

Abstract
Purpose
Tumor endothelial marker 8 (TEM8) has been reported to be upregulated in both tumor cells and tumor-associated endothelial cells in several cancer types. TEM8 antagonists and TEM8-targeted delivery of toxins have been developed as effective cancer therapeutics. The ability to image TEM8 expression would be of use in evaluating TEM8-targeted cancer therapy.
Methods
A 13-meric peptide, KYNDRLPLYISNP (QQM), identified from the small loop in domain IV of protective antigen of anthrax toxin was evaluated for TEM8 binding and labeled with 18F for small-animal PET imaging in both UM-SCC1 head-and-neck cancer and MDA-MB-435 melanoma models.
Results
A modified ELISA showed that QQM peptide bound specifically to the extracellular vWA domain of TEM8 with an IC50 value of 304 nM. Coupling 4-nitrophenyl 2-18F-fluoropropionate with QQM gave almost quantitative yield and a high specific activity (79.2±7.4 TBq/mmol, n=5) of 18F-FP-QQM at the end of synthesis. 18F-FP-QQM showed predominantly renal clearance and had significantly higher accumulation in TEM8 high-expressing UM-SCC1 tumors (2.96±0.84 %ID/g at 1 h after injection) than TEM8 low-expressing MDA-MB-435 tumors (1.38±0.56 %ID/g at 1 h after injection).
Conclusion
QQM peptide bound specifically to the extracellular domain of TEM8. 18F-FP-QQM peptide tracer would be a promising lead compound for measuring TEM8 expression. Further efforts to improve the affinity and specificity of the tracer and to increase its metabolic stability are warranted.
Keywords: Tumor endothelial marker 8 (TEM8), Small-animal PET, 18F, Peptide
Introduction
Antiangiogenesis is a promising strategy for the treatment of cancer and targeting the endothelial cells that line tumor-infiltrating blood vessels has gained widespread attention and support [1-3]. The upregulation of tumor endothelial marker 8 (TEM8) on the endothelial cells of blood vessels has been observed in several tumor types in both humans and mice, making this receptor a particularly promising new candidate for future tumor angiogenesis targeting studies [4, 5].
TEM8, also known as anthrax toxin receptor 1 (ANTXR1), is one of the two cell surface anthrax toxin receptors. It shares the extracellular von Willebrand factor type A (vWA) domain with the other receptor for anthrax toxin called capillary morphogenesis protein 2 (CMG2/ANTXR2) [6, 7]. TEM8 is reported to be selectively overexpressed on both tumor endothelial and cancer cells during tumor angiogenesis, whereas CMG2 is more widely expressed in normal tissues [8]. TEM8 links extracellular matrix components with the actin cytoskeleton to promote cell adhesion and cell spreading [9]. Moreover, TEM8 appears to regulate endothelial cell migration and tubule formation [10].
It has been reported that TEM8 may be a useful marker to identify tumor-associated microvessels and that elevated levels of TEM8 are associated with disease progression of breast cancer [11]. Cullen et al. [12] found that host-derived TEM8 promotes the growth of certain tumors and suggested that TEM8 antagonists may have utility in the development of new anticancer therapies. Fernando and Fletcher [13] successfully developed a fusion protein that targets TEM8 and disrupts tumor vasculature by promoting localized thrombosis.
Anthrax toxin, which contains protective antigen (PA), lethal factor, and edema factor, enters target cells through a multistep mechanism. The full-length PA (PA83, 83 kDa) binds cell surface receptors and is cleaved to PA protein (PA63, 63 kDa) by cellular furin-like enzymes [8, 14, 15]. The crystal structure of PA protein contains four domains that, according to mutational and biochemical analyses [16, 17], are associated with different functions. Among these, domain IV (residues 596–735) is believed to be a critical receptor-binding region for the binding of the cellular receptor (either TEM8 or CMG2) of PA [15, 16, 18]. A MIDAS motif present in the integrin-like inserted (I) domain of ANTXR1/TEM8 mediates the binding of the D683 residue of PA domain IV.
Several studies have been conducted based on the interaction between PA and TEM8 or CMG2. Chen et al. [8] used phage display to select PA mutants that preferentially bind to TEM8 over CMG2 in order to target tumors using modified anthrax toxin PA. Duan et al. [19] engineered an antibody-like protein, TEM8-Fc, that consists of the PA-binding domain of human TEM8 linked to the Fc portion of human immunoglobulin G1. They then showed that TEM8-Fc can protect J774A.1 macrophage-like cells against anthrax toxin challenge in a dose-dependent manner, and it could suppress the growth and metastasis of xenografted human tumors in athymic nude mice.
In this study, we designed a peptide 13-mer based on the small loop (amino acids 680–692) [16] in domain IV of PA (Fig. 1). The sequence was designed initially as K680YNDK684LPLYISNP692. We replaced K684 with R684, however, to avoid 18F labeling on this amino-acid residue. The final 13-mer peptide is KYNDRLPLYISNP (denoted as “QQM”). In vitro cell binding and ELISAs (described below) demonstrated that this peptide binds specifically to the extracellular domain of TEM8, and we labeled it with 18F for positron emission tomography (PET) imaging of TEM8 expression patterns in tumor xenograft models.
Fig. 1.
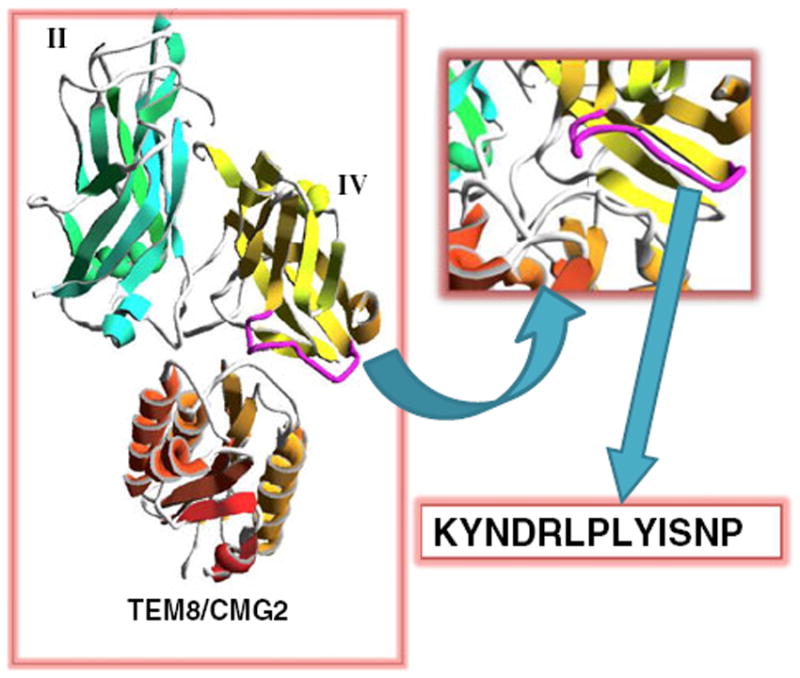
Schematics of the KYNDRLPLYISNP (QQM) peptide design. The small loop (purple) of domain IV (yellow) is packed in the interface between PA domains II (green) and TEM8/CMG2 (orange). The 13-mer QQM peptide was designed from the sequence of the small loop
Materials and methods
General
The linear 13-meric peptide QQM was prepared via standard solid-phase Fmoc chemistry with an automated peptide synthesizer (CS Bio, Menlo Park, CA). No-carrier-added 18F-F− was produced from an in-house GE Healthcare PETtrace cyclotron. Reversed-phase extraction C18 Sep-Pak cartridges (Waters) were pretreated with ethanol (5 ml) and water (10 ml) before use. The syringe filter and polyethersulfone membranes (pore size 0.22 mm, diameter 13 mm) were purchased from Nalge Nunc International. Analytical and semipreparative reversed-phase high-performance liquid chromatography (RP-HPLC) were performed on a Waters 600 chromatography system with a Waters 996 photodiode array detector and Beckman 170 radioisotope detector. A C18 Vydac protein and peptide column (218TP510; 5 μm, 250×10 mm) was used for peptide purification. The flow was set at 5 ml/min using a gradient system starting from 95% solvent A (0.1% trifluoroacetic acid in water) and 5% solvent B (0.1% trifluoroacetic acid in acetonitrile) (0–2 min) and ramped to 35% solvent A and 65% solvent B at 32 min. The flow rate of the analytical HPLC was set at 1 ml/min using the same gradient system, but with a different C18 Vydac column (218TP54; 5 μm, 250×4.6 mm). The UV absorbance was monitored at 218 nm and the identification of the peptides was confirmed based on the UV spectrum acquired (using a photodiode array detector) and mass spectrometry.
Synthesis of FP-QQM
A solution of O-(N-succinimidyl)-1,1,3,3-tetramethyluronium tetrafluoroborate (0.37 mg, 1 μmol) in dimethylformamide (DMF, 30 μl) and diisopropylethylamine (10 μl) was added to 2-fluoropropionic acid (92 μg, 1 μmol) in DMF (9.2 μl). The reaction mixture was heated at 60°C for 20 min and then added to QQM (1.6 mg, 1 μmol) in DMF (120 μl). After 15 min at room temperature, the reaction mixture was quenched with 20 μl of acetic acid. The crude product was purified by RP-HPLC on a semipreparative C18 column. The desired fractions containing FP-QQM conjugate were harvested and lyophilized to give a white fluffy powder (yield >90%, purity >99%). The identity of the product was confirmed by mass spectrometry (ESI-MS): m/z 1666.96 for [MH]+ (C76H116FN19O22, calculated MW 1666.85).
Synthesis of 18F-FP-QQM
The 18F-labeling precursor, 4-nitrophenyl 2-18F-fluoropropionate (18F-NFP), was synthesized as previously reported [20, 21]. The QQM labeling procedure (Fig. 2) was as follows: QQM (300 μg) dissolved in 150 μl anhydrous dimethyl sulfoxide (DMSO) was added to dried 18F-NFP in a 1 ml reaction vial, followed by adding 20 μl of diisopropylethylamine. The reaction mixture was allowed to stand at room temperature for 10 min and then quenched with 800 μl of 5% aqueous acetic acid solution. The labeled peptide was purified by RP-HPLC on a semipreparative C18 column. The desired fractions containing 18F-FP-QQM were collected and diluted with 20 ml of water. The identity of 18F-FP-QQM was confirmed by HPLC by coinjection with the cold standard FP-QQM. After trapping with a preactivated C18 Sep-Pak cartridge, the product was washed with 2 ml of water and eluted with 2 ml of ethanol. The ethanol solution was then blown dry with a slow stream of nitrogen at 60°C. The 18F-labeled peptide was redissolved in phosphate-buffered saline (PBS) solution and passed through a 0.22-μm Millipore filter into a sterile multidose vial for in vitro and in vivo experiments. The labeling yield of 18F-FP-QQM based on 18F-NFP was over 95%. The chemical purity and radiochemical purity were over 99%. The specific activity was 79.2±7.4 TBq/mmol (n=5) at the end of synthesis.
Fig. 2.
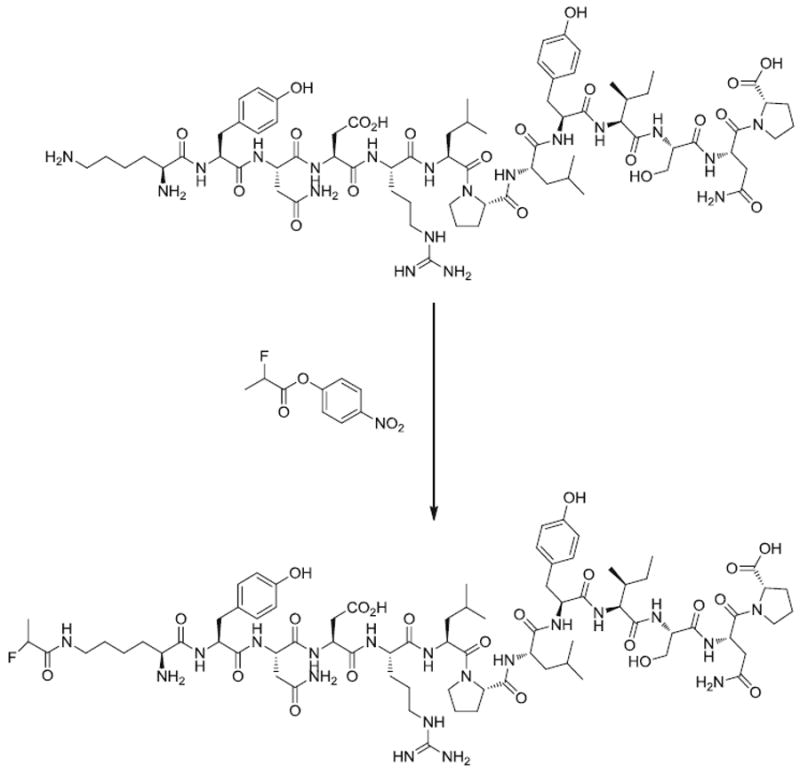
Synthesis of 18F-FP-QQM
Cell culture and animal models
The MDA-MB-435 human melanoma cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA). The UM-SCC1 cell line was obtained from the University of Michigan. Both cell lines were grown in high glucose Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with fetal bovine serum (10%, volume percentage) and 1% antibiotics (penicillin 100 U/ml, plus streptomycin 100 μg/ml) at 37°C in a humidified incubator containing 5% CO2.
The MDA-MB-435 tumor model was generated by subcutaneous injection of 5×106 cells into the right front flank of female athymic nude mice (Harlan Laboratories). The UM-SCC1 tumor model was established by injection of 6×106 cells into the right front flank of the mice with a 100 μl total volume consisting of a 1:1 ratio of sterile PBS and Matrigel (Becton Dickinson). The mice were used for PET studies when the tumor volume reached about 200–500 mm3 (about 2–3 weeks for MDA-MB-435 and about 5–6 weeks for UM-SCC1). All animal studies were conducted in accordance with the principles and procedures outlined in the NRC Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (IACUC) of the Clinical Center, National Institutes of Health.
Expression and purification of recombinant TEM8 vWA domain
The pGEX4T-1 plasmid encoding the human TEM8 vWA domain was a generous gift from Dr. Jianping Xiong (Massachusetts General Hospital, Charlestown, MA). The protein expression and purification were performed following previously reported procedures [6]. Briefly, the pGEX4T-1 plasmid was transformed to TOP10 chemically competent E. coli (Invitrogen), and selected single colonies were shaken in 1 l lysogeny broth medium containing 100 μg/ml ampicillin at 37°C and 250 rpm. After induction with 0.1 mM isopropyl-d-thiogalactopyranoside for 4 h, the E. coli cells were harvested for protein purification. All subsequent purification steps were performed on ice or at 4°C. After being resuspended in buffer containing 100 mM Tris, 300 mM NaCl and 5 mM EDTA (pH 8.2), cells were disrupted by sonication, and cell debris was removed by centrifugation at 14,000 rpm for 30 min. The supernatant was incubated with 2 ml pre-equilibrated glutathione Sepharose 4B beads (GE Healthcare, Piscataway, NJ) for 2 h. Then the beads were washed with elution buffer (2, 15, 40, and 100 mM reduced glutathione in 50 mM Tris-HCl, pH 8.0). The protein eluates were collected separately and pooled after SDS-PAGE analysis.
In vitro cell binding assay
TEM8 binding assay was performed to determine binding affinities of QQM peptide. Briefly, UM-SCC1 cells (Sibtech, Brookfield, CT) were trypsinized and resuspended in PBS containing 50 mM HEPES, 1 mM CaCl2, 5 mM MgCl2, 0.5% w/v bovine serum albumin (BSA) and 0.3 mM NaN3. Incubation was conducted in 96-well plates with each well containing 1 × 105 cells, 500 nCi (18.5 KBq) of 18F-FP-QQM, and 0–2,000 nM of unlabeled QQM in a total volume of 200 μl for 45 min on a shaker at room temperature. After incubation and washing with PBS, hydrophilic polyvinylidene fluoride filters were collected and the radioactivity was determined using a gamma counter (1480 Wizard 3, Perkin-Elmer). Binding results were expressed as the percent of total counts, and IC50 values were calculated using Prism software (GraphPad, La Jolla, CA).
ELISA
A modified ELISA was used to examine the binding affinity of QQM peptide to the extracellular domain of TEM8. In brief, a PVC microtiter plate (Millipore, Billerica, MA) was coated with 100 μl per well of the captured QQM peptide, at a concentration of 100 μg/ml in carbonate/bicarbonate buffer (pH 9.6), with overnight incubation at 4°C in a humidified chamber. The coated wells were then blocked for 2 h with 5% BSA/PBS and incubated with the vWA domain of TEM8 protein at different concentrations, from 5 nM to 3 μM, at 37°C for 90 min. After washing with PBST (0.1% Tween-20 in PBS) four times, the wells were incubated with goat anti-human TEM8 antibody (1:500, R&D Biosciences) and then with horseradish peroxidase-conjugated donkey anti-goat secondary antibody (1:10,000; Jackson ImmunoResearch Laboratories, West Grove, PA). The signal was measured colorimetrically using a 96-well plate reader (BioTek Synergy 2, Winooski, VT) at a wavelength of 450 nm after adding tetra-methylbenzidine and hydrogen peroxide.
Tumor tissue TEM8 expression by western blotting
For western blotting, the total protein from each of the UM-SCC1 and MDA-MB-435 tumor samples was extracted in NP40 cell lysis buffer (Invitrogen). The concentration of total protein was determined using a microBCA protein assay kit (Pierce Biotechnology, Rockford, IL). After SDS-PAGE separation of 10 μg of total protein, the protein was transferred to a polyvinylidene fluoride membrane (Invitrogen, Carlsbad, CA) and incubated at room temperature with 5% non-fat milk blocking buffer. The blots were then incubated with either a goat anti-human TEM8 antibody (1:200, R&D) or a rabbit polyclonal antibody against β-actin (1:1,000, ab8227; Abcam) primary antibody for 1 h and subsequently labeled using appropriate horseradish peroxidase-conjugated donkey anti-goat or donkey antirabbit secondary antibodies (1:6,000; Jackson ImmunoResearch Laboratories, West Grove, PA), respectively, for 1 h. The bands were detected using an ECL western blotting detection system (GE Healthcare, Piscataway, NJ).
Fluorescent immunohistochemical staining
In order to determine the different expression patterns of TEM8 in tumors, frozen UM-SCC1 and MDA-MB-435 tumor tissue sections (5–8 μm) were fixed with cold acetone for 20 min and dried in the air. All the sections were blocked with 1% BSA for 30 min, and then incubated with goat anti-human TEM8 antibody (1:200, R&D) and rat anti-mouse CD31 antibody (1:500) for 1 h at room temperature. They were then visualized with Daylight 488-conjugated bovine anti-goat secondary antibody (1:500) or Cy3-conjugated donkey anti-rat secondary antibody (1:1,000; Jackson ImmunoResearch Laboratories), respectively. After five washing steps, the tissue slices were mounted with mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) and all slices were viewed by epifluorescence microscopy (Olympus X81).
Cell uptake of 18F-FP-QQM
The UM-SCC1 and MDA-MB-435 cells were seeded into 24-well plates at a density of 1×105 cells per well and incubated overnight. The cells were incubated with about 37 kBq (1 μCi) per well of 18F-FP-QQM at 37°C for 15, 30, or 60 min. In an experiment to block specific binding of 18F-FP-QQM, the QQM peptide was added to UM-SCC1 cells in 24-well plates at a final concentration of 1 μM. The cells were then incubated at 37°C for 15, 30, and 60 min. To determine the internalized activity, additional samples of UM-SCC1 cells were washed with acid (50 mM glycine-HCl/100 mM NaCl, pH 2.8) at different time points (15, 30, and 60 min) after incubation with 18F-FP-QQM at 37°C to remove the radioactivity bound to the cell surface. These tumor cells were then washed three times with chilled PBS and lysed by 0.1 N NaOH. The cell lysate was collected and measured using a γ counter (Packard, Meriden, CT). The cell uptake was expressed as a percentage of the decay-corrected total input radioactivity. All the experiments were performed twice with triplicate wells.
Small-animal PET imaging
PET scans and image analysis were performed using an Inveon microPET scanner (Siemens). Each UM-SCC1 or MDA-MB-435 tumor-bearing mouse was injected intravenously with 3.7 MBq (100 μCi) of 18F-FP-QQM under isoflurane anesthesia (n=6/group). Static scans of duration 5 min were acquired at 15, 30, and 60 min after injection (p.i.), and 10-min scans were acquired at 2 h p.i. For the TEM8 receptor-blocking experiment, QQM (500 μg in 100 μl PBS per mouse) was coinjected with 3.7 MBq of 18F-FP-QQM peptide into UM-SCC1 tumor mice, and 5-min static microPET scans were acquired at 1 h p.i. Images were reconstructed using the 2-dimensional ordered subsets expectation maximum algorithm without attenuation or scatter correction. Regions of interest (ROIs) over the tumor, normal tissue, and major organs were drawn on decay-corrected whole-body coronal images for each PET scan using the vendor software (ASI Pro 5.2.4.0). The radioactivity concentration (accumulation) within a tumor or an organ was obtained from mean pixel values within the multiple ROI volume, after conversion of the values to megabecquerels per milliliter per minute using a conversion factor assuming a tissue density of 1 g/ml. Imaging ROI-derived percentage injected dose per gram of tissue (%ID/g) was calculated by dividing the ROI radioactivity concentration by the administered activity [22].
Ex vivo biodistribution
Immediately after imaging at 1 or 2 h p.i. the tumor-bearing mice were killed. Blood, tumor, major organs, and tissues were collected and weighed wet. The radioactivity in the wet whole tissue was measured using a γ counter (Packard). The results are presented as %ID/g. For each mouse, the radioactivity of the tissue samples was calibrated against a known aliquot of the injected tracer and normalized to the mean body mass of each group. The results are expressed as means±SD (n=4 per group).
Statistical analysis
Quantitative data are expressed as means±SD. Means were compared using Student’s t-test. P values of <0.05 were considered statistically significant.
Results
QQM specifically binds to TEM8
We performed competitive cell binding assays and ELISAs to confirm the specific binding of QQM peptide to TEM8 protein. Since there was no commercially available TEM8-specific radioligand, we used 18F-FP-QQM for homologous displacement. The IC50 of QQM peptide binding to 293-TEM8 cells was 33.04 nM (Fig. 3a) In order to further confirm the binding affinity and specificity, we also expressed and purified the extracellular domain of TEM8 and performed an ELISA with the purified protein. As shown in Fig. 3b, the QQM peptide showed gradual binding saturation with increasing concentrations of the purified TEM8 protein, which suggests a specific binding of QQM to TEM8. The Kd value was around 304 nM.
Fig. 3.
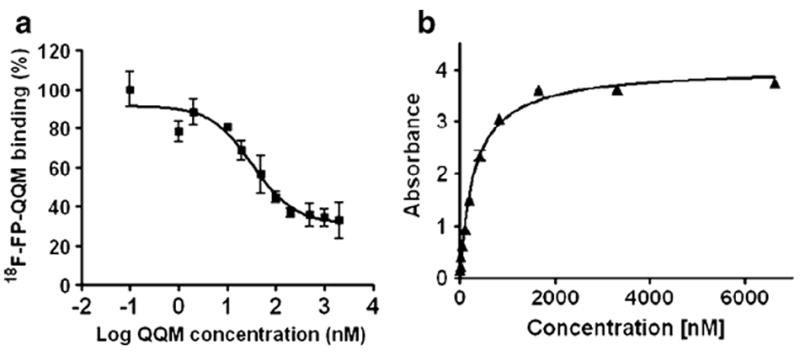
QQM binding assays. a Competitive cell binding assay. The IC50 value of QQM peptide binding to 293-TEM8 cells using 18F-FP-QQM for homologous displacement was 33.04 nM. b ELISA. The QQM peptide specifically binds to the extracellular domain of TEM8 receptor in a dose-dependent manner (Kd 304 nM)
Vasculature-related TEM8 expression
The expression of TEM8 in UM-SCC1 and MB-MDA-435 tumors was assessed at the protein level by both western blotting and fluorescent immunohistochemical staining (Fig. 4). Western blotting showed a distinct 60-kDa molecular weight band representing TEM8. As seen in Fig. 4a, the homogenized UM-SCC1 tumor tissue extract had much higher TEM8 expression than the MDA-MB-435 tumor tissue extract. The TEM8 distribution in the tumor was also evaluated by ex vivo fluorescent immunohistochemical staining. Representative images of CD31 and TEM8 receptor staining of UM-SCC1 and MDA-MB-435 tumor sections are shown in Fig. 4b. The intensity of TEM8 expression in UM-SCC1 tumor was substantially higher than that in MDA-MB-435 tumor, using the same scale. The green fluorescence of the TEM8 receptor was concentrated around the vessel-rich region and was partially overlaid with the red fluorescence of CD31, a vasculature marker. Much lower staining signals were observed in the MDA-MB-435 tumor sections.
Fig. 4.
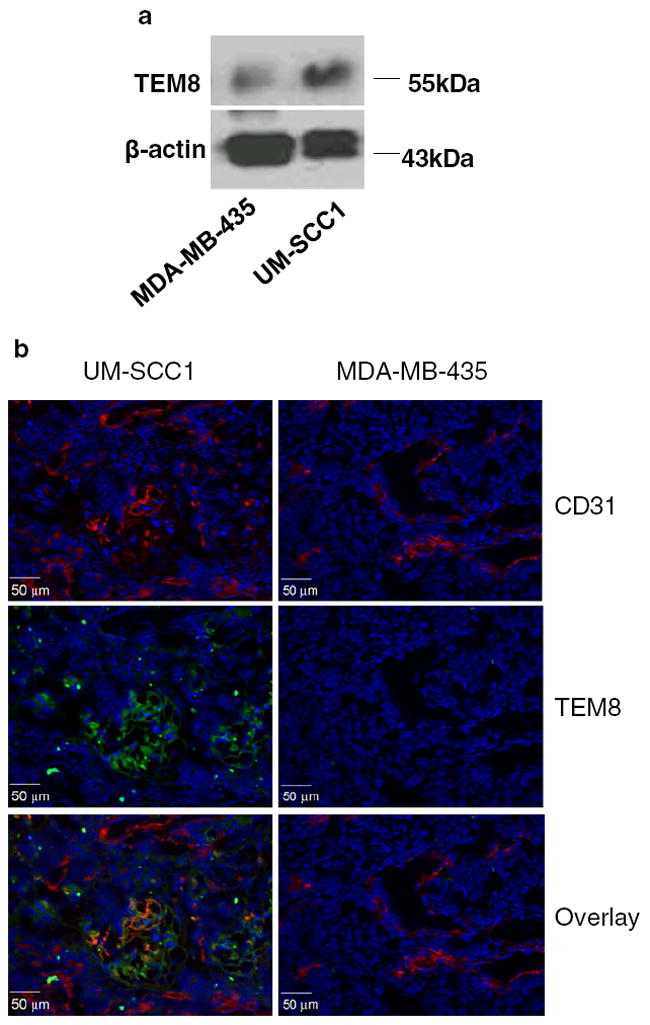
TEM8 expression in UM-SCC1 and MB-MDA-435 tumors, a Western blotting. β-actin was used as the loading control. b Fluorescent immunostaining of tumor sections. The nuclei were counterstained with DAPI (blue)
Cell uptake and internalization of 18F-FP-QQM
Additionally, we developed QQM peptide as a PET imaging tracer by labeling it with a positron emitting radioisotope, 18F (t1/2 109.8 min). 18F-FP-QQM was characterized in vitro by cell uptake assays in UM-SCC1 and MDA-MB-435 cells. 18F-FP-QQM had significantly higher cell uptake efficiency in UM-SCC1 cells than in MDA-MB-435 cells (Fig. 5). The UM-SCC1 cell uptake percentages of total input radioactivity were 6.21±0.21%, 8.35±0.17% and 8.34±0.64% at 15, 30, and 60 min, respectively. By contrast, the MDA-MD-435 cells showed only minimal uptake percentages of 0.49±0.23%, 0.65±0.20%, and 0.50±0.13% at 15, 30, and 60 min, respectively. The uptake of 18F-FP-QQM in UM-SCC1 was also effectively blocked in the presence of non-radiolabeled QQM peptide (0.48±0.06%, 0.63±0.07%, and 1.08±0.13% at 15, 30, and 60 min, respectively). We used the acid wash experiment to remove 18F-FP-QQM bound to the cell surface and determine the effectiveness of the internalization process. It was clear that 18F-FP-QQM was almost completely internalized into UM-SCC1 cells after 15 min incubation at 37°C.
Fig. 5.
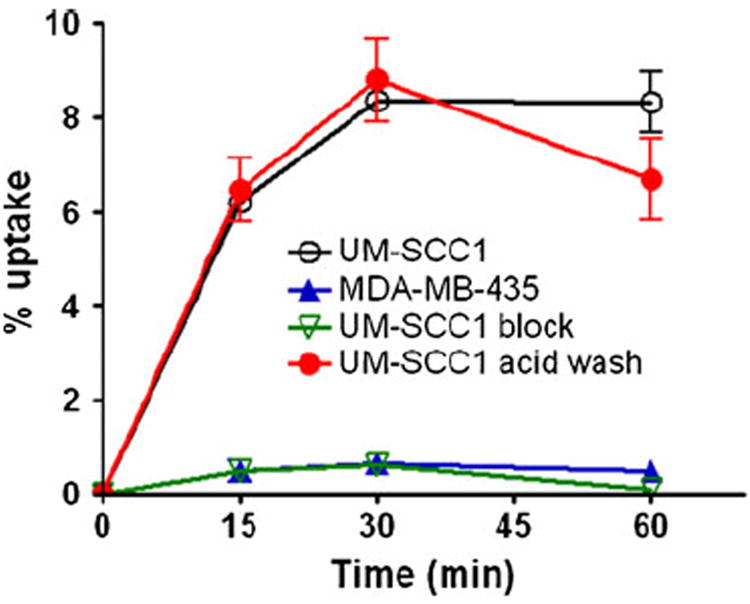
Cell uptake assay of 18F-FP-QQM in tumor cells. For the blocking experiment, the QQM peptide was added to UM-SCC1 cells in 24-well plates at a final concentration of 1 μM to block the specific binding of 18F-FP-QQM. To determine the internalized activity, extra groups of UM-SCC1 cells were washed with acid (50 mM glycine-HCl/100 mM NaCl, pH 2.8) to remove the radioactivity bound to the cell surface. The cell uptake is expressed as the percentage of decay-corrected total input radioactivity. All the experiments were performed twice with triplicate wells
MicroPET imaging
Representative coronal microPET images of UM-SCC1 and MDA-MB-435 tumor-bearing mice (n=6/group) at 15, 30, 60 and 120 min after intravenous injection of 3.7 MBq (100 μCi) of 18F-FP-QQM are shown in Fig. 6a. A plot of the ROI analysis results in UM-SCC1 and MDA-MB-435 tumors is shown in Fig. 6b. The UM-SCC1 tumors were clearly contoured with high contrast in relation to the MDA-MB-435 tumors at all time points after injection (3.73±0.56 vs. 1.64 ±0.24 %ID/g at 30 min, 2.96 ±0.84 vs. 1.38 ±0.56 %ID/g at 60 min, and 2.04±0.22 vs. 1.00±0.44%ID/g at 60 min; P<0.01 at 30 min, P<0.05 at 60 min and 120 min). 18F-FP-QQM was excreted mainly through the renal/urinary route, as reflected by very minimal liver uptake (8.47±1.19 %ID/g at 15 min, 4.23±3.20 %ID/g at 30 min, 3.84±2.49%ID/g at 60 min, and 1.63±0.51 %ID/g at 120 min), and an initially high radioactive signal in the kidneys followed by rapid clearance from the kidneys (16.36±2.69 %ID/g at 15 min, 11.42±5.7 %ID/g at 30 min, 6.00±2.92 %ID/g at 60 min, and 2.06±0.36 %ID/g at 120 min) in the UM-SCC1 tumor model. The in vivo TEM8 binding specificity of 18F-FP-QQM was also confirmed by an imaging study following administration of a blocking dose of QQM peptide. The uptake of 18F-FP-QQM in UM-SCC1 tumors with coinjection of 500 μg QQM peptide dropped from 2.96±0.84 to 1.12±1.01 %ID/g (P<0.01) at 60 min (Fig. 6c). No bone uptake was observed, suggesting no defluoridation during the course of imaging studies.
Fig. 6.
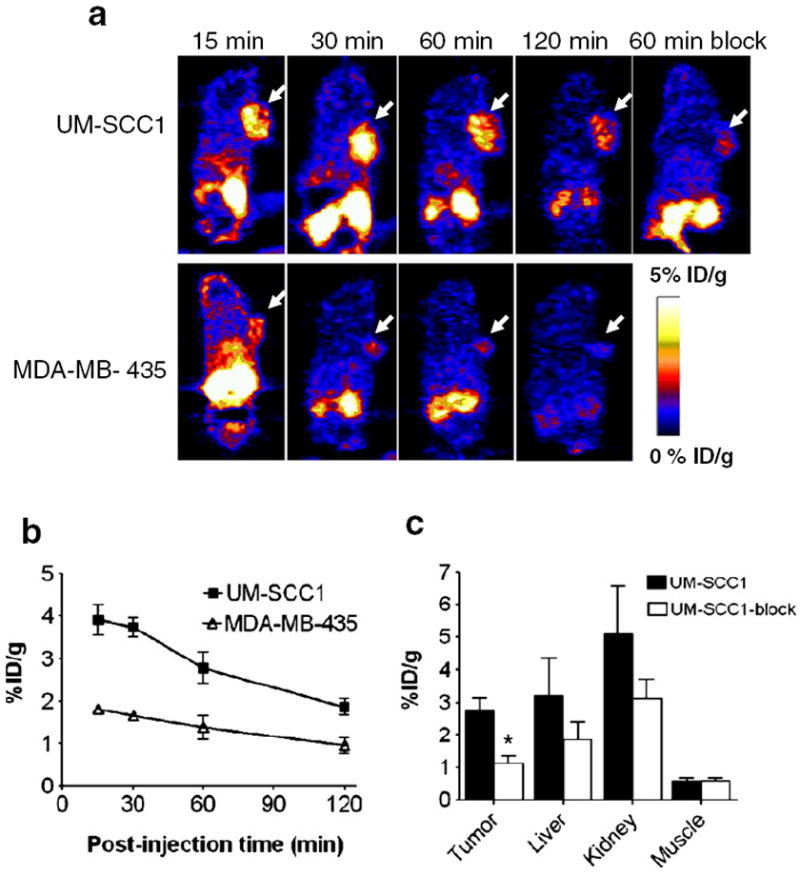
In vivo PET imaging of xenografted mice treated with 18F-FP-QQM. a Decay-corrected whole-body coronal microPET images of UM-SCC1 and MDA-MB-435 tumor–bearing mice at 15, 30, 60, and 120 min after injection of 3.7 MBq (100 μCi) of 18F-FP-QQM. The tumors are indicated by arrows. b ROI analysis of tumor uptake of 18F-FP-QQM at 15, 30, 60, and 120 min in UM-SCC1 and MDA-MB-435 tumor–bearing mice (n=6/group) as derived from static microPET images. c Quantification of 18F-FP-QQM uptake in UM-SCC1 tumor, liver, kidneys and muscle with and without the presence of a blocking dose of QQM peptide (n=6/group). Uptake values are shown as mean %ID/g±SD. *P<0.05
Ex vivo biodistribution study
After microPET imaging at 1 h and 2 h p.i., the animals were killed for biodistribution studies and the results are shown in Fig. 7. Consistent with the non-invasive microPET imaging results, the tumor uptake percentages were 3.45±0.95 %ID/g and 2.03±0.29 %ID/g at 60 min and 120 min p.i., respectively. The kidneys, lung and pancreas also showed prominent radioactivity accumulation, with uptakes of 5.21±2.67, 3.19±1.44 and 5.24±2.66 %ID/g, respectively, at 60 min.
Fig. 7.
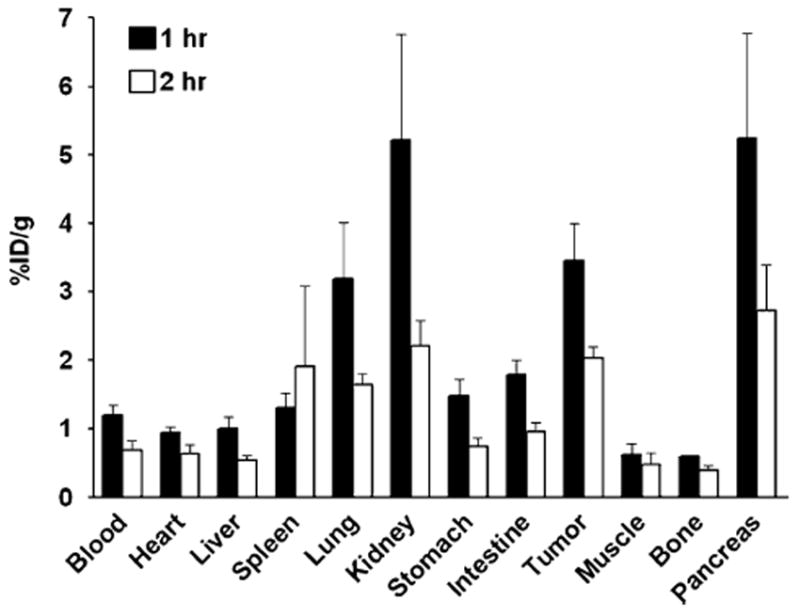
Ex vivo biodistribution of 18F-FP-QQM (3.7 MBq per mouse) in UM-SCC1 tumor-bearing nude mice at 1 h and 2 h p.i. after microPET scans. The values are shown as mean %ID/g±SD (n=3 per group); bars SD
Discussion
The fact that TEM8 is differentially expressed on tumor endothelium but shows limited expression in normal tissues [4] makes this receptor an attractive target for cancer imaging and therapy. Protective antigen is an essential part of anthrax toxin, serving as a cellular binding moiety and a delivery vehicle for translocation of lethal factor and edema factor into the cytosol of cells. It has been confirmed that the small loop of amino acids 680–692 within domain IV of PA is required for PA binding to cells [23, 24]. By taking advantage of the specific interaction between this small loop of PA and TEM8, we synthesized and developed a 13-mer peptide (QQM) probe for TEM8 imaging (Fig. 2). It has been reported that Y681, N682, D683 and P686 are most important amino acid residues for receptor binding [16]. Thus, conjugation of the 18F-containing prosthetic group on the lysine residue at the N-terminal of the peptide will not affect the receptor binding ability of this peptide. Indeed, competitive binding assays with homologous displacement indicates the QQM peptide possesses a high binding affinity for TEM8. In an ELISA assay using purified extracellular domain of TEM8, we also confirmed the specific interaction between QQM and TEM8.
The binding specificity was further confirmed by the fact that the in vitro cell uptake of 18F-FP-QQM could be effectively blocked by the presence of an extra amount of “cold” QQM peptide. QQM peptide was rapidly internalized into TME8-expressing UM-SCC1 cells (Fig. 5). It has been reported that oligomerization of PA63 triggers receptor-mediated endocytosis [25]. However, it is domain II (residues 259–487), not the domain IV, of PA that is involved in oligomerization [18, 26]. Thus, the mechanism of QQM-induced internalization after binding with TEM8 receptor remains to be elucidated.
It has been found that most of the genes expressed differentially in tumor endothelium are also expressed during angiogenesis of corpus luteum formation and wound healing. However, TEM8 expression is not detectable in developing corpus luteum, suggesting that it can be used as a tumor endothelial marker to differentiate between tumor angiogenesis and normal angiogenesis [4]. However, TEM8 has also been detected in a variety of tumor cell lines and in a human fibroblast cell line (MRC5) at both the mRNA and protein levels [11, 27]. TEM8 has also been found in stimulated endothelial progenitor cells [28]. Fernando and Fletcher [13] found that TEM8 protein expression colocalizes with both endothelial cells and CD34-positive cells. With fluorescent immunohistostaining, we found that TEM8 expression in UM-SCC1 tumor was substantially higher than in MDA-MB-435 tumor. The green fluorescence of the TEM8 receptor was concentrated around the vessel-rich region and was partly overlain with CD31, a vasculature marker.
When labeled with 18F through a 18F-NFP prosthetic group, the resulting 18F-FP-QQM peptide tracer showed enhanced uptake in vivo in UM-SSC1 tumors expressing high levels of TEM8 and only background uptake levels in vivo in MB-MDA-435 tumors expressing low levels of TEM8. The receptor specificity of 18F-FP-QQM was further confirmed by effective blocking studies, both in vitro and in vivo. The results demonstrated that 18F-FP-QQM can be used as a specific tracer to monitor TEM8 expression by non-invasive PET imaging. The 18F-FP-QQM was excreted mainly through the renal/urinary route, as indicated by initially high radioactive signal in the kidneys and rapid clearance. Besides the kidneys, the lung and pancreas also showed relatively high radioactivity accumulation. PA of anthrax toxin binds to both TEM8 and another anthrax receptor, CMG2, and the latter has been reported to have approximately 1,000-fold higher affinity for PA [29]. We cannot exclude the possibility that these organs show relatively high expression of CMG2 [8] and thus 18F-FP-QQM accumulation. Future investigations will involve the design of probes with more selectivity for TEM8 over CMG2.
In vivo PET imaging showed that the uptake of 18F-FP-QQM in UM-SCC1 tumors reached a peak between 15 and 30 min after tracer injection and then decreased with time. The rapid clearance of the peptide from the tumor area was probably due to both the metabolic instability of the linear peptide and washout of the peptide from the circulation and the body. Our future work will seek to stabilize the QQM peptide with different molecular scaffolds.
Conclusion
With a rationally designed 13-mer peptide (QQM) based on the critical structure involved in PA-TEM8 interaction, we successfully imaged TEM8 overexpressing xenografts in a mouse model. To the best of our knowledge, this is the first time that TEM8 expression has been visualized with PET using a peptidic probe. The results demonstrate the feasibility of TEM8-based imaging.
Acknowledgments
This project was supported by the Intramural Research Program of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), and the International Cooperative Program of the National Science Foundation of China (NSFC) (81028009). Q.Q. is partially supported by the China Scholarship Council (CSC). G.N. currently is an Imaging Sciences Training Program (ISTP) Fellow jointly supported by the Radiology and Imaging Sciences Department, NIH Clinical Center, and the Intramural Research Program, NIBIB, NIH.
Footnotes
Conflicts of interest None.
Contributor Information
Qimeng Quan, Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, 9 Memorial Drive, 9/1 W111, Bethesda, MD 20892, USA; Department of Radiology, Shanghai First People’s Hospital, Shanghai Jiaotong University, Shanghai 200080, China.
Min Yang, Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, 9 Memorial Drive, 9/1 W111, Bethesda, MD 20892, USA.
Haokao Gao, Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, 9 Memorial Drive, 9/1 W111, Bethesda, MD 20892, USA.
Lei Zhu, Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, 9 Memorial Drive, 9/1 W111, Bethesda, MD 20892, USA.
Xin Lin, Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, 9 Memorial Drive, 9/1 W111, Bethesda, MD 20892, USA.
Ning Guo, Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, 9 Memorial Drive, 9/1 W111, Bethesda, MD 20892, USA.
Gang Niu, Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, 9 Memorial Drive, 9/1 W111, Bethesda, MD 20892, USA; Imaging Sciences Training Program, Radiology and Imaging Sciences, Clinical Center and National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, MD 20892, USA, niug@mail.nih.gov.
Guixiang Zhang, Department of Radiology, Shanghai First People’s Hospital, Shanghai Jiaotong University, Shanghai 200080, China.
Henry S. Eden, Intramural Research Program, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, MD 20892, USA
Xiaoyuan Chen, Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, 31 Center Dr, 31/1 C22, Bethesda, MD 20892, USA, shawn.chen@nih.gov.
References
- 1.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–74. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 2.Nanda A, St Croix B. Tumor endothelial markers: new targets for cancer therapy. Curr Opin Oncol. 2004;16:44–9. doi: 10.1097/00001622-200401000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Niu G, Chen X. Why integrin as a primary target for imaging and therapy. Theranostics. 2011;1:30–47. doi: 10.7150/thno/v01p0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, et al. Genes expressed in human tumor endothelium. Science. 2000;289:1197–202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- 5.Herrmann D, Ferrer-Vaquer A, Lahsnig C, Firnberg N, Leibbrandt A, Neubuser A. Expression and regulation of ANTXR1 in the chick embryo. Dev Dyn. 2010;239:680–7. doi: 10.1002/dvdy.22194. [DOI] [PubMed] [Google Scholar]
- 6.Ding Z, Bradley KA, Amin Arnaout M, Xiong JP. Expression and purification of functional human anthrax toxin receptor (ATR/TEM8) binding domain from Escherichia coli. Protein Expr Purif. 2006;49:121–8. doi: 10.1016/j.pep.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Ramey JD, Villareal VA, Ng C, Ward SC, Xiong JP, Clubb RT, et al. Anthrax toxin receptor 1/tumor endothelial marker 8: mutation of conserved inserted domain residues overrides cytosolic control of protective antigen binding. Biochemistry. 2010;49:7403–10. doi: 10.1021/bi100887w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen KH, Liu S, Bankston LA, Liddington RC, Leppla SH. Selection of anthrax toxin protective antigen variants that discriminate between the cellular receptors TEM8 and CMG2 and achieve targeting of tumor cells. J Biol Chem. 2007;282:9834–45. doi: 10.1074/jbc.M611142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hotchkiss KA, Basile CM, Spring SC, Bonuccelli G, Lisanti MP, Terman BI. TEM8 expression stimulates endothelial cell adhesion and migration by regulating cell-matrix interactions on collagen. Exp Cell Res. 2005;305:133–44. doi: 10.1016/j.yexcr.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 10.Cryan LM, Rogers MS. Targeting the anthrax receptors, TEM-8 and CMG-2, for anti-angiogenic therapy. Front Biosci. 2011;16:1574–88. doi: 10.2741/3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies G, Rmali KA, Watkins G, Mansel RE, Mason MD, Jiang WG. Elevated levels of tumour endothelial marker-8 in human breast cancer and its clinical significance. Int J Oncol. 2006;29:1311–7. [PubMed] [Google Scholar]
- 12.Cullen M, Seaman S, Chaudhary A, Yang MY, Hilton MB, Logsdon D, et al. Host-derived tumor endothelial marker 8 promotes the growth of melanoma. Cancer Res. 2009;69:6021–6. doi: 10.1158/0008-5472.CAN-09-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernando S, Fletcher BS. Targeting tumor endothelial marker 8 in the tumor vasculature of colorectal carcinomas in mice. Cancer Res. 2009;69:5126–32. doi: 10.1158/0008-5472.CAN-09-0725. [DOI] [PubMed] [Google Scholar]
- 14.Backer MV, Patel V, Jehning BT, Claffey KP, Karginov VA, Backer JM. Inhibition of anthrax protective antigen outside and inside the cell. Antimicrob Agents Chemother. 2007;51:245–51. doi: 10.1128/AAC.00983-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scobie HM, Young JA. Interactions between anthrax toxin receptors and protective antigen. Curr Opin Microbiol. 2005;8:106–12. doi: 10.1016/j.mib.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Rosovitz MJ, Schuck P, Varughese M, Chopra AP, Mehra V, Singh Y, et al. Alanine-scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for binding to the cellular receptor and to a neutralizing monoclonal antibody. J BiolChem. 2003;278:30936–44. doi: 10.1074/jbc.M301154200. [DOI] [PubMed] [Google Scholar]
- 17.Lacy DB, Wigelsworth DJ, Melnyk RA, Harrison SC, Collier RJ. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc Natl Acad Sci U S A. 2004;101:13147–51. doi: 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santelli E, Bankston LA, Leppla SH, Liddington RC. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature. 2004;430:905–8. doi: 10.1038/nature02763. [DOI] [PubMed] [Google Scholar]
- 19.Duan HF, Hu XW, Chen JL, Gao LH, Xi YY, Lu Y, et al. Antitumor activities of TEM8-Fc: an engineered antibody-like molecule targeting tumor endothelial marker 8. J Natl Cancer Inst. 2007;99:1551–5. doi: 10.1093/jnci/djm132. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Liu Z, Chen K, Yan Y, Watzlowik P, Wester HJ, et al. 18F-labeled galacto and PEGylated RGD dimers for PET imaging of αvβ3 integrin expression. Mol Imaging Biol. 2010;12:530–8. doi: 10.1007/s11307-009-0284-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang M, Gao H, Zhou Y, Ma Y, Quan Q, Lang L, et al. 18F-labeled GRPR agonists and antagonists: a comparative study in prostate cancer imaging. Theranostics. 2011;1:220–9. doi: 10.7150/thno/v01p0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun X, Niu G, Yan Y, Yang M, Chen K, Ma Y, et al. Phage display-derived peptides for osteosarcoma imaging. Clin Cancer Res. 2010;16:4268–77. doi: 10.1158/1078-0432.CCR-10-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leppla SH, Robbins JB, Schneerson R, Shiloach J. Development of an improved vaccine for anthrax. J Clin Invest. 2002;110:141–4. doi: 10.1172/JCI16204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maynard JA, Maassen CB, Leppla SH, Brasky K, Patterson JL, Iverson BL, et al. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat Biotechnol. 2002;20:597–601. doi: 10.1038/nbt0602-597. [DOI] [PubMed] [Google Scholar]
- 25.Liu S, Leppla SH. Cell surface tumor endothelium marker 8 cytoplasmic tail-independent anthrax toxin binding, proteolytic processing, oligomer formation, and internalization. J Biol Chem. 2003;278:5227–34. doi: 10.1074/jbc.M210321200. [DOI] [PubMed] [Google Scholar]
- 26.Fu S, Tong X, Cai C, Zhao Y, Wu Y, Li Y, et al. The structure of tumor endothelial marker 8 (TEM8) extracellular domain and implications for its receptor function for recognizing anthrax toxin. PLoS One. 2010;5:e11203. doi: 10.1371/journal.pone.0011203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rmali KA, Al-Rawi MA, Parr C, Puntis MC, Jiang WG. Upregulation of tumour endothelial marker-8 by interleukin-1beta and its impact in IL-1beta induced angiogenesis. Int J Mol Med. 2004;14:75–80. [PubMed] [Google Scholar]
- 28.Bagley RG, Rouleau C, St Martin T, Boutin P, Weber W, Ruzek M, et al. Human endothelial precursor cells express tumor endothelial marker 1/endosialin/CD248. Mol Cancer Ther. 2008;7:2536–46. doi: 10.1158/1535-7163.MCT-08-0050. [DOI] [PubMed] [Google Scholar]
- 29.Scobie HM, Thomas D, Marlett JM, Destito G, Wigelsworth DJ, Collier RJ, et al. A soluble receptor decoy protects rats against anthrax lethal toxin challenge. J Infect Dis. 2005;192:1047–51. doi: 10.1086/432731. [DOI] [PubMed] [Google Scholar]