

Abstract
Inherent or therapy-induced drug resistance is a major clinical setback in cancer treatment. The extensive usage of cytotoxic nucleobases and nucleoside analogs in chemotherapy also results in the development of specific mechanisms of drug resistance; such as nucleoside transport or activation deficiencies. These drugs are prodrugs; and being converted into the active mono-, di- and triphosphates inside cancer cells following administration, they affect nucleic acid synthesis, nucleotide metabolism, or sensitivity to apoptosis. Previously, we have actively promoted the idea that the nanodelivery of active nucleotide species, e.g. 5′-triphosphates of nucleoside analogs, can enhance drug efficacy and reduce nonspecific toxicity. In this study we report the development of a novel type of drug nanoformulations, polymeric conjugates of nucleoside analogs, which are capable of the efficient transport and sustained release of phosphorylated drugs. These drug conjugates have been synthesized, starting from cholesterol-modified mucoadhesive polyvinyl alcohol or biodegradable dextrin, by covalent attachment of nucleoside analogs through a tetraphosphate linker. Association of cholesterol moieties in aqueous media resulted in intramolecular polymer folding and the formation of small nanogel particles containing 0.5 mmol/g of a 5′-phosphorylated nucleoside analog, e.g. 5-fluoro-2′-deoxyuridine (floxuridine, FdU), an active metabolite of anticancer drug 5-fluorouracyl (5-FU). The polymeric conjugates demonstrated rapid enzymatic release of floxuridine 5′-phosphate and much slower drug release under hydrolytic conditions (pH 1.0–7.4). Among the panel of cancer cell lines, all studied polymeric FdU-conjugates demonstrated an up to 50 times increased cytotoxicity in human prostate cancer PC-3, breast cancer MCF-7 and MDA-MB-231 cells, and more than 100 times higher efficacy against cytarabine-resistant human T-lymphoma (CEM/araC/8) and gemcitabine-resistant follicular lymphoma (RL7/G) cells as compared to free drugs. In the initial in vivo screening, both PC-3 and RL7/G subcutaneous tumor xenograft models showed enhanced sensitivity to sustained drug release from polymeric FdU-conjugate after peritumoral injections and significant tumor growth inhibition. All these data demonstrate a remarkable clinical potential of novel polymeric conjugates of phosphorylated nucleoside analogs, especially as new therapeutic agents against drug-resistant tumors.
Introduction
Despite several decades of research and drug development, progress in the clinical struggle against cancer has been only moderate. Development of resistance by cancer cells to chemotherapeutic agents has currently become a major clinical problem, limiting the effectiveness of the treatment of hematological malignancies as well as solid tumors. Multiple studies have helped to identify many mechanisms of drug resistance, but in general terms they can all be reduced to the prevention of a drug from entering cells, deactivation of drug molecules, or the enhanced resistance of cancer cells to apoptosis. The first group includes deficiencies in membrane nucleoside transporters, or the overexpression of ATP-dependent drug efflux transporters like P-glycoprotein (MDR) and other membrane proteins (MRP) responsible for drug efflux. In the second group, the drug metabolism/degradation and reduced levels of enzymatic drug activation should be mentioned. In the third group, the induction of anti-apoptotic mechanisms, as well as the suppression of pro-apoptotic pathways plays important roles in drug resistance1.
Cytotoxic nucleoside analogs belong to the important class of anticancer drugs, which are currently used as the first-line treatment of hematological malignancies and certain solid tumors2. These drugs act as antimetabolites by interfering with nucleic acid synthesis and enzymes of the nucleotide metabolism. The clinical efficacy of these drugs depends on higher metabolic activity and drug activation in rapidly proliferating cancer cells compared to normal cells. Activation of therapeutic nucleoside analogs occurs through the de novo synthesis of 5′-mono-, di- and triphosphate derivatives, which interfere with the cellular pool of natural nucleosides. Nucleoside analogs require participation of specialized nucleoside transporter proteins such as hENT1, hENT2, hCNT1 in order to accumulate in the cells. The integral drug uptake depends on the proper balance of the nucleoside transporters and drug efflux proteins presented on cellular membrane. Therefore, the drug accumulation is substantially reduced when the expression of such nucleoside transporters is deficient3,4, or the activity of drug efflux transporter proteins is elevated5,6. After entering cells, nucleoside analogs undergo phosphorylation into 5′-monophosphates with deoxycytidine kinase (dCK) or thymidine kinase (TK), a rate-limiting step in the intracellular activation of nucleosides, and are subsequently converted into active 5′-diphosphates and 5′-triphosphates by other nucleoside kinases3. The efficacy of nucleoside analogs may further be limited by additional factors such as metabolic deamination and intracellular dephosphorylation7,8.
In order to increase tumor accumulation of nucleoside analogs, various prodrug and drug delivery approaches have been developed; for example, the application of lipophilic nucleoside derivatives with an enhanced cellular membrane affinity. Many prodrugs with degradable lipophilic masking groups demonstrate the enhanced cell membrane permeability9. Early phase clinical trials have shown some improvements in the treatment of hematological malignancies, but these prodrugs were not effective in the treatment of solid tumors10. Moreover, lipophilic prodrugs have a reduced half-life in circulation due to the fast accumulation in liver. Various nanocarriers such as liposomes, biodegradable nanoparticles, polymeric micelles and nanocapsules have been extensively for tumor delivery of chemotherapeutic drugs11. Many of these nanocarriers demonstrated advanced features, but have also shown serious shortcomings limiting their clinical usefulness. For example, liposomal formulations were unable to achieve effective drug concentration inside tumors because many anticancer drugs (cytarabine, 5-FU, etc) diffused rapidly through the liposome bilayer12.
Recently, we introduced an alternative tumor treatment strategy using formulation and nanodelivery of activated drugs, nucleoside 5′-triphosphates, encapsulated in cationic nanogels17. Previously, several laboratories have attempted to achieve in vivo delivery of bioactive 5′-triphosphates of nucleoside analog through encapsulation in liposomes13,14, nanoparticles15, or red blood cells16. Evidently, the success of this strategy depends on the advantages of nanoformulations in drug protection in biological milieu, controlled drug release, and the specific tumor targeting. Nanogel carriers have dramatically improved the delivery of activated phosphorylated nucleoside analogs into cancer cells and tumor growth inhibition effect18,19. However, the non-covalent nature of the encapsulation of anionic 5′-triphosphates in cationic nanogels was the reason of relatively fast drug release kinetics. Here, we report synthesis of novel types of drug-loaded nanogels, containing polymer network with covalently linked phosphorylated nucleoside analogs, which are capable of sustained drug release and structurally different from the previously studied polymeric nanogels. Covalent drug attachment is an important factor of the controlled drug release, because it allows using specific chemistries. Various hydrolytically or enzymatically-sensitive linkers such as peptides, carboxylates, etc have been previously evaluated with polymeric drug delivery systems20–22. In this study, drug conjugates were synthesized by the attachment of nucleoside analogs through a biodegradable tetraphosphate linker starting from amphiphilic polymers such as cholesterol-modified polyvinyl alcohol (PVA) or dextrin (DEX). The linker has strong advantage over other linkers, because the polymeric drug conjugates are able to release nucleoside analogs in active phosphorylated form in the result of its hydrolytic or enzymatic degradation, eventually showing an enhanced tumor growth inhibition efficacy against normal and drug-resistant cancer cells. These drug-containing polymer conjugates can form stable nanogels with a small hydrodynamic diameter after ultrasonication in aqueous media as demonstrated in Figure 1. Selection of biodegradable or mucoadhesive biocompatible polymers for preparation of polymeric conjugates might also reduce toxicity of chemotherapy and open potentials for oral administration of these nanoformulations.
Figure 1.
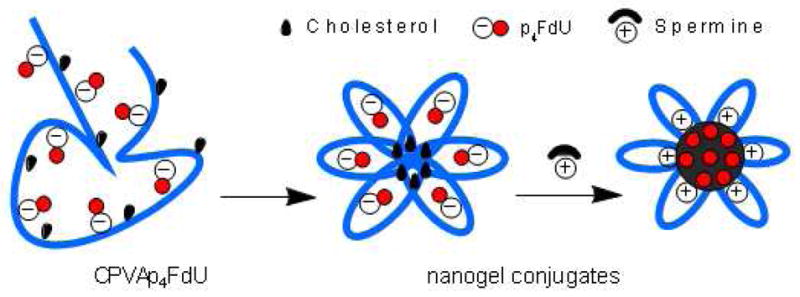
Formation of compact nanogels from polymer drug conjugates
Materials and Methods
Materials
Most reagents, solvents, and polymers were purchased from Sigma Aldrich (St. Louis, MO) and Alfa Aesar (Wardhill, MA) with the highest available purity and used without purification unless otherwise stated. Thymidine, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) and snake venom phosphodiesterase 1, type VI, from Carotalus adamanteus were purchased from Sigma (St. Louis, MO). Nucleoside analogs: 5-fluro-2′-deoxyuridine (Floxuridine, FdU) was from SynQuest Laboratories (Alachua, FL), 2, 2′-difluorocytidine (dFdC, Gemcitabine) was from Beta Pharma, Inc (Branford, CT), and arabinosylcytosine (araC, Cytarabine) was from 3B Medical Systems, Inc (Libertyville, IL). Centrifuge filter devices (MWCO 5,000 Da) were purchased from Millipore (Bedford, MA).
All NMR spectra were recorded using a 500 MHz Varian NMR- spectrometer. All chemical shift values are given in parts per million (ppm) and are referenced to a signal from (CH3)4Si (0 ppm) for 1H, DMSO-d6 (39.7 ppm) for 13C, and 85% phosphoric acid (0 ppm) for 31P spectra at 25°C. Hydrodynamic diameter, polydispersity, and zeta-potential of nanogels and polymeric conjugates were measured using a dynamic light scattering instrument, the Zetasizer Nano-ZS90 (Malvern Instruments, Southborough, MA) at 25°C. Monodisperse polystyrene dispersions were used as standards. UV absorbance of samples was measured by Biophotometer (Eppendorf, Hamburg, Germany). IR spectra were recorded using a Nicolet IR-200 FT-IR spectrometer (Thermo Scientific, Waltham, MA).
Cells
Human breast carcinoma MCF-7, human hepatocellular carcinoma HepG2, and human prostate adenocarcinoma PC-3 cells were obtained from ATCC (Rockville, MD). These cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine and 2% penicillin-streptomycin at 37°C in a humidified atmosphere containing 5% CO2. Human breast carcinoma MDA-MB-231 cell line was a gift from Dr. R. Singh (UNMC). These cells were maintained in DMEM: Nutrient mixture F-12 (DMEM/F12) with similar supplements and serum as above. Gemcitabine-resistant human follicular lymphoma RL7/G cell line, which is characterized by a reduced level of dCK enzyme23, was a gift from Dr. F. Bontemps (De Duve Institute, Bruxelles, Belgium). They were grown in the presence of 2 μM gemcitabine. Nucleoside transport-deficient cytarabine-resistant human leukemic lymphoblast CEM/araC/8 cell line24 was obtained from Dr. C. Galmarini (UFR Lyon-Sud, Oullins, France). The cells were grown in the presence of 0.5 μM cytarabine (araC). Both drug-resistant cell lines were grown in RPMI medium supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, and 2% penicillin-streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
Synthesis of cholesterol conjugates
PVA was grafted with cholesterol moieties according to the procedures described below. Briefly, 2.1 g of PVA (M.w. 13 kDa) was dried over phosphorus pentoxide in vacuo and dissolved in 50 mL of anhydrous DMSO at 70 °C. Triethylamine (0.8 mmol) was added to the cooled solution (25°C) followed by 0.3g (0.68 mmol) of cholesteryl chloroformate, and the final solution was stirred overnight at 25°C. The reaction mixture was concentrated in vacuo and dialyzed (MWCO 3.5 kDa) against 20% aqueous ethanol three times for 24 hours. The product (CPVA) was isolated after concentration in vacuo and freeze-drying with a yield of 80%. In another method, 2.1 g of PVA (M.w. 13 kDa) was dissolved in 50 mL of anhydrous N-methyl pyrrolidone at 70°C, then 0.3g (0.68 mmol) of cholesteryl chloroformate was added, and the mixture was stirred for 4h at 70°C. The substituted CPVA was precipitated in diethyl ether (0.5L) and dried in vacuo; the light yellow precipitate was obtained with a yield of 70%. 1H-NMR: 0.63 (s, 18H), 0.83 (m, 36H), 0.88 (m, 18H), 0.92 (s, 18H), 1.11–1.95(m, 764H), 3.84–3.90(m, 295H), 4.31(brs, OH), 4.40(s, 12H), 5.25(s, 6H). Following the same protocol for PVA (M.w. 31 kDa), we obtained CPVA with a yield of 85%. 1H-NMR: 0.65 (s, 18H), 0.83–0.85(dd, J=5.0, 1.6 Hz, 36H), 0.89(d, J=4.8 Hz, 18H), 0.94(s, 18H), 1.07–1.98(m, 1582H), 3.84(m, 704H), 4.35(12H and OH), 5.28(s, 6H). IR: 3264, 2897, 1642, 1409, 1323, 1082, 915, 829.
The dextrin-cholesterol nanogel (CDEX) was synthesized as follows. The water-soluble fraction of dextrin (M.w. 9 kDa) was isolated by dialysis in a SpectraPor membrane tube (MWCO 2 kDa) followed by centrifugation. The supernatant was freeze-dried and used for nanogel synthesis. 1.0 g of the purified dextrin was dried over phosphorus pentoxide in vacuo and dissolved in 15 mL of anhydrous DMSO at 70°C. After 0.3g (0.68 mmol) of cholesteryl chloroformate was added, the reaction mixture was stirred for 24h at 25°C, concentrated in vacuo and dialyzed (MWCO 3.5 kDa) against 20% aqueous ethanol three times for 24 hours. The product (CDex) was isolated after concentration in vacuo and freeze-drying with a yield of 76%. 1H-NMR: 0.65 (s, 18H), 0.83–0.85(dd, J=5.0, 1.6 Hz, 36H), 0.89–1.51(m, 211H), 3.24–3.64(m, 333H), 4.28–5.10 (m, 122H), 5.23(s, 6H), 5.37–5.62(m, 55H).
Synthesis of phosphorylating reagent, CNEtOP(O)Im2
The intermediate product CNEtOP(O)Cl2 was synthesized by dissolving 18.6 mL (30.6 g, 0.2 mol) phosphorus(V) oxychloride and 20.6 mL (14.84 g, 0.147 mol) triethylamine in 40 mL anhydrous tetrahydrofuran (THF) at 0°C. It was treated with 10 mL (10.45 g, 0.147 mol) 2-cyanoethanol in 5mL THF while stirring at 0°C. Stirring was continued for 30 minutes until white precipitate formed. The precipitate was carefully filtered with exclusion of moisture, and the resulting solution was concentrated in vacuo and distilled under argon. The product, CNEtOP(O)Cl2 was recovered at 90°C/1 mm with a yield of 60%.
The product, CNEtOP(O)Im2 was synthesized by mixing 2.69 g (0.015 mol) CNEtOP(O)Cl2 and 5.25g (0.037 mol) N-trimethylsilyl-imidazole in 40 mL cold anhydrous toluene. The solutions were then incubated for 2h at room temperature, concentrated in vacuo to a half-volume and placed in a freezer for 2 h at −20 °C. The precipitate of CNEtOP(O)Im2 was recovered after centrifugation with a yield of 70%25.
Preparation of polymeric drug conjugates
A solution of 3.3 g of dried cholesterol-polymer conjugates in 33 mL DMF was treated with a 2M solution of CNEtOP(O)Im2 in anhydrous DMF (2 mL) for 30 min at 25°C. Then, a 1M solution of tetra-n-butylammonium salt of pyrophosphate (PPi-TBA) in anhydrous DMF (4 mL) was added, and the reaction mixture was incubated for 1 h at 25°C. In a separate flask, floxuridine (FdU, 490 mg, 2 mmol) was treated with a 2M solution of CNEtOP(O)Im2 in anhydrous DMF (1 mL) and allowed to stand for 20 min at 25°C. Both solutions were then mixed and stirred for 40 min at 25°C. The reaction mixture was treated with 1 mL of methanol and left overnight at 4°C. Insoluble material was removed by filtration; the nanogel conjugate was purified three times over 24 h by dialysis (MWCO 3,500 Da) against 20% aqueous ethanol, concentrated in vacuo and precipitated as a sodium salt in acetone. FdU content in the nanogel conjugate was measured by UV absorbance (ε260 = 7,570). Drug loading: CPVA31, 0.51 μmol/mg; CPVA13, 0.50 μmol/mg; and CDex9, 0.44 μmol/mg.
Particle size and zeta-potential measurements
The hydrodynamic diameter and polydispersity of nanogels and polymeric conjugates were measured by dynamic light scattering (DLS) using a Zetasizer Nano-ZS90 with a 15 mV solid state laser operated at a wavelength of 635 nm. In brief, dry samples were resuspended in filtered de-ionized water, then sonicated for 1 h at 4°C to form a uniform dispersion of nanoparticles and centrifuged for 4 min at 10,000 ×g. The size distribution in samples was characterized by polydispersity index. Zeta-potential was calculated based on electrophoretic mobility measurements performed with an electrical field strength of 15–18 V cm−1 at 25°C using the instrument software. The data reported in Table 1 represents an average of three measurements.
Table 1.
Particle characteristics of polymeric conjugates
| Polymeric conjugate* | dh, nm (volume- averaged) | PDI | ζ, mV |
|---|---|---|---|
| CPVA31 | 35.00 ± 1.30 | 0.361 ± 0.01 | 0.00 ± 3.70 |
| CPVA31-p4FdU | 42.12 ± 6.41 | 0.405 ± 0.04 | −8.47 ± 3.60 |
| CPVA13 | 12.52 ± 5.12 | 0.596 ± 0.03 | −2.57 ± 3.40 |
| CPVA13-p4FdU | 34.95 ± 5.43 | 0.417 ± 0.01 | −34.0 ± 4.67 |
| CDEX9 | 44.53 ± 8.34 | 0.458 ± 0.02 | −8.00 ± 4.02 |
| CDEX9-p4FdU | 26.23 ± 4.12 | 0.440 ± 0.03 | −34.80 ± 5.06 |
| CDEX9-p4T | 18.27 ± 2.01 | 0.508 ± 0.00 | 0.00 ± 4.52 |
Particle size (dh), polydispersity index (PDI) and zeta potential (ζ) were measured in 1% solutions in water after 2 h-sonication. The results are average values ± SD of three measurements.
Enzymatic hydrolysis
Enzymatic stability and drug release from polymeric conjugates was assayed in 50μL reaction mixtures containing: 100 mM Tris-HCl (pH 8.75), 2 mM MgCl2, 0.5 mg of snake venom phosphodiesterase (VPDE) and 0.5 mg nanogel sample (FdU, 0.25 μmol). The reaction mixture was incubated at 37°C and, at appropriate times, 5 μL aliquots were taken out and quenched with 1.5 μL of 1M HCl. Nucleotide content was analyzed by ion-pair HPLC using an Ascentis C18 column (10 μm, 15cm × 4.6mm) at a flow rate of 1 mL/min. The elution was performed with buffer A: 40 mM KH2PO4, 0.2% tetrabutylammonium hydroxide, pH 7.0, and buffer B: 30% acetonitrile, 40 mM KH2PO4, 0.2% tetrabutylammonium hydroxide, pH 7.0, in a linear gradient mode (100% B in 20 min).
In vitro drug release
In vitro drug release was investigated under different pH values. In short, 18 mg of CPVA31-p4FdU or CDex9-p4FdU conjugates was dissolved in 20 ml of PBS solution at pH 7.4, 4.0 and 1.0. These solutions were incubated at 37°C and 0.5 mL-aliquots were taken out every 24 h. The released nucleotides were separated from the rest of the polymeric conjugates by centrifugation at 7,500 rpm for 25 min using an Amicon Ultra-0.5 centrifuge filter device (MWCO 3,000 Da). The pH of the filtrate was adjusted to 7.4 and UV absorbance was measured at 260 nm.
Cytotoxicity studies
Cytotoxicity of the polymeric conjugates was analyzed in different cancer cell lines by a standard MTT assay. Briefly, MCF-7, PC-3, HepG2, and MDA-MB-231 cells were seeded at a density of 10,000 cells/200 μL growth medium/well in flat-bottom 96-well plates; the corresponding suspensions of RL7/G and CEM/araC/8 cells were placed in round-bottom 96-well plates. Cells were allowed to grow overnight and appropriate amounts of drug, nanogels or polymeric conjugates were added. Samples were incubated in full medium for 72 h at 37 °C, and the metabolic activity of each sample was determined by adding 20 μL of a 5 mg/mL of MTT stock solution in sterile PBS buffer to each well. The samples were then incubated for 2 h at 37°C, the medium and the MTT dye were washed out by PBS, and 100 μL of extraction buffer (20% w/v SDS in DMF/water, 1:1, pH 4.7) was added to each well. Samples were incubated for 24 h at 37°C. Optical absorbance was measured at 560 nm using a Model 680 microplate reader (BioRad, Hercules, CA) and cytotoxicity was expressed as a percentage of survived cells relative to non-treated control cells. All samples were analyzed by an average of eight measurements (means ± SEM). These data were plotted versus drug/nanogel concentrations and converted into IC50 values (concentration of the 50% cell survival).
In vivo tumor growth inhibition assay
These experiments were performed using female nu/nu mice (RL7/G cells) or male nu/nu mice (PC-3 cells), aged 6–8 weeks (Charles River Laboratories, Wilmington, MA). Animal studies were carried out according to the Principles of Animal Care outlined by the National Institutes of Health, and protocols were approved by the Institutional Animal Care and Use Committee at the University of Nebraska Medical Center. The animals were randomly divided into groups of five per cage and maintained under sterile conditions and 12-h light/dark cycle in a temperature-controlled environment. All manipulations with animals were performed in a sterile laminar hood using sterile solutions. PC-3 and RL7/G cell suspensions of 5× 106 cells/400 μL of medium containing 20% Matrigel (Becton-Dickinson, San Diego, CA) were injected subcutaneously in the right flank areas of mice. After tumors could be palpitated, the treatment solutions of CPVA31-p4FdU were injected peritumorally (2 × 100 μL) twice a week at a dose of 12 mg FdU/kg. Tumor volume was measured by digital calipers and calculated based on the equation: TV=L/2 × W2, where L and W are length and width of tumor (mm).
Results and Discussion
Synthesis of nanogel conjugates
Cholesterol is a well-known hydrophobic moiety used in many drug delivery applications in order to enhance the interactions of modified macromolecules or nanocarriers with the cellular membrane26,27. In our design of polymeric conjugates, we have exploited polymer modification with cholesterol for several reasons: (i) to render hydrophilic polymers soluble in organic solvents, (ii) to compel the modified polymers to form compact nanogels, and (iii) to increase membranotropic properties and ease transport of hydrophilic drug molecules across the cellular membrane. In aqueous solutions at ultrasonication cholesterol-modified polymers form ‘flower-type’ micelles with internally aggregated cholesterol moieties with the least association numbers 4–628. We synthesized the cholesterol-modified polyvinyl alcohol (CPVA) containing six hydrophobic moieties per polymer chain by reaction of the corresponding PVA, M.w. 13 and 31 kDa, with cholesterol chloroformate in dry DMSO in the presence of triethylamine at room temperature. The cholesterol-modified PVA polymers were isolated with a yield of 80–85%. In an alternative method, PVA was modified in N-methyl pyrrolidone at 70°C, yielding the corresponding cholesterol-modified polymer at 70–75 % as a white solid after precipitation in diethyl ether29. The cholesterol-modified PVA polymers were designated as CPVA13 and CPVA31 (Scheme 1). Similarly, as shown in Scheme 2, we synthesized a cholesterol-modified dextrin (CDEX) with a high yield starting from dextrin (M.w. 9 kDa). Four cholesterol moieties were attached to the smaller dextrin molecule. The flexibility of charged polymer chains was restricted by hydrophobic cholesterol groups aggregated in the core of nanogels. The nanogels bearing negative charged phosphate groups formed even smaller spherical compacted particles following the addition of positively charged spermine molecules (Figure 1).
Scheme 1.

Synthetic steps in the preparation of CPVA conjugates
Scheme 2.

Synthesis of CDex-conjugates (compounds 1–11, see Scheme 1).
1H NMR spectra showed that the cholesterol modification was nearly quantitative and amounted for six moieties per polymer chain in CPVA13 and CPVA31 and four per polymer chain in CDex9. Similarly, the formation of micelles in aqueous solutions by these polymers was also demonstrated by 1H NMR spectroscopy. Figures S1 and S2 represent the spectra of CPVA13 and CDex9 in (a) D2O and (b) DMSO-d6, respectively (see Supporting Materials). As shown in the spectra, proton signals of the cholesterol moiety (δ=0.6–2.4 ppm) appeared in DMSO-d6 (b), but completely disappeared or wide broadening of signals was observed in D2O (a). This indicates the restricted molecular motion of cholesterol moieties upon self-aggregation. Our results confirmed the formation of a rigid core of hydrophobic cholesterol moieties and a relatively mobile shell consisting of hydrophilic PVA or Dex molecules in aqueous medium. The degree of cholesterol substitution (DS) was evaluated by calculating the ratio between the integrals of the protons in terminal CH3-groups of cholesterol and methylene protons in CPVA or the protons of sugar monomers in CDex.
The hydroxyl functional groups in the cholesterol-modified polymers have been used as sites for the conjugation of the active phosphorylated nucleoside analog, floxuridine (FdU), resulting in the formation of polymeric conjugates as anticancer drug carriers (Scheme 1 and 2). The 5′-hydroxyl group of the nucleoside analog was chemically attached via a biodegradable tetraphosphate linker to nanogels using a 2-cyanoethyl-bis(imidazolyl)phosphate, CNEtOP(O)Im2, as a phosphorylating reagent. As shown in Scheme 1 and 2, the polymers were phosphorylated with CNEtOP(O)Im2 in DMF and then reacted efficiently with inorganic pyrophosphate in the form of tetra-n-butylammonium salt PPi-TBA(6) in order to form the polymeric triphosphate 7. Separately, the 5′-hydroxyl group of the nucleoside was phosphorylated by CNEtOP(O)Im2, and the activated 5′-phosphorylated nucleoside 8 was reacted in the next step with the polymeric triphosphate 7.
In our preliminary experiments, we found that a similar phosphorylating agent, methyl-bis(imidazolyl)phosphate, MeOPOIm2, reacted efficiently with the primary hydroxyl groups of nucleosides in the formation of activated 5′-monophosphates, but compared with CNEtOP(O)Im2, this reaction was much slower. The relative efficacy of phosphorylation using these two reagents was compared based on the yields of nucleoside 5′-monophosphate analyzed by ion-pair HPLC. During the first hour of reaction, CNEtOP(O)Im2 yielded 80% of monophosphate compared with only 10% when MeOPOIm2 was used.. The electron donor effect of the methoxy group makes the phosphorous atom less electrophilic, which resulted in longer reaction times, especially with secondary hydroxyl groups. However, both phosphorylating agents formed the same final activated 5′-monophosphorylated nucleoside in our synthesis. Next, the activated imidazolyl-phosphate moiety 8 readily reacted with polymer-triphosphate 7 and converted into nucleoside 5′-tetraphosphate anchored to CPVA, (9–11) or CDex (14, 15). Initially, we used thymidine (T) as a model drug in this study. Polymeric conjugates were purified by extensive dialysis to remove all reactants. CPVA- and CDex-conjugated 5′-tetraphosphates of FdU were designated as CPVA13-p4FdU, CPVA31-p4FdU, CDex9-p4FdU and CDex9-p4T and their properties are shown in Table 1. The amount of nucleoside attached to the polymer was determined by UV absorbance. We observed a high degree of nucleoside loading in nanogels, which was equal to 0.4–0.5 μmol/mg.
Covalent conjugation of the nucleoside to nanogels via a tetraphosphate linker was further verified by 31P-NMR and IR spectroscopy. 31P-NMR spectra confirmed the formation of tetraphosphate structures along with trace amounts of a triphosphate and polyphosphates such as a pentaphosphate (Figure 2). According to published chemical shifts30,31, upper field region of 31P-NMR spectra at −20 to −25 ppm corresponds to β- and γ-phosphates (peaks c, d and e), while signals between −8 to −12 ppm correspond to terminal α- and δ-phosphates (peaks a and b). IR spectra of CPVA-p4FdU have allowed us to observe conjugation of the FdU nucleoside to the CPVA nanogel (Figure S3). As is clearly shown in the spectra, the peak at 899 cm−1 corresponding to P-OR stretch, the peak at 1234 cm−1 corresponding to P=O stretch of phosphate, and the peak at 1708 cm−1 corresponding to C=O stretch of amide are clearly increased in the CPVA-p4FdU products, while peaks at 1311–1417 cm−1 corresponding to O-H bonding of CPVA were reduced, confirming the formation of a polyphosphate linker between CPVA and FdU.
Figure 2.
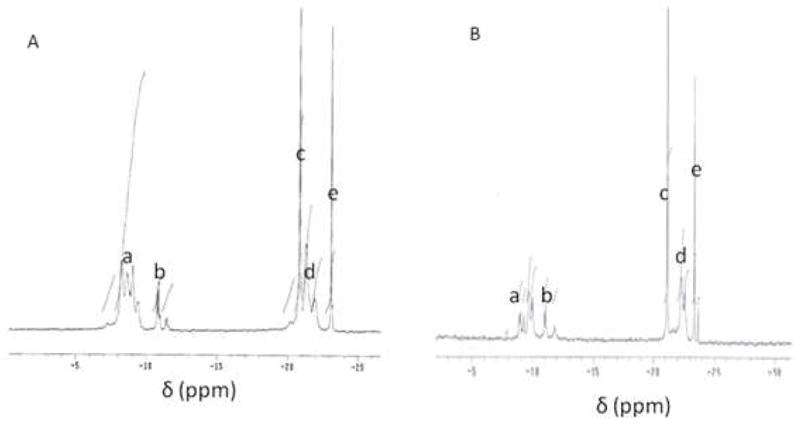
31P-NMR spectrum of polymeric conjugates, CPVA31-p4FdU (A) and CDex9-p4FdU (B) (phosphorus signals a-e are described in the text)
Particle size and zeta-potential
Compact nanogel conjugates could be successfully formed as the result of self-organization of CPVA/CDex-p4FdU molecules during ultrasonication in aqueous solutions. When sufficient energy was applied, the cholesterol moieties formed compact intramolecular clusters surrounded by a hydrophilic polymeric shell containing the embedded negatively-charged drug molecules. The particle size, homogeneity, and morphology of CPVA/CDex-p4FdU structures were measured by dynamic light scattering (DLS) and transmission electron microscopy (TEM). A single sharp peak in DLS profiles with a hydrodynamic diameter in the range of 12–45 nm implied the presence of a single population of small particles with a relatively low polydispersity index of 0.30–0.59 (Table 1). The high negative zeta- potential of CPVA/CDex-p4FdU confirmed the presence of phosphates in the surface layer of polymeric conjugates. TEM pictures showed the spherical particle morphology with hydrodynamic diameters in the range of 38–58 nm (Figure 3). We have also studied the change in particle size after neutralization of the negative charge in polymeric conjugates with the polyamine spermine (Figure 1). The addition of positively-charged spermine at physiological conditions resulted in a 2–3-fold reduction in hydrodynamic and TEM-observed diameters and the change in morphology of particles, e.g. the appearance of a thicker electron-dense exterior layer surrounding these compacted polymeric conjugates32.
Figure 3.
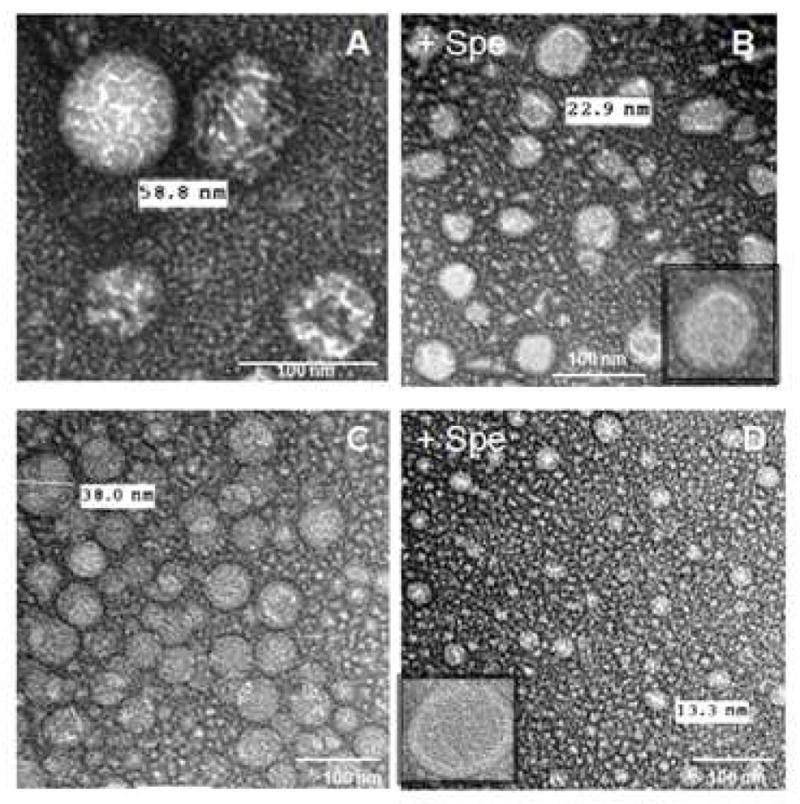
Transmission electron microscopy (TEM) images of nanogels formed from polymer conjugates: (A) CPVA31-p4FdU, (B) its spermine complex (Spe), (C) CDex9-p4FdU, and (D) its spermine complex. Samples were stained with vanadate.
Enzymatic hydrolysis
The formation of natural nucleoside 5′-phosphate bonds between nucleoside and nanogel backbone was further confirmed using enzymatic hydrolysis by snake venom phosphodiesterase I (VPDE). VPDE was able to catalyze the hydrolysis of esterified nucleoside 5′-phosphates into a nucleoside 5′-phosphate and also the cleavage of nucleoside 5′-phosphate from oligonucleotides with a free 3′-end33,34. This property of VPDE allows us to investigate the enzymatic stability of nanogel-bound nucleoside phosphates. Nanogels CPVA-p4FdU and CDex-p4FdU were incubated with VPDE at 37 °C and then analyzed by ion-pair HPLC (Figure 4). We observed the gradual disappearance of the initial wide peak with the elution time of 25–34 min (a) and the formation of a sharp peak at the elution time of ca. 5 min (b), which corresponded to control nucleoside 5′-phosphates (TMP or FdUMP). Structure of the released nucleoside products was further confirmed by comparison of UV spectra with initial nucleoside analogs. Our data demonstrated a nearly quantitative enzymatic release of nucleotide 5′-phosphate from polymeric conjugates within 12 to 24 h.
Figure 4.

Enzymatic hydrolysis of polymeric conjugates by snake venom phosphodiesterase I (VPDE). (A) The enzyme hydrolyzes the P-O bond at α-phosphate group in nanogel conjugate resulting in nucleoside 5′-phosphate (2). (B) Ion-pair HPLC profiles of initial (a) and hydrolyzed (b) CPVA31-p4FdU. (C) Ion-pair HPLC profiles of initial (a) and hydrolyzed (b) CDex9-p4T after 24 h-incubation with 0.01 units of VPDE enzyme.
In vitro drug release
The in vitro release of FdU was monitored at 37 °C at different pH values (1.0, 4.0 and 7.4) in order to assess the stability of nanogel conjugates in the environments inside of the stomach, endosomal vesicles, and blood, respectively. Each nanogel, CPVA-p4FdU or CDEX-p4FdU, was placed into the appropriate buffer solution and incubated at 37 °C. Serial aliquots were removed at the appropriate times during hydrolysis, and the cleaved nucleoside/nucleotide was separated by ultrafiltration and quantified by UV absorbance. The release profiles at different pH values are shown in Figure 5. In general, polymeric conjugates displayed linear first-order reaction kinetics of hydrolysis with slower drug release at pH 7.4 and pH 4.0 than at pH 1.0. At pH 4.0 and 7.4, drug release was 1–2% per day, while at pH 1.0 drug release reached 4% per day. These results are consistent with the pH-dependent degradation behavior of other dinucleoside polyphosphates, such as diadenosine-P1,P3-triphosphate and P1,P4-tetraphosphate35. However, CPVA/CDex-p4FdU conjugates showed much slower drug release compared to diadenosine-P1,P4-tetraphosphate (25 days vs. 3 days). The pH-dependent hydrolysis can be facilitated by a hydronium ion through the nucleophilic attack of protonated phosphate groups at lower pH, which results in an SN(P) type substitution, and then the formation of a penta-coordinated phosphorus transition state and nucleoside 5′-phosphates as final products (Scheme 3). Highly hydrated polymer coils surrounding the tetraphosphate linkers evidently create a steric hindrance and significantly slow down the process.
Figure 5.
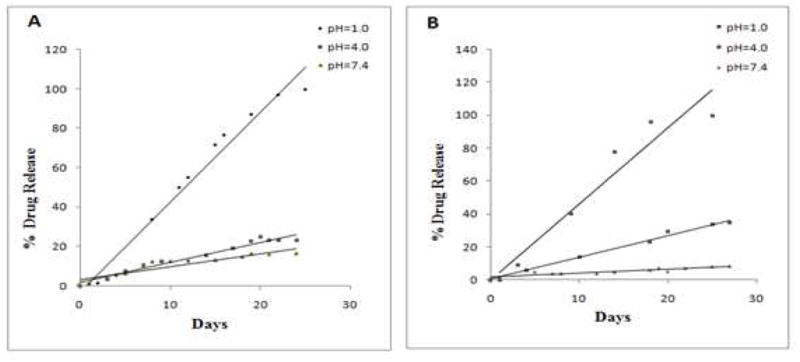
In vitro drug release from polymeric conjugates CPVA31-p4FdU (A) and CDex9-p4FdU (B) at different pH in buffered saline.
Scheme 3.
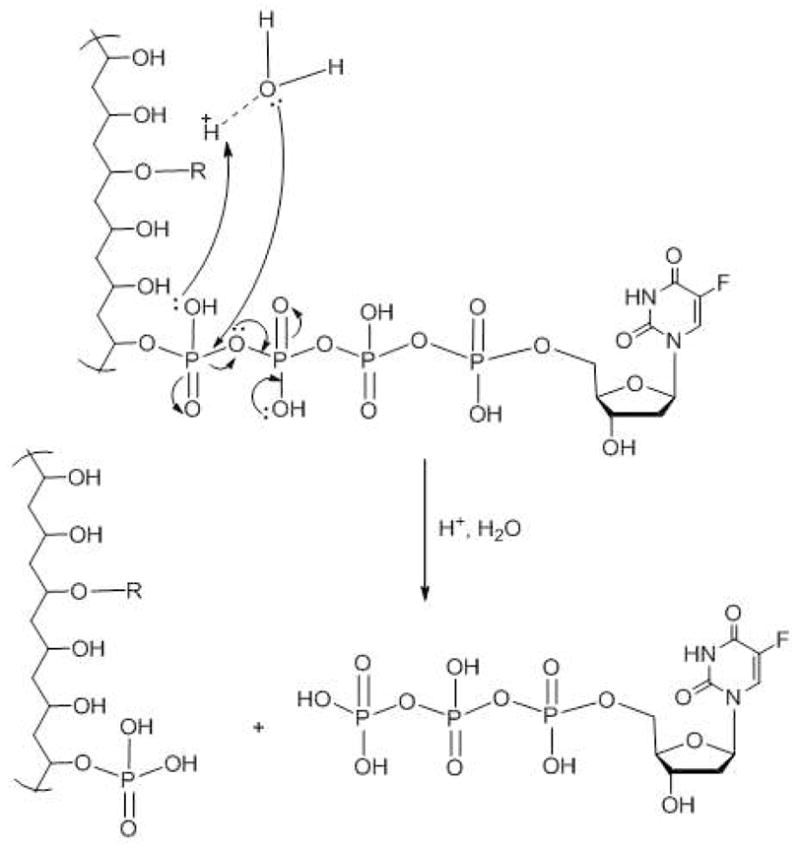
Mechanism of acidic hydrolysis of polymeric conjugates and release of phosphorylated nucleosides (R = cholesterol).
Evidently, the release of phosphorylated nucleosides in cancer cells would provide a strong therapeutic advantage to these polymeric conjugates, because the drug component doesn’t have to pass through the phosphorylation step, which is known to be a rate-limiting step in biological activation of nucleoside analogs36. Our data show that this type of polymeric conjugates is capable of the sustained drug release during the extended period of time and can serve as active drug depot, significantly enhancing therapeutic effect against cancer cells. Peritumoral injections or systemic administration of polymeric conjugates would result in only minimal initial drug burst, a shortcoming of many drug delivery systems at systemic administration. Additionally, accumulation of nanocarriers from blood circulation through the enhanced permeability and retention (EPR) effect in leaking tumor neovasculature can potentially enhance the tumor growth inhibitory effect. Polymeric conjugates can also be considered as potential oral therapeutic formulations due to the observed slow drug release at low pH in the digestive tract (in vivo experiments are underway). We have also demonstrated that enzymatic hydrolysis of polymeric conjugates is at least 20–25 times faster than hydrolytic hydrolysis. Therefore, these polymeric conjugates, which have a slow sustained drug release in tumor tissue and other organs in normal conditions in vivo, might be quickly activated by enzymatic activities present in the cytosol or subcellular compartments of proliferating cancer cells37. The most common type of hydrolytic enzymes in mammalian cells are cytosolic phosphodiesterases and nucleotide phosphatases, which can release active 5′-nucleotides from the polymeric conjugates38.
Cytotoxicity Assay
Drug resistance to nucleoside analogs is known to be an important clinical problem in the treatment of cancer. Therapeutic effects can be achieved with nucleoside analogs as a single agent or in combination with other drugs only at the increasingly higher dosage. Here, we have studied the cytotoxicity of several polymeric conjugates of activated analogs of 5-fluorouracyl as a model drug in various cancer cell lines including ones that are resistant to nucleoside analogs. The cytotoxicity of polymeric floxuridine conjugates CPVA13-p4FdU, CPVA31-p4FdU and CDEX9-p4FdU was determined in human prostate adenocarcinoma PC-3, breast carcinoma MCF-7 and MDA-MB-231, hepatic carcinoma HepG2, gemcitabine-resistant follicular lymphoma RL7/G and cytarabine-resistant T-lymphoma CEM/araC/8 cells using a thiazolyl blue (MTT) dye reduction assay39. As shown in Figure 6 and Table 2, nanoconjugates showed considerably enhanced cytotoxicity and lower IC50 values (drug concentration resulting in 50% cell death) compared to free floxuridine in all of these cell lines. The enhancement factor (EF), which is equal to IC50(drug)/IC50(conjugate), was used as a measure of the increase in cytotoxicity of polymeric conjugates compared to free drug. Nanogels without conjugated drug demonstrated no cytotoxicity (IC50 > 10 mg/mL). All of these polymeric conjugates showed a higher EF in drug resistant cell lines, CEM/araC/8 and RL7/G, compared to other tumor cells. Specifically, CPVA13-p4FdU and CDex9-p4FdU exhibited an EF of 100 and 85 in CEM/araC/8 cells, while the EF showed by other cells was normally in the lower range of 3.5–50.
Figure 6.

Cytotoxicity of polymeric conjugates in drug-resistant human T-lymphoma CEM/araC/8 cells (A–C) and prostate carcinoma PC-3 cells (D).
Table 2.
Cytotoxicity of cytotoxic drugs, polymeric conjugates and drug conjugates in cancer cells
| Drug formulation | IC50 values (μM)* |
|||||
|---|---|---|---|---|---|---|
| PC-3 | MCF-7 | HepG2 | MDA-MB-231 | CEM/araC/8 | RL7/G | |
| Floxuridine (FdU) | 6.5 | 12195 | 2195 | 14.2 | ||
| Cytarabine (araC) | 1234 | |||||
| Gemcitabine (G) | 19011 | |||||
| CPVA31 | >10 | >10 | >10 | >10 | >10 | >10 |
| CPVA13 | >10 | >10 | >10 | >10 | >10 | >10 |
| CDEX9 | >10 | >10 | ||||
| CPVA31-p4FdU | 0.4 (EF=16) | 2032 (EF=6) | 487 (EF=4.5) | 0.28 (EF=50) | 12.3 (EF=100) | 3802 (EF=5) |
| CPVA13-p4FdU | 81.3 | 813 (EF=15) | 609.7 (EF=3.6) | 0.6 (EF=23) | 102 (EF=12) | 950 (EF=20) |
| CDEX9-p4FdU | 14.5 (EF=85) | |||||
Cytotoxicity was measured after 72-h treatment. IC50 of CPVA31, CPVA13 and CDEX9 in each cell line are given in mg/mL. EF = enhancement factor showing the efficacy compared to free drug.
In vivo tumor growth inhibition
The therapeutic efficacy of polymeric conjugates was evaluated in subcutaneous (s.c.) human prostate adenocarcinoma PC-3 and gemcitabine-resistant follicular lymphoma RL7/G tumor xenograft mouse models. These tumors were established by s.c. injection of tumor cells in the lower flank areas of athymic nu–nu mice. After the observation of the initial tumors, animals were randomly separated into control and treatment groups (n = 5–6). Median tumor volume was measured by digital calipers twice a week simultaneously with peritumoral injections of polymeric conjugates with a dose of 80 mg/kg, which is equivalent to 10 mg FdU/kg. This way of administration allowed a subcutaneous storage of the viscous polymer-drug conjugate and the sustained release of activated drug into the tumor. We observed a strong 6.5-fold tumor growth inhibition in human prostate carcinoma PC-3 model that confirms the high therapeutic potential of polymeric conjugates (Figure 7A). Although the observed 2-fold growth inhibition was lower in drug-resistant human follicular lymphoma RL7/G tumors compared to PC-3 tumors, the effect of the CPVA31-p4FdU conjugate was statistically significant (P < 0.05) (Figure 7B). The significance of the experimental data was determined by a two tailed Student’s t-test. No significant weight loss or acute toxicity of polymeric conjugates was observed during the entire period of treatment. Tumors removed from experimental animals at the end of the experiments were clearly smaller than tumors in the control group (Figure 7C).
Figure 7.

Tumor growth inhibition in mice with subcutaneous human prostate carcinoma PC-3 (A) and gemcitabine-resistant follicular lymphoma RL7/G (B) tumors following the peritumoral injections of the polymeric conjugate CPVA31-p4FdU (dose 80 mg/kg, or 10 mg FdU/kg). The data were statistically significant with P<0.05 between (a) control and (b) treatment groups. (C) Tumor photographs in the end of the experiment B taken from control and treatment groups.
Conclusions
We have developed a novel type of covalently-bound polymeric phosphorylated nucleoside analogs, polymeric conjugates, for a sustained delivery of the activated anticancer drugs into tumors. These carriers combine attractive properties of biocompatible polymers with an enhanced cytotoxic efficacy of activated nucleoside analogs and form compact drug-loaded polymeric nanoparticles. In vitro evaluation against various cancer cell lines, including drug-resistant cancer cells, demonstrated that the activated floxuridine conjugate has 50–100 times stronger cytotoxicity compared to free nucleoside analog. Furthermore, the observed sustained drug release was a potential cause of the increased tumor growth inhibition following the peritumoral administration of polymeric conjugates in subcutaneous tumor xenograft models. This class of anticancer drug formulations also has features, which makes them a very promising vehicle for oral administration of activated phosphorylated nucleoside analogs.
Supplementary Material
Acknowledgments
The financial support from National Cancer Institute (R01 CA136921 for S.V.V.) is gratefully acknowledged. The authors thank Tom Bargar of the Electron Microscopy Core Facility, Ed Ezell of the NMR Core Facility and Marina Sokolsky-Papkov from Center for Drug Delivery and Nanomedicine (University of Nebraska Medical Center) for providing assistance with TEM microscopy, NMR and IR spectroscopy, respectively. We are grateful to Trevor Gerson for his help in the revision of the manuscript.
Footnotes
Supporting information available: Analytical characteristics (1H-NMR and IR spectra, Figures S1–S3) of synthesized products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 2.Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002;3:415–424. doi: 10.1016/s1470-2045(02)00788-x. [DOI] [PubMed] [Google Scholar]
- 3.Galmarini CM, Thomas X, Calvo F, Rousselot P, Rabilloud M, El Jaffari A, Cros E, Dumontet C. In vivo mechanisms of resistance to cytarabine in acute myeloid leukaemia. Br J Haematol. 2002;117:860–868. doi: 10.1046/j.1365-2141.2002.03538.x. [DOI] [PubMed] [Google Scholar]
- 4.Ward JL, Sherali A, Mo ZP, Tse CM. Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J Biol Chem. 2000;275:8375–8381. doi: 10.1074/jbc.275.12.8375. [DOI] [PubMed] [Google Scholar]
- 5.Crawford CR, Ng CY, Belt JA. Isolation and characterization of an L1210 cell line retaining the sodium-dependent carrier cif as its sole nucleoside transport activity. J Biol Chem. 1990;265:13730–13734. [PubMed] [Google Scholar]
- 6.Crawford CR, Ng CY, Noel LD, Belt JA. Nucleoside transport in L1210 murine leukemia cells. Evidence for three transporters. J Biol Chem. 1990;265:9732–9736. [PubMed] [Google Scholar]
- 7.Funato T, Satou J, Nishiyama Y, Fujimaki S, Miura T, Kaku M, Sasaki T. In vitro leukemia cell models of Ara-C resistance. Leuk Res. 2000;24:535–541. doi: 10.1016/s0145-2126(00)00016-3. [DOI] [PubMed] [Google Scholar]
- 8.Dumontet C, Fabianowska-Majewska K, Mantincic D, Callet Bauchu E, Tigaud I, Gandhi V, Lepoivre M, Peters GJ, Rolland MO, Wyczechowska D, Fang X, Gazzo S, Voorn DA, Vanier-Viornery A, MacKey J. Common resistance mechanisms to deoxynucleoside analogues in variants of the human erythroleukaemic line K562. Br J Haematol. 1999;106:78–85. doi: 10.1046/j.1365-2141.1999.01509.x. [DOI] [PubMed] [Google Scholar]
- 9.Meier C, Balzarini J. Application of the cycloSal-prodrug approach for improving the biological potential of phosphorylated biomolecules. Antiviral Res. 2006;71:282–292. doi: 10.1016/j.antiviral.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Galmarini CM, Popowycz F, Joseph B. Cytotoxic nucleoside analogues: different strategies to improve their clinical efficacy. Curr Med Chem. 2008;15:1072–1082. doi: 10.2174/092986708784221449. [DOI] [PubMed] [Google Scholar]
- 11.Zamboni WC. Liposomal, nanoparticle, and conjugated formulations of anticancer agents. Clin Cancer Res. 2005;11:8230–8234. doi: 10.1158/1078-0432.CCR-05-1895. [DOI] [PubMed] [Google Scholar]
- 12.Crosasso P, Brusa P, Dosio F, Arpicco S, Pacchioni D, Schuber F, Cattel L. Antitumoral activity of liposomes and immunoliposomes containing 5-fluorouridine prodrugs. J Pharm Sci. 1997;86:832–839. doi: 10.1021/js9604467. [DOI] [PubMed] [Google Scholar]
- 13.Duzgunes N, Simoes S, Slepushkin V, Pretzer E, Flasher D, Salem, Steffan G, Konopka K, Pedroso de Lima MC. Delivery of antiviral agents in liposomes. Methods Enzymol. 2005;391:351–373. doi: 10.1016/S0076-6879(05)91020-3. [DOI] [PubMed] [Google Scholar]
- 14.Oussoren C, Magnani M, Fraternale A, Casabianca A, Chiarantini L, Ingebrigsten R, Underberg WJ, Storm G. Liposomes as carriers of the antiretroviral agent dideoxycytidine-5′-triphosphate. Int J Pharm. 1999;180:261–270. doi: 10.1016/s0378-5173(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 15.Vinogradov SV, Poluektova LY, Makarov E, Gerson T, Senanayake MT. Nano-NRTIs: efficient inhibitors of HIV type-1 in macrophages with a reduced mitochondrial toxicity. Antivir Chem Chemother. 2010;21:1–14. doi: 10.3851/IMP1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magnani M, Rossi L, Fraternale A, Casabianca A, Brandi G, Benatti U, De Flora A. Targeting antiviral nucleotide analogues to macrophages. J Leukoc Biol. 1997;62:133–137. doi: 10.1002/jlb.62.1.133. [DOI] [PubMed] [Google Scholar]
- 17.Vinogradov SV, Zeman AD, Batrakova EV, Kabanov AV. Polyplex Nanogel formulations for drug delivery of cytotoxic nucleoside analogs. J Control Release. 2005;107:143–157. doi: 10.1016/j.jconrel.2005.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vinogradov SV, Kohli E, Zeman AD. Cross-linked polymeric nanogel formulations of 5′-triphosