain poorly understood. Several converging lines of evidence implicate the mitochondrion as a primary target of MeHg-induced injury. Early studies by Yoshino and coworkers (Yoshino et al., 1966) reported rapid accumulation of MeHg in the mitochondrion; subsequent studies documented a spectrum of mitochondrial effects of MeHg in vivo and in vitro, including alterations in Complex III of the mitochondrial electron transport chain (ETC) (Yee and Choi, 1996), depression of respiration and ATP production (Sone et al., 1977; Verity et al., 1975) and swelling of the mitochondrial matrix (Fowler and Woods, 1977b).
MeHg induces loss of mitochondrial membrane potential (Ψm) (Bondy and McKee, 1991; Hare and Atchison, 1992; Limke and Atchison, 2002), with subsequent release of cytochrome c, a pro-apoptotic mediator (Shenker et al., 1999, 2002). Loss of Ψm appears to be a result of opening of the mitochondrial permeability transition (MPT) pore, as it could be prevented by treatment with the pore-blocking agents cyclosporin A (CsA) or bongkrekic acid (for reviews, see Bernardi et al, 1994; Halestrap 2009; Lemasters et al, 2009; Nieminen et al., 1996). In several studies CsA, but not FK506, which is similar to CsA but without MPT inhibitory activity, provided a degree of neuroprotection (Friberg et al., 1998; Limke and Atchison, 2002; Sullivan et. al., 2005). MPT is a phenomenon in which the outer or both the inner and the outer mitochondrial membranes become non-selectively permeable to solutes less than 1500 Da, permitting passage of most metabolites, inorganic anions and calcium (Ca2+) (Hunter and Haworth, 1979). Existing predominantly in the closed position, the pore’s transient opening under normal conditions may serve as a physiological means of releasing excess metabolites and ions, especially Ca2+, from mitochondria (Jordan et al., 2003). Mitochondria release several apoptotic mediators, including cyt c, Apaf-1 and apoptosis-inducing factor (AIF) (Ravagnan et al., 2002), and are a primary target of others, especially the Bcl-2 family proteins (Robertson and Orrenius, 2000; Ward et al., 2004). Thus, aside from their role in energy regulation, mitochondria are directly involved in the regulation of cell death pathways.
MeHg stimulates a dose- and time-dependent rise in lipid peroxidation and oxygen radical (ROS) formation in cerebellar granule cell cultures (Oyama et al., 1994; Verity and Sarafian, 1991) and astrocytes (Shanker and Aschner, 2003), and significantly increases ROS formation in cerebellar synaptosomal preparations from MeHg-treated rats (LeBel et al., 1990). Using the ROS-sensitive dye dichlorofluoroscin (DCFH) LeBel and coworkers (Ali et al., 1992; LeBel et al., 1990) demonstrated increases of cellular ROS following MeHg treatment. The importance of increased ROS is supported by reports of protection against MeHg cytotoxicity by vitamin E, a major cellular antioxidant or by radical scavengers such as selenium (Chang et al., 1978; Welsh and Soares, 1976; Yip and Chang, 1982), glutathione peroxidase (Polunas et al., 2004) or superoxide dismutase (Yee and Choi, 1994). Administration of the antioxidants vitamin E, propyl gallate, or Trolox (a water-soluble vitamin E analog), ameliorate MeHg-induced neural cell injury in rat brain in vivo (Usuki et al., 2001), in vitro in rat cerebellar granule and human cerebral cortical neurons (Gasso et al., 2001) and in cerebral astrocytes in culture (Shanker and Aschner, 2003; Shanker et al., 2005). Since mitochondrial respiration is a major source of ROS in normal and toxicant-damaged cells (Brookes et al., 2002; Nohl et al., 2005; Turrens, 2003), it has been speculated that MeHg-induced mitochondrial stress and injury (Bondy and McKee, 1991; Sarafian and Bredesen, 1994), coupled with increased MPT pore opening and cytochrome c release, might increase oxidative damage and induce cell death via apoptosis (Dreiem et al., 2005; Limke et al., 2004).
While there is abundant evidence for the involvement of mitochondrial-mediated oxidative stress in MeHg neurotoxicity, the majority of studies have been performed using samples from whole adult brain or fully differentiated neurons in culture (Bondy et al., 1990; Sarafian et al., 1994; Yee and Choi, 1994, 1996). The potential role of mitochondria and ROS generation in developmental MeHg neurotoxicity is less well understood. There is clear agreement that ROS participate in developmental toxicity in numerous organ systems, including the CNS (Allen and Balin, 1989; Fantel, 1996), and that the developing nervous system may be more susceptible to oxidative insult due to its high content of saturated lipids and lack of fully developed antioxidant systems (de Haan et al, 1994; Mavelli et al., 1982; McBurney et al, 1988; Philbert et al, 1995). Tsatmali and colleagues (2005) reported enhanced basal generation of ROS in embryonic rat cortical neurons compared to their neural precursors and to glial cells; these neurons also contained higher levels of several mitochondrial respiratory chain components than their progenitors. Further, Mundy and Freudenrich (2000) described enhanced sensitivity of immature neurons (day 1 in vitro) to MeHg-induced ROS production compared to functionally mature neurons (day 7 in vitro) in cerebellar granule cells and cortical neurons. However, few studies have directly compared the response of undifferentiated and fully differentiated cells to MeHg.
The present study utilized the P19 embryonal carcinoma cell line, a well-characterized model for the study of neuronal differentiation (eg., Bain et al., 1994; Jones-Villeneuve et al., 1982; MacPherson and McBurney, 1995; McBurney et al., 1988), to examine whether undifferentiated cells and differentiated neurons are equally susceptible to MeHg-induced changes in ROS production and to MeHg-induced alterations in mitochondrial membrane potential/pore state. The study also examined whether the observed mitochrondrial effects trigger subsequent cytotoxic events, such as release of the apoptotic mediator cytochrome c from the intermembrane space. CsA, a short-acting MPT pore blocking compound, was used to better define the relative contribution and timing of mitochondrial dysfunction and ROS production, and to identify potential mechanisms of MeHg-induced toxicity in undifferentiated cells and their D5 neuronal derivatives.
2. Materials and methods
2.1 Cell Culture
P19 murine embryonal carcinoma (EC) cells (Jones-Villeneuve et al., 1982, 1983) were maintained using the method of McBurney et al. (1988). Briefly, undifferentiated P19 cells (a generous gift of Dr. Michael McBurney) were maintained in exponential growth phase in 60 mm tissue culture dishes containing alpha-modified eagle medium (α-MEM) supplemented with 10% fetal bovine serum and 1% antibiotic/antimycotic in 5% CO2 at 37°C. All cell culture reagents were obtained from Gibco/Invitrogen Corp. (Carlsbad, CA).
Neuronal differentiation was performed using a modification of the protocol of MacPherson and McBurney (1995). Near-confluent plates of exponentially growing EC cells were dissociated as for routine passage, resuspended in α-MEM containing 1.0 μM retinoic acid (RA) in 100 mm tissue culture dishes, and left undisturbed at 37°C for 48 hrs. Monolayers were again dissociated and resuspended in α-MEM containing 1.0 μM RA in 60 mm petri dishes. Cells were aggregated in suspension for 24 hrs, transferred to 15 ml conical tubes, centrifuged at 900 × g for 3 min, rinsed with Dulbecco’s modified eagle medium (DMEM, Gibco/Invitrogen Corp.) and repelleted as above. Each pellet was resuspended in 0.5 ml cell dissociation buffer, incubated at 37°C for 5 min, then vigorously pipetted with a flame-polished Pasteur pipette to disperse the aggregates. Suspensions were rinsed again in DMEM and repelleted as above. Finally, pellets were resuspended in serum-free medium and plated onto laminin-coated coverslips at a concentration of approximately 1 × 105 cells/ml. This protocol reproducibly generates cultures containing up to 90% neurons by day 5 after RA addition; the remaining cells are astrocytes and a small proportion of fibroblast-like cells.
Fresh stocks of 3.0 mM methylmercury chloride (ICN, Aurora, OH) were prepared in distilled water 24-48 hrs before use and diluted to appropriate working solutions in medium immediately prior to use. Undifferentiated EC cells or day 5 neurons (5 days post-RA addition) were incubated in 1.5 μM MeHg for 50 min for live cell studies and 4 hrs for immunofluorescent localization of cyt c. A subset of cells was pre-incubated (prior to dye loading) in 10 μM CsA (Sigma, St. Louis, MO) for 30 min to block MPT pore opening.
2.2 Confocal microscopy
Confocal microscopy was used to detect early MeHg-induced ROS production and alterations of mitochondrial membrane potential in living cells. Images were captured on a Noran OZ laser scanning confocal microscope (Middleton, WI) equipped with a Krypton-Argon 50 mW multi-line laser. A 60x oil-immersion objective was used at a slit width setting of 15 mm. Experiments subsequently were replicated using a comparably equipped Leica TCS-SP2 laser scanning confocal microscope (Heitelburg, Germany) with similar results.
2.2.1 Detection of ROS production
Cells were incubated in 2.0 μM 6-carboxy-dichlorofluoroscin diacetate (DCFH-DA, Molecular Probes, Eugene, OR) (ex. 488, em. 525) at 37 °C for 30 min, rinsed in fresh medium, and placed on a 37 °C-warmed microscope stage in Hank’s balanced salt solution (HBSS). MeHg (1.5 μM) or an equal volume of HBSS for controls was added immediately after the first image acquisition (0 min, t0). Images were subsequently captured at 10 min intervals for 50 min. DCFH-DA, a stable non-fluorescent molecule which readily crosses cell membranes, is hydrolyzed by esterases to DCFH, trapped inside the cell and oxidized by ROS to the fluorescent DCF, providing a semi-quantitative measure of ROS generation.
2.2.2 Detection of mitochondrial membrane potential (Ψm) loss
Cells were loaded with 150 nM tetramethylrhodamine methyl ester (TMRM) (ex. 568) at 37 °C for 15 min, followed by addition of 1.0 μM calcein acetoxymethyl ester (AM) (ex. 488) to the TMRM incubate for the final 15 min. Both dyes were obtained from Molecular Probes (Eugene, OR). Cells were then rinsed in fresh medium and mounted on the stage in HBSS containing 15 nM TMRM, to which 1.5 μM MeHg or vehicle was added immediately after the first image acquisition (0 min). Images were captured at 5 min intervals for 50 min. The red-fluorescent TMRM is cleaved intracellularly; this positively charged form accumulates electrophoretically within mitochondria and is released upon depolarization of the inner mitochondrial membrane. The lipid soluble calcein ester readily enters the cell and is hydrolyzed to its negatively-charged free acid which becomes trapped in the cytoplasm (green fluorescence).
2.2.3 Immunofluorescent localization of cytochrome c
Release of cyt c from the mitochondrial intermembrane space is widely regarded as an early step in the apoptotic cell death pathway (Ravagnan et al., 2002; Robertson and Orrenius, 2000; Spierings et al., 2005). Undifferentiated P19 cells and day 5 neurons were plated on coverslips and incubated in fresh medium or in 1.5 μM MeHg in medium for 4 hrs, with or without pre-incubation with 1.0 μM CsA. Parallel controls were run for each treatment group. Following treatment, cells were rinsed in PBS, fixed in 4% paraformaldehyde for 20 min, and incubated for 5 min in cold methanol. After rinsing in PBS, cells were blocked in goat serum, incubated (1:500) in the primary antibody, anti-mouse cytochrome c (BD-Pharmingen monoclonal, 65971A) for 30 min and in Texas Red-linked goat anti-mouse IgG secondary (ex. 568) for 30 min (1:2000 dilution). Cells were rinsed 3 times in PBS (pH 7.6) and mounted on glass slides using the Prolong Anti-fade kit (Molecular Probes, P-7481). Images of the fixed cells were collected on the laser scanning confocal microscope.
2.4 Statistical analysis
For each cell, fluorescence values were first divided by background AOI fluorescence and converted to percent fluorescence of t0. For the purpose of the analyses, the average fluorescence of each separate experiment (3-20 cells per experiment) was calculated; these values represented the ‘n’ for each group. Separate analyses were run for undifferentiated vs. neurons and for DCF and TMRM, resulting in 4 main analyses. The ‘n’ for each group and analysis is specified in the figure legends. A repeated measures ANOVA was used to detect effect of time, treatment and interaction of treatment over time. Significant effects were followed up by Fisher’s PLSD post hoc with significance set at p<0.05.
3. Results
3.1 DCF fluorescence
Laser confocal microscopy of dichlorofluorescein (DCF) fluorescence was used to semi-quantitatively assess oxidative stress. The reduced non-fluorescent DCFH is oxidized by intracellular peroxides (and potentially other ROS) to the fluorescent DCF, which has been commonly used as an index of ROS production (Lebel and Bondy, 1990; Oyama et al, 1994; Voie and Fonnum, 2000).
Undifferentiated P19 cells demonstrated a significant increase in DCF levels over time (F(5,50)=6.3, p<0.001). The increase in DCF levels was most evident in control cells which rose to about 142% of t0 levels during the 50 minutes of incubation (Figure 1b). This increase was statistically significant from 30-50 minutes. MeHg seemed to suppress this increase compared to vehicle control cells to about 114% of t0 fluorescence (significant at 50 minutes). P19 cells pretreated with CsA-only tended to increase over their own t0 values, but this was significant only at 50 min (127% of their t0 levels) and was not statistically different from the vehicle control at any time point. P19 cells treated with MeHg + CsA were relatively unaffected over this exposure period and essentially indistinguishable (with an average increase at 50 minutes to 121% of their t0) from the MeHg-only group (Figure 1b).
Figure 1.
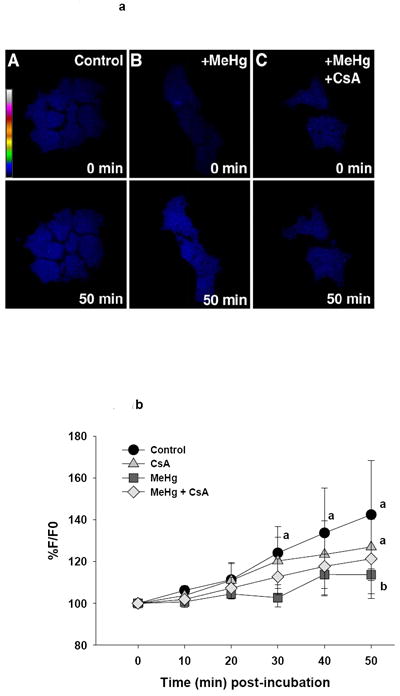
a (top). Representative confocal images of ROS production in live undifferentiated P19 cells as visualized by DCF fluorescence. Pseudocolor scale bars indicates fluorescence intensity, with white most intense and blue least intense. Treatment groups are listed on the image.
b (bottom). Graphical representation of change in DCF fluorescence in undifferentiated P19 cells relative to baseline. Each line represents the average fluorescence of separate experiments where n=3 for control and CsA, and n=4 for MeHg and MeHg + CsA.
a=significantly different from t0
b=significantly different from control treated cells
c=significantly different from CsA treated cells
d=MeHg + CsA significantly different from MeHg-only treated cells, using a Fisher’s PLSD, p<0.05 following a repeated measures ANOVA with time and treatment as independent variables.
In D5 P19-derived neurons, there was a significant increase in DCF levels over time (F(5, 70)=18.8, p<0.001). There was also an effect of treatment (F(3, 14)=3.6, p=0.04), and an interaction of treatment and time after incubation (F(15, 70)=4.3, p<0.001). As seen in Figure 2b, neurons treated with MeHg exhibited an increase in DCF fluorescence from 20-50 minutes post-incubation. This increase from its own t0 value became significant at 20 minutes and reached 249% of baseline at 50 minutes, while the increase over vehicle control and CsA-only controls became significant from 30-50 minutes. Pre-incubation with the MPT pore-blocking agent CsA largely protected cells against this MeHg-induced increase (statistically significant at 30-50 minutes). DCF fluorescence in MeHg + CsA treated neurons (144% of t0) was similar to that of vehicle controls and CsA-only controls (approximate 117% and 132% of t0, respectively, figure 2b). The ability of CsA to protect against neuronal ROS production suggests that MeHg-induced mitochondrial dysfunction may occur prior to the significant increase in cellular ROS in D5 neurons. The CsA-only group was not significantly different from vehicle control neurons at any time point, and while MeHg + CsA had a slight, gradual increase over its t0 value, this was significant at 50 minutes only.
Figure 2.
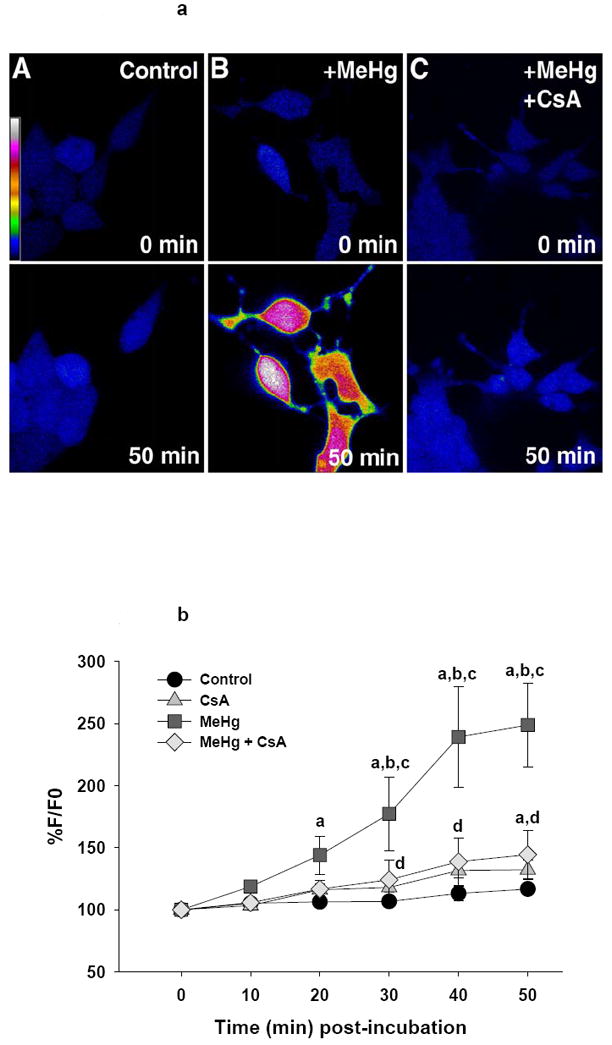
a (top). Representative confocal images of ROS production in live D5 neurons visualized by DCF fluorescence. Pseudocolor scale bar indicates fluorescence intensity. Treatment groups are listed on the image.
b (bottom). Graphical representation of change in DCF fluorescence in these cells relative to baseline. Each line represents the average fluorescence of separate experiments where n=5 for control, n=3 for CsA, n=7 for MeHg and n= 3 for MeHg + CsA.
a=significantly different from t0
b=significantly different from control treated cells
c=significantly different from CsA treated cells
d=MeHg + CsA significantly different from MeHg-only treated cells, using a Fisher’s PLSD, p<0.05 following a repeated measures ANOVA with time and treatment as independent variables.
3.2 TMRM fluorescence
Tetramethylrhodamine methyl ester (TMRM) accumulates electrophoretically in mitochondria and is used as an index of membrane potential (Ψm). Control undifferentiated P19 cells showed little change in Ψm throughout the 50 minute incubation period. MeHg induced a gradual and almost linear decrease in TMRM fluorescence, reaching 57% of baseline by 50 minutes of incubation, indicating a progressive and statistically significant loss of Ψm (20-50 minutes) (F(3,12)=3.7, p=0.04) (Figure 3b). Pre-incubation of MeHg-treated cells with CsA protected against this effect at 20-50 minutes post-incubation, exhibited by a significant treatment × time interaction (F(15, 60)=3.6, p<0.001) (Figure 3b). Further, vehicle control, CsA only, and CsA + MeHg were not significantly different from each other at any time point except at 30 minutes, where MeHg + CsA peaked at 117% of its baseline level. Protection against loss of Ψm by CsA suggests that the MeHg-stimulated loss of Ψm was associated, at least in part, with the opening of the MPT pore (Figure 3b).
Figure 3.
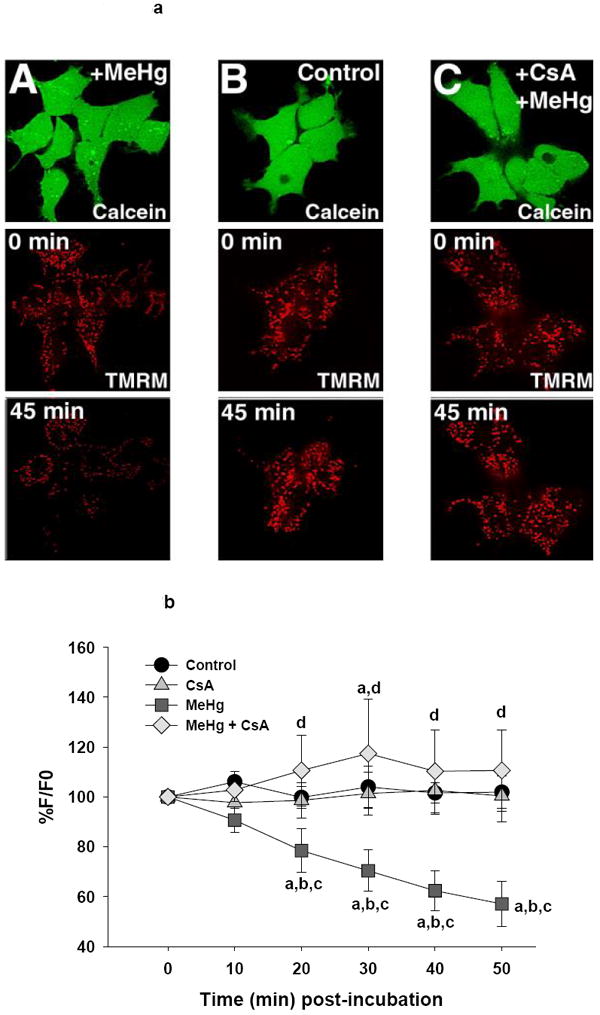
a (top). Representative confocal images of Ψm in live undifferentiated P19 cells as visualized by TMRM fluorescence. Pseudocolor scale bar indicates fluorescence intensity. Treatment groups are listed on the image.
b (bottom). Graphical representation of change in TMRM fluorescence in the undifferentiated P19 cells relative to baseline. Each line represents the average fluorescence of separate experiments where n=4 for control, n=5 for CsA, n=4 for MeHg and n= 3 for MeHg + CsA.
a=significantly different from t0
b=significantly different from control treated cells
c=significantly different from CsA treated cells
d=MeHg + CsA significantly different from MeHg-only treated cells, using a Fisher’s PLSD, p<0.05 following a repeated measures ANOVA with time and treatment as independent variables.
P19-derived neurons demonstrated a markedly different pattern of TMRM fluorescence than undifferentiated cells (Figure 4a). There was a significant effect of time (F(5,55)=11.1, p<0.001), treatment (F(3,11)=11.9, p<0.001) and an interaction of time by treatment (F(15, 55)=8.1, p<0.001). A graphical representation of the effects of MeHg and CsA is shown in Figure 4b. While the control neurons showed an early increase in TMRM fluorescence to approximately 119% of baseline by 20 minutes, MeHg treatment induced a significant immediate (10 minutes) and progressive decrease in fluorescence, falling to 39% of the t0 baseline by 30 minutes (then gradually falling to 31% by the end of the study). In contrast, neurons pre-incubated with CsA showed an initial resistance to MeHg-induced loss of mitochondrial TMRM. However, these neurons abruptly began to lose fluorescence between 20-30 minutes of MeHg exposure and showed a gradual decline over the ensuing 20 minutes. By 40 minutes, they had lost 50% of their t0 TMRM staining and were not significantly different from neurons treated with MeHg alone (Figure 4b). The TMRM fluorescence of CsA only-pretreated neurons was stable, remaining near 100% of t0 throughout the 50 minute incubation period.
Figure 4.
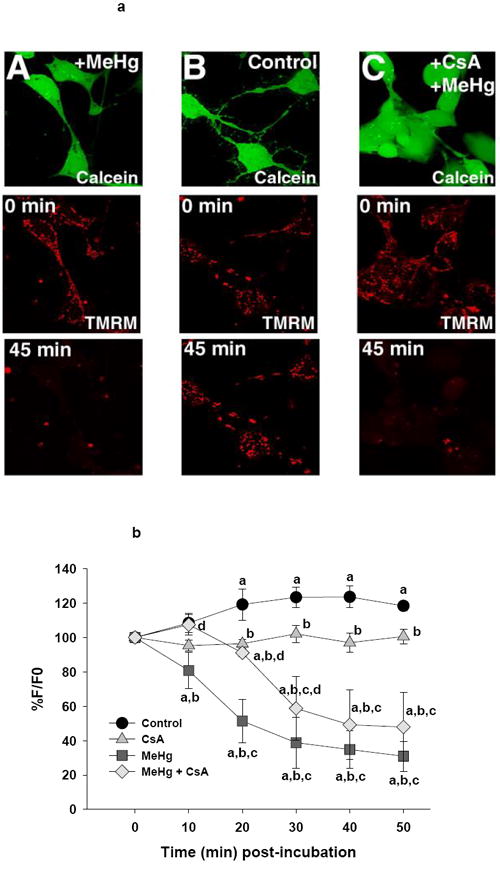
a (top). Representative confocal images of Ψm in live D5 neurons cells as visualized by TMRM fluorescence. Pseudocolor scale bar indicates fluorescence intensity. Treatment groups are listed on the image.
b (bottom). Graphical representation of change in TMRM fluorescence over time in these cells relative to baseline. Each line represents the average fluorescence of separate experiments where n=3 for control, n=4 for CsA, n=5 for MeHg and n= 3 for MeHg + CsA.
a=significantly different from t0
b=significantly different from control cells
c=significantly different from CsA treated cells
d=MeHg + CsA significantly different from MeHg-only treated cells, using a Fisher’s PLSD, p<0.05 following a repeated measures ANOVA with time and treatment as independent variables.
3.3 Cytochrome c immunohistochemistry
Release of cytochrome c from the mitochondria is an early step in the apoptotic sequence. Under normal physiological conditions, immunocytochemical staining of cyt c is evident as bright, punctuate dots reflecting its localization to the mitochondria. With toxic insult or other apoptosis-inducing stimuli, cyt c staining becomes diffuse and dim, reflecting its release from the mitochondria and dispersion into the cytoplasm. In the present study, both control P19 cells and day 5 neurons showed bright, punctuate staining of cyt c (Figure 5). Following 4hr incubation with 1.5 μM MeHg, undifferentiated P19 cells showed a slight and variable loss of cyt c with little discernable effect of 1.0 μM CsA pre-treatment. In contrast, neurons showed a dimming and diffusion of cyt c; pre-incubation with CsA largely protected neurons against this change.
Figure 5.
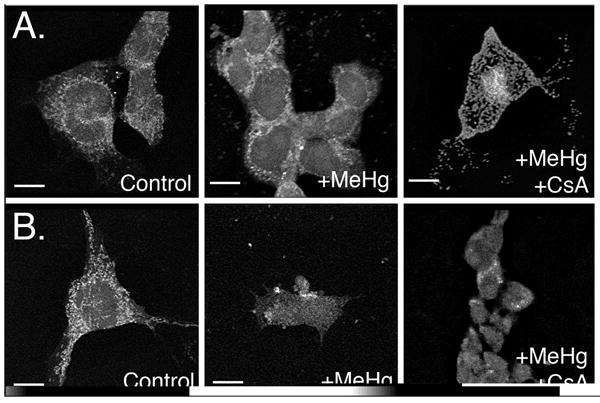
Representative immunofluorescent confocal images of cytochrome c distribution. Treatment groups are listed on the image. (A) Undifferentiated EC cells exposed to MeHg or MeHg + CsA. (B) Differentiated D5 neurons exposed to MeHg or MeHg and CsA. Scale bars =10 μm.
4. Discussion
Numerous investigators have described greater sensitivity of the developing brain to MeHg, but the mechanisms underlying this phenomenon remain obscure. Intrinsic physiological and biochemical differences between precursor cells and mature neurons have been postulated to underlie MeHg-induced developmental neurotoxicity (for review, see Costa et al., 2004; Limke et al., 2004). A common link among many proposed pathogenetic mechanisms is the depletion of intracellular GSH and critical protein thiols and inhibition of critical antioxidant enzymes (Kromidas et al., 1990; Ou et al., 1999; Sarafian and Verity, 1991; Yee and Choi, 1994; Yonaha et al., 1983). This would lead to increased vulnerability of neural cells to reactive oxygen species (ROS) which may originate from numerous cellular sites, including cytochrome p450 isozymes (Parke and Sapota, 1996), auto-oxidation of various flavin coenzymes, low molecular weight thiols, thiol-containing macromolecules, ascorbic acid, and reactive nitrogen species (Chandra et al., 2000).
The mitochondrion has long been recognized as an important target of MeHg poisoning (Bondy and McKee, 1991; Fowler and Woods, 1977a, 1977b; Sone et al., 1977; Verity et al., 1975; Yoshino et al., 1966), as well as a major source of constitutive cellular ROS (Jezek and Hlavata, 2005; Turrens, 2003). Evidence exists supporting the contribution of mitochondrial-mediated oxidative stress to MeHg neurotoxicity, but the majority of studies have been performed in adult animals or mature cells (Bondy et al., 1990; Sarafian et al., 1994; Yee and Choi, 1994, 1996), and the potential role of ROS in developmental MeHg neurotoxicity is poorly understood. Developing/immature CNS neurons (cerebellar granule and cortical neurons) exhibit greater MeHg-induced ROS production compared to mature neurons (Mundy and Freudenrich, 2000), and Tsatmali and coworkers (2005) showed that embryonic rat cortical neurons exhibit enhanced basal generation of ROS and higher levels of several mitochondrial respiratory chain components compared to their neural precursors and to glial cells. However, broad generalizations with respect to putative roles of enhanced ROS production in developing neurons based upon extrapolation from studies of mature cells/tissues should be interpreted with caution.
ROS generated by mitochondria may initiate or augment important mitochondrial alterations, including loss of Ψm and stimulation of the mitochondrial permeability transition (MPT). Cytochrome c, an electron carrier in the electron transport chain and key mediator in the mitochondrial pathway of cell death, is released from mitochondria subsequent to MPT activation (Lemasters et al., 2002; Ravagnan et al., 2002), although the precise mechanism of release is unresolved and likely involves several different pathways (Gogvadze et al., 2004). Cyclosporin A (CsA), which blocks the MPT pore, delays MeHg-induced increases in intracellular Ca2+ and loss of Ψm, and protects against MeHg-induced cell mortality in cerebellar granule cell cultures (Limke and Atchison, 2002; Maldonado et al., 2000). These studies strongly implicate a role for the MPT in certain models of cell death, yet MeHg-induced alterations of Ψm or cyt c release and CsA protection in developing neurons (in vivo or in vitro) remain unexplored.
4.1 ROS/oxidative stress in MeHg toxicity
Studies demonstrating amelioration of MeHg neurotoxicity with vitamin E (Chang et al., 1978; Ganther, 1978; Welsh and Soares, 1976) and other antioxidants such as the iron chelator desferoxamine, selenium, and vitamins A and C indicate a role for oxidative stress in MeHg-mediated neurotoxicity (LeBel et al., 1992; Prasad and Ramanujam, 1980; Sarafian and Verity, 1991). In our studies, undifferentiated cells showed lower levels of ROS production in response to 1.5 μM MeHg for 50 min relative to their controls. MeHg alone and MeHg + CsA showed similar elevations of their t0 DCF level (114 and 121%, respectively, at 50 min). The reason for this apparent MeHg-induced inhibition is unclear. D5 neurons, however, responded to MeHg challenge with a robust surge of ROS generation that began at about 20-30 min after addition of 1.5 μM MeHg and increased progressively until the experiment was terminated.
Nohl et al. (2005) suggest that as undifferentiated cells rely more on glycolysis, they should generate more ROS as measured by DCF, and others have found constitutively high ROS levels in neural stem cells (Le Belle et al, 2011). It is possible that undifferentiated P19s have slightly elevated levels of basal ROS production in comparison to their neuronal derivatives, but this may not necessarily appear as a greater stimulation of MeHg-induced ROS production in comparison to D5 neurons. Alternatively, the P19s may have different antioxidant defenses, including different antioxidant molecules, enzyme levels, or enzyme activities. Indeed, Oh and coworkers (2005) found three antioxidant proteins expressed only in undifferentiated neuroblastoma cells and eight that were detected exclusively following neuronal differentiation. Further, studies in our laboratory indicate that undifferentiated P19s have high levels of GSH and cytosolic GSH levels are reported to be significantly lower in mature neurons than other neural types such as astrocytes (Aschner et al., 1994; Huang and Philbert, 1996; Lowndes et al., 1994; Philbert et al., 1991).
Pretreatment of P19 cells with CsA had essentially no effect on the relative ROS production compared to P19 cells administered MeHg only. Interestingly, while CsA treatment had only limited protection against diminishing Ψm in MeHg-treated neurons, it largely prevented the neuronal burst of ROS production, with DCF levels essentially equivalent to vehicle-treated controls. Protection of neurons by CsA against ROS production suggests that, at least early in MeHg exposure, MPT pore opening is delayed, and the early burst of ROS is prevented by this delay. However, MeHg may still cause a diminution of Ψm by other mechanisms (i.e., by affecting other mitochondrial membrane channels, ion transporters or pore formation by Bcl-2 family members) (see Zoratti et. al, 2005).
It is worth noting that the exact nature of intracellular DCFH-oxidizing substances is not known. DCFH has been proposed to be oxidized by H2O2, alkyl hydroperoxides and potentially by the hydroxy free radical, but is not likely to be directly oxidized by superoxide anion (LeBel and Bondy, 1990). Thus, prevention of the major increase in DCF fluorescence cannot be interpreted as protection against elevation of all species of cellular ROS. Furthermore, whole cell DCF fluorescence does not specifically distinguish mitochondrial ROS. Superoxide anion, a major product of normal and disrupted mitochondrial metabolism, is unlikely to travel far from its site of generation and while some superoxide will be converted to H2O2 by MnSOD, there is high potential for mitochondrial-generated ROS to interact with the mitochondrial membrane. In theory, such locally generated ROS would not produce a large increase in DCF fluorescence if the majority reacts with nearby mitochondrial membranes rather than the DCFH molecule. In fact, using the mitochondrial specific ROS-sensitive mitotracker dye CM-H2XRos (chloromethyl derivative of dihydro X-rosamine) in MeHg-treated astrocytes, Shanker et al. (2004) demonstrated that elevation of mitochondrial ROS production preceded elevation of cellular ROS as measured by DCF fluorescence. Consequently, it may be possible to observe nearly complete protection by exogenous antioxidants against ROS formation measured by DCF fluorescence at the whole cellular level, yet miss the biologically significant ROS produced within mitochondria.
4.2 Loss of mitochondrial membrane potential (Ψm) in MeHg toxicity
Mitochondria not only contribute to cellular energy production, but significantly affect the distribution of ATP for execution of cellular functions, the division of electron flow to O2 and to reductive biosynthetic reactions, and the integration of metabolic pathways (Pendersen, 1978). Further, maintenance of ATP production is required for cells to undergo apoptotis-like cell death (Lemasters et al., 2002). ATP is also required in the maintenance of the mitochondrial membrane potential; loss of Ψm has been reported to be a stimulator of the mitochondrial permeability transition (MPT) (Petronilli et al., 2001).
Under normal physiological conditions, the mitochondrial matrix is highly negatively charged relative to the cytoplasm. Prolonged loss of Ψm is associated with MPT and is held to be an early indicator of compromised mitochondrial function. In undifferentiated P19 cells, MeHg exposure caused a gradual decline in Ψm to approximately 57% of control levels (statistically significant from 20-50 min); CsA pre-treatment completely inhibited loss of Ψm over the 50 min exposure period, suggesting that this change in membrane potential was consequent to opening of the CsA-sensitive permeability transition pore. In contrast, in D5 P19-derived neurons, MeHg induced a rapid and sustained decrease in mitochondrial membrane potential to 39% of baseline by 30 min. The enhanced sensitivity of the neurons may represent developmental differences in the MPT pore itself or the presence of other channels regulating membrane potential. Alternatively, the undifferentiated cells may be more efficient at sequestering or exporting MeHg from the cell, which would be expected to decrease the magnitude of several MeHg-induced toxic responses and is generally consistent with the previously described effects on ROS production. However, we have no direct evidence of such a difference. CsA initially (significant from 10-30 min) inhibited the loss of neuronal Ψm, yet by 40 min there was no longer a protective effect compared to the MeHg-only group. Limke and Atchison (2002) observed a similar time course in MeHg +/- CsA-treated cerebellar granule cells. Thus, in D5 neurons the CsA-sensitive MTP may be initially responsible for alterations in membrane potential but with continued MeHg-induced insult, other mechanisms (possibly another membrane channel formed by Bax/Bak, or MPT pores lacking cyclophilin D) may contribute to the eventual observed loss of TMRM staining (Friberg and Wieloch, 2002; Zoratti et al., 2005).
The D5 neurons were protected by CsA pretreatment against the MeHg-induced stimulation of cellular ROS, even while undergoing major loss of Ψm. The time-course of these two processes (decrease in TMRM preceding the increase in DCF fluorescence by approximately 15 min) suggests that either 1) the elevation of ROS production is only delayed in neurons and would eventually occur with longer incubation in MeHg even with CsA pretreatment; 2) that downstream effects of mediators released from non-CsA sensitive pores are largely responsible for the increased ROS in D5 neurons; or 3) that the observed increase in ROS is largely independent of the MPT and perhaps due to stimulation of other sources of ROS in the cytoplasm. A combination of the latter two mechanisms is possible; nevertheless, if protection is relatively complete and not simply delayed beyond the timepoints we examined, this implies that CsA has other, non-MPT mediated, protective effects in the D5 neurons that ameliorate the MeHg-stimulated cellular ROS production. Further, the formation and activation of an MPT pore is highly complex process, and pores even within the same mitochondrion exhibit differing composition, conformation, and sensitivity to both cellular stressors and protective agents (Zoratti et al., 2005). Possible heterogeneity of the MPT pore could have significant impact on mitochondrial responses to toxicants and would be a fruitful avenue for future research.
4.3 Alteration in cytochrome c localization in MeHg toxicity
The mitochondrion plays a central role in the regulation of cell death pathways through direct interactions with pro- and anti-apoptotic molecules (Bcl-2 family) and/or release of apoptogenic proteins such as AIF, endonuclease G, and cyt c (Ravagnan et al., 2002; Robertson and Orrenius, 2000). Cyt c, an important electron carrier in the electron transport chain, is intimately involved in many models of cell death. Under normal physiological conditions, cyt c is localized to the mitochondrial intermembrane space. Following many toxicological insults that induce the MPT, cyt c is released from the mitochondria and dispersed into the cytoplasm.
In the present study, both control P19 cells and D5 neurons showed bright, punctate staining of cyt c, reflecting its normal mitochondrial localization. Following a 4 hr incubation with 1.5 μM MeHg, P19 cells showed only modest dimming of cyt c fluorescence, whereas this change was clearly evident in treated neurons. While few investigators have studied MeHg-induced cyt c release from neuronal mitochondria, Shenker et al. (2002) similarly observed cyt c accumulation in the cytosol 4 to 8 hrs following MeHg-induced decline of Ψm in human T lymphocytes. In agreement with TMRM studies, D5 neurons appeared to be both more sensitive to MeHg (greater diffusion and dimming of cyt c label) than their undifferentiated counterparts and only partially protected by CsA pre-treatment. This suggests that CsA is only delaying MPT pore opening in the neurons, or that cyt c is, at least in part, leaving the mitochondria by some mechanism other than MPT pore opening.
The present studies reveal several fundamental points. In this model, major cellular ROS production measured by DCF fluorescence appears to be a downstream consequence of early MeHg-induced mitochondrial perturbations, first affecting mitochondrial redox status, then cellular ROS production and signaling pathways. While our data support the mitochondrion to be an early target of MeHg neurotoxicity, the magnitude and specific consequences of mitochondrial dysfunction likely depend on the dose, cell type and, particularly, the stage of development. D5 neurons were more sensitive to MeHg-induced ROS generation and loss of Ψm than undifferentiated P19 cells. Maturational changes in mitochondrial content, metabolism or pore dynamics, as well as differences in antioxidant systems/activity during development, likely contribute to the differential response to low-dose MeHg insult and to the degree of production afforded by CsA.
It is important to note that our measure of membrane potential is an average of many mitochondria with pores in various stages of opening: some transient, some prolonged. This average measure of Ψm is useful in estimating the overall status of the cell, but oversimplified in characterizing the mechanism of MeHg influence on single transmembrane potentials and individual pore opening. Similarly, potential limitations of using the DCF assay to measure ROS production include the undefined nature of the species that oxidize DCFH and the manner in which the whole cell appears to fluoresce, which obscures the precise site(s) of ROS production. Thus, conservative interpretation of the relative changes in fluorescence of these dyes must be enforced and future studies are indicated to more clearly delineate the mechanisms and time-course of MeHg-induced mitochondrial disruption, MPT, and enhanced generation of ROS.
RESEARCH HIGHLIGHTS.
The role of differentiation state on methylmercury (MeHg) toxicity was studies in P19 cells and differentiated neurons.
MeHg induced greater ROS production in neurons than P19 cells.
Mitochondrial depolarization and cytochrome c release were greater in neurons than P19 cells.
Cyclosporin A was more protective in P19 cells than neurons.
Differentiation state influences mitochondrial transition pore responses to MeHg.
Acknowledgments
This work was supported by NIH ES 011256, ES 007148, ES 005022 (MP, KR) and ES 08846 (MAP). The authors gratefully acknowledge the assistance of Dr. Carol Gardner.
Footnotes
Conflict of interest The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali SF, LeBel CP, Bondy SC. Reactive oxygen species formation as a biomarker of methylmercury and trimethyltin neurotoxicity. Neurotoxicology. 1992;13:637–48. [PubMed] [Google Scholar]
- Allen RG, Balin AK. Oxidative influence on development and differentiation: an overview of a free radical theory of development. Free Radic Biol Med. 1989;6:631–61. doi: 10.1016/0891-5849(89)90071-3. [DOI] [PubMed] [Google Scholar]
- Aschner M, Mullaney KJ, Wagoner D, Lash LH, Kimelberg HK. Intracellular glutathione (GSH) levels modulate mercuric chloride (MC)- and methylmercuric chloride (MeHgCl)-induced amino acid release from neonatal rat primary astrocytes cultures. Brain Res. 1994;664:133–40. doi: 10.1016/0006-8993(94)91963-1. [DOI] [PubMed] [Google Scholar]
- Bain G, Ray WJ, Yao M, Gottlieb DI. From embryonal carcinoma to neurons: the P19 pathway. Bioessays. 1994:343–48. doi: 10.1002/bies.950160509. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Brekemeier KM, Pfeiffer DR. Recent progress on regulation of the mitochondrial trasition pore; a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr. 1994;26:509–17. doi: 10.1007/BF00762735. [DOI] [PubMed] [Google Scholar]
- Bondy SC, McKee M. Disruption of the potential across the synaptosomal plasma membrane and mitochondria by neurotoxic agents. Toxicol Lett. 1991;58:13–21. doi: 10.1016/0378-4274(91)90185-9. [DOI] [PubMed] [Google Scholar]
- Bondy SC, McKee M, Davoodbhoy YM. Prevention of chemically induced changes in synaptosomal membrane order by ganglioside GM1 and alpha-tocopherol. Biochim Biophys Acta. 1990;2:213–9. doi: 10.1016/0005-2736(90)90066-w. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Levonen AL, Shiva S, Sarti P, Darley-Usmar VM. Mitochondria: regulators of signal transduction by reactive oxygen and nitrogen species. Free Radic Biol Med. 2002;33:755–64. doi: 10.1016/s0891-5849(02)00901-2. [DOI] [PubMed] [Google Scholar]
- Chandra J, Samali J, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med. 2000;29:323–33. doi: 10.1016/s0891-5849(00)00302-6. [DOI] [PubMed] [Google Scholar]
- Chang L, Gilbert M, Sprecher J. Modification of methylmercury neurotoxicity by vitamin E. Environ Res. 1978;17:356–66. doi: 10.1016/0013-9351(78)90040-3. [DOI] [PubMed] [Google Scholar]
- Costa LG, Aschner M, Vitalone A, Syversen T, Soldin OP. Developmental neuropathology of environmental agents. Annu Rev Pharmacol Toxicol. 2004;44:87–110. doi: 10.1146/annurev.pharmtox.44.101802.121424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan JB, Tymms MJ, Cristiano F, Kola I. Expression of copper/zinc superoxide dismutase and glutathione peroxidase in organs of developing mouse embryos, fetuses, and neonates. Pediatr Res. 1994;35:188–96. doi: 10.1203/00006450-199402000-00013. [DOI] [PubMed] [Google Scholar]
- Dreiem A, Gertz CC, Seegal RF. The effects of methylmercury on mitochondrial function and reactive oxygen species formation in rat striatal synaptosomes are age-dependent. Toxicol Sci. 2005;87:156–62. doi: 10.1093/toxsci/kfi224. [DOI] [PubMed] [Google Scholar]
- Fantel AG. Reactive oxygen species in developmental toxicity: review and hypothesis. Teratology. 1996;6:196–217. doi: 10.1002/(SICI)1096-9926(199603)53:3<196::AID-TERA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Fowler B, Woods J. The transplacental toxicity of methylmercury to fetal rat liver mitochondria. Lab Invest. 1977a;36:122–30. [PubMed] [Google Scholar]
- Fowler BA, Woods JS. Ultrastructural and biochemical changes in renal mitochondria during chronic oral methyl mercury exposure: the relationship to renal function. Exp Mol Pathol. 1977b;27:403–12. doi: 10.1016/0014-4800(77)90010-7. [DOI] [PubMed] [Google Scholar]
- Friberg H, Ferrand-Drake M, Bengtsson F, Halestrap AP, Wieloch T. Cyclosporin A, but not FK506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. J Neurosci. 1998;18:5151–59. doi: 10.1523/JNEUROSCI.18-14-05151.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg H, Wieloch T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie. 2002;84:241–50. doi: 10.1016/s0300-9084(02)01381-0. [DOI] [PubMed] [Google Scholar]
- Ganther H. Modification of methylmercury toxicity and metabolism by selenium and vitamin E: possible mechanisms. Environ Health Perspect. 1978;25:71–6. doi: 10.1289/ehp.782571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasso S, Cristofol RM, Selema G, Rosa R, Rodriguez-Farre E, Sanfeliu C. Antioxidant compounds and Ca(2+) pathway blockers differentially protect against methylmercury and mercuric chloride neurotoxicity. J Neurosci Res. 2001;66:135–45. doi: 10.1002/jnr.1205. [DOI] [PubMed] [Google Scholar]
- Gogvadze V, Robertson JD, Enoksson M, Zhivotovsky B, Orrenius S. Mitochondrial cytochrome c release may occur by volume-dependent mechanisms not involving permeability transition. Biochem J. 2004;378:213–7. doi: 10.1042/BJ20031193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP. What is the mitochondrial transition pore? J Mol Cell Cardiol. 2009;46:821–31. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Hare MF, Atchison WD. Comparative action of methylmercury and divalent inorganic mercury on nerve terminal and intraterminal mitochondrial membrane potentials. J Pharmacol Exp Ther. 1992;261:166–72. [PubMed] [Google Scholar]
- Huang J, Philbert MA. Cellular responses of cultured cerebellar astrocytes to ethacrynic acid-induced perturbation of subcellular glutathione homeostasis. Brain Res. 1996;711:184–92. doi: 10.1016/0006-8993(95)01376-8. [DOI] [PubMed] [Google Scholar]
- Hunter D, Haworth R. The Ca2+ -induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch Biochem Biophys. 1979;195:468–77. doi: 10.1016/0003-9861(79)90373-4. [DOI] [PubMed] [Google Scholar]
- Jezek P, Hlavata L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol. 2005;37:2478–2503. doi: 10.1016/j.biocel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Jones-Villeneuve E, McBurney M, Rogers K, Kalins V. Retinoic acid induces embryonal carcinoma cells to differentiate into neurons and glial cells. J Cell Biol. 1982;94:253–62. doi: 10.1083/jcb.94.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Villeneuve E, Rudnicki M, Harris S, McBurney M. Retinoic acid-induced neural differentiation of embryonal carcinoma cells. Mol Cell Biol. 1983;3:2271–9. doi: 10.1128/mcb.3.12.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J, Cena V, Prehn JH. Mitochondrial control of neuron death and its role in neurodegenerative disorders. J Physiol Biochem. 2003;59:129–41. doi: 10.1007/BF03179878. [DOI] [PubMed] [Google Scholar]
- Kromidas L, Trombetta L, Jamall I. The protective effects of glutathione against methylmercury cytotoxicity. Toxicol Lett. 1990;51:67–80. doi: 10.1016/0378-4274(90)90226-c. [DOI] [PubMed] [Google Scholar]
- LeBel CP, Ali SF, Bondy SC. Deferoxamine inhibits methyl mercury-induced increases in reactive oxygen species formation in rat brain. Toxicol Appl Pharmacol. 1992;112:161–5. doi: 10.1016/0041-008x(92)90292-z. [DOI] [PubMed] [Google Scholar]
- LeBel CP, Ali SF, McKee M, Bondy SC. Organometal-induced increases in oxygen reactive species: the potential of 2’,7’-dichlorofluorescin diacetate as an index of neurotoxic damage. Toxicol Appl Pharmacol. 1990;104:17–24. doi: 10.1016/0041-008x(90)90278-3. [DOI] [PubMed] [Google Scholar]
- LeBel CP, Bondy SC. Sensitive and rapid quantitation of oxygen reactive species formation in rat synaptosomes. Neurochem Int. 1990;17:435–40. doi: 10.1016/0197-0186(90)90025-o. [DOI] [PubMed] [Google Scholar]
- Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, Wu H, Kornblum HI. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a P13K/Akt-dependent manner. Cell Stem Cell. 2011;8:59–71. doi: 10.1016/j.stem.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, et al. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4:769–81. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- Limke TL, Atchison WD. Acute exposure to methylmercury opens the mitochondrial permeability transition pore in rat cerebellar granule cells. Toxicol Appl Pharmacol. 2002;178:52–61. doi: 10.1006/taap.2001.9327. [DOI] [PubMed] [Google Scholar]
- Limke TL, Heidemann SR, Atchison WD. Disruption of intraneuronal divalent cation regulation by methylmercury: are specific targets involved in altered neuronal development and cytotoxicity in methylmercury poisoning? Neurotoxicology. 2004;25:741–60. doi: 10.1016/j.neuro.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Lowndes HE, Beiswanger CM, Philbert M, Reuhl KR. Substrates for neural metabolism of xenobiotics in adult and developing brain. Neurotoxicology. 1994;15:61–73. [PubMed] [Google Scholar]
- MacPherson P, McBurney M. P19 embryonal carcinoma cells: a source of cultured neurons amenable to genetic manipulation. Methods: a companion to Methods in Enzymology. 1995;7:238–52. [Google Scholar]
- Maldonado B, Autio D, Marty M, Atchison W. Protective effect of cyclosporin A on methylmercury (MeHg)-induced cell death in cerebellar granule cells. The Toxicologist. 2000;54:241. [Google Scholar]
- Mavelli I, Rigo A, Federico R, Ciriolo MR, Rotilio G. Superoxide dismutase, glutathione peroxidase and catalase in developing rat brain. Biochem J. 1982;204:535–40. doi: 10.1042/bj2040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBurney MW, Reuhl KR, Ally AI, Nasipuri S, Bell JC, Craig J. Differentiation and maturation of embryonal carcinoma-derived neurons in cell culture. J Neurosci. 1988;8:1063–73. doi: 10.1523/JNEUROSCI.08-03-01063.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy WR, Freudenrich TM. Sensitivity of immature neurons in culture to metal-induced changes in reactive oxygen species and intracellular free calcium. Neurotoxicology. 2000;21:1135–44. [PubMed] [Google Scholar]
- Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Cyclosporin A delays mitochondrial depolarization induced by N-Methyl-D-aspartate in cortical neurons: evidence of the mitochdrial permeability transition. Neuroscience. 1996;75:993–97. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- Nohl H, Gille L, Stankiek K. Intracellular generation of reactive oxygen species by mitochondria. Biochem Pharmacol. 2005;69:719–23. doi: 10.1016/j.bcp.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Oh J-E, Raja KK, Shin J-H, Hengstschlager M, Pollak A, Lubec G. The neuronal differentiation process involves a series of antioxidant proteins. Amino Acids. 2005;29:273–82. doi: 10.1007/s00726-005-0214-9. [DOI] [PubMed] [Google Scholar]
- Ou YC, White CC, Krejsa CM, Ponce RA, Kavanagh TJ, Faustman EM. The role of intracellular glutathione in methylmercury-induced toxicity in embryonic neuronal cells. Neurotoxicology. 1999;20:793–804. [PubMed] [Google Scholar]
- Oyama Y, Tomiyoshi F, Ueno S, Furukawa K, Chikahisa L. Methylmercury-induced augmentation of oxidative metabolism in cerebellar neurons dissociated from the rats: its dependence on intracellular Ca2+ Brain Res. 1994;660:154–7. doi: 10.1016/0006-8993(94)90849-4. [DOI] [PubMed] [Google Scholar]
- Parke DV, Sapota A. Chemical toxicity and reactive oxygen species. Int J Occup Med Environ Health. 1996;9:331–41. [PubMed] [Google Scholar]
- Pendersen PL. Tumor mitochondria and the bioenergetics of cancer cells. Prog Exp Tumor Res. 1978;22:190–274. doi: 10.1159/000401202. [DOI] [PubMed] [Google Scholar]
- Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem. 2001;276:12030–4. doi: 10.1074/jbc.M010604200. [DOI] [PubMed] [Google Scholar]
- Philbert MA, Beiswanger CM, Manson MM, Green JA, Novak RF, Primiano T, et al. Glutathione S-transferases and gamma-glutamyl transpeptidase in the rat nervous systems: a basis for differential susceptibility to neurotoxicants. Neurotoxicology. 1995;16:349–62. [PubMed] [Google Scholar]
- Philbert MA, Beiswanger CM, Waters DK, Reuhl KR, Lowndes HE. Cellular and regional distribution of reduced glutathione in the nervous system of the rat: histochemical localization by mercury orange and o-phthaldialdehyde-induced histofluorescence. Toxicol Appl Pharmacol. 1991;107:215–27. doi: 10.1016/0041-008x(91)90204-r. [DOI] [PubMed] [Google Scholar]
- Polunas M, Tjalkens R, Philbert M, Reuhl K. Neuronal differentiation state influences susceptibity to methylmercury-induced oxidative stress and mitochondrial perturbations. Neurotoxicology; 21st Internat Neurotoxicol Conf; Honolulu, HI. 2004. [Google Scholar]
- Prasad K, Ramanujam S. Vitamin E and vitamin C alter the effect of methylmercuric chloride on neuroblastoma and glioma cells in culture. Environ Res. 1980;21:343–51. doi: 10.1016/0013-9351(80)90036-5. [DOI] [PubMed] [Google Scholar]
- Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol. 2002;192:131–7. doi: 10.1002/jcp.10111. [DOI] [PubMed] [Google Scholar]
- Robertson JD, Orrenius S. Molecular mechanisms of apoptosis induced by cytotoxic chemicals. Crit Rev Toxicol. 2000;30:609–27. doi: 10.1080/10408440008951122. [DOI] [PubMed] [Google Scholar]
- Sarafian T, Verity MA. Oxidative mechanisms underlying methyl mercury neurotoxicity. Int J Dev Neurosci. 1991;9:147–53. doi: 10.1016/0736-5748(91)90005-7. [DOI] [PubMed] [Google Scholar]
- Sarafian TA, Bredesen DE. Is apoptosis mediated by reactive oxygen species? Free Radic Res. 1994;21:1–8. doi: 10.3109/10715769409056549. [DOI] [PubMed] [Google Scholar]
- Sarafian TA, Vartavarian L, Kane DJ, Bredesen DE, Verity MA. bcl-2 expression decreases methyl mercury-induced free-radical generation and cell killing in a neural cell line. Toxicol Lett. 1994;74:149–55. doi: 10.1016/0378-4274(94)90093-0. [DOI] [PubMed] [Google Scholar]
- Shanker G, Aschner JL, Syversen T, Aschner M. Free radical formation in cerebral cortical astrocytes in culture induced by methylmercury. Brain Res Mol Brain Res. 2004;128:48–57. doi: 10.1016/j.molbrainres.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Shanker G, Aschner M. Methylmercury-induced reactive oxygen species formation in neonatal cerebral astrocytic cultures is attenuated by antioxidants. Brain Res Mol Brain Res. 2003;110:85–91. doi: 10.1016/s0169-328x(02)00642-3. [DOI] [PubMed] [Google Scholar]
- Shanker G, Syversen T, Aschner JL, Aschner M. Modulatory effect of glutathione status and antioxidants on methylmercury-induced free radical formation in primary cultures of cerebral astrocytes. Brain Res Mol Brain Res. 2005;137:11–22. doi: 10.1016/j.molbrainres.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Shenker BJ, Guo TL, InSug O, Shapiro IM. Induction of apoptosis in human T-cells by methyl mercury: temporal relationship between mitochondrial dysfunction and loss of reductive reserve. Toxicol Appl Pharmacol. 1999;157:23–35. doi: 10.1006/taap.1999.8652. [DOI] [PubMed] [Google Scholar]
- Shenker BJ, Pankoski L, Zekavat A, Shapiro IM. Mercury-induced apoptosis in human lymphocytes: caspase activation is linked to redox status. Antioxid Redox Signal. 2002;4:379–89. doi: 10.1089/15230860260196182. [DOI] [PubMed] [Google Scholar]
- Sone N, Larsstuvold MK, Kagawa Y. Effect of methyl mercury on phosphorylation, transport, and oxidation in mammalian mitochondria. J Biochem. 1977;82:859–68. doi: 10.1093/oxfordjournals.jbchem.a131762. [DOI] [PubMed] [Google Scholar]
- Spierings D, McStay G, Saleh M, Bender C, Chipuk J, Maurer U, et al. Connected to death: the (unexpurgated) mitochondrial pathway of apoptosis. Science. 2005;310:66–7. doi: 10.1126/science.1117105. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in cell trauma: cause or effect of neuronal cell death? J Neurosci Res. 2005;79:231–39. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- Tsatmali M, Walcot EC, Crossin KL. Newborn neurons acquire high levels of reactive oxygen species and increased mitochondrial proteins upon differentiation from progenitors. Brain Res. 2005;1040:137–50. doi: 10.1016/j.brainres.2005.01.087. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usuki F, Yasutake A, Umehara F, Tokunaga H, Matsumoto M, Eto K, et al. In vivo protection of a water-soluble derivative of vitamin E, Trolox, against methylmercury-intoxication in the rat. Neurosci Lett. 2001;304:199–203. doi: 10.1016/s0304-3940(01)01764-5. [DOI] [PubMed] [Google Scholar]
- Verity M, Sarafian T. Role of oxidative injury in the pathogenesis of methylmercury neurotoxicity. In: Clarkson T, editor. Advances in mercury toxicology. T. Clarkson. New York: Plenum; 1991. pp. 209–22. [Google Scholar]
- Verity MA, Brown WJ, Cheung M. Organic mercurial encephalopathy: in vivo and in vitro effects of methyl mercury on synaptosomal respiration. J Neurochem. 1975;25:759–66. doi: 10.1111/j.1471-4159.1975.tb04405.x. [DOI] [PubMed] [Google Scholar]
- Voie OA, Fonnum F. Effect of polychlorinated biphenyls on production of reactive oxygen species (ROS) in rat synaptosomes. Arch Toxicol. 2000;73:588–93. doi: 10.1007/s002040050012. [DOI] [PubMed] [Google Scholar]
- Ward MW, Kogel D, Prehn JH. Neuronal apoptosis: BH3-only proteins the real killers? J Bioenerg Biomembr. 2004;36:295–8. doi: 10.1023/B:JOBB.0000041756.23918.11. [DOI] [PubMed] [Google Scholar]
- Welsh S, Soares J. The protective effect of vitamin E and selenium against methylmercury toxicity in the Japanese quail. Nutr Reports International. 1976;13:43–51. [Google Scholar]
- Yee S, Choi BH. Methylmercury poisoning induces oxidative stress in the mouse brain. Exp Mol Pathol. 1994;60:188–96. doi: 10.1006/exmp.1994.1017. [DOI] [PubMed] [Google Scholar]
- Yee S, Choi BH. Oxidative stress in neurotoxic effects of methylmercury poisoning. Neurotoxicology. 1996;17:17–26. [PubMed] [Google Scholar]
- Yip RK, Chang LW. Protective effects of vitamin E on methylmercury in the dorsal root ganglion. Environ Res. 1982;28:84–95. doi: 10.1016/0013-9351(82)90156-6. [DOI] [PubMed] [Google Scholar]
- Yonaha M, Saito M, Sagai M. Stimulation of lipid peroxidation by methylmercury in rats. Life Sci. 1983;32:1507–14. doi: 10.1016/0024-3205(83)90917-7. [DOI] [PubMed] [Google Scholar]
- Yoshino Y, Mozai T, Nakao K. Distribution of mercury in the brain and its subcellular units in experimental organic mercury poisoning. J Neurochem. 1966;13:397–406. [Google Scholar]
- Zoratti M, Szabo I, De Marchi U. Mitochondrial permeability transitions: how many doors to the house? Biochim Biophys Acta. 2005;1706:40–52. doi: 10.1016/j.bbabio.2004.10.006. [DOI] [PubMed] [Google Scholar]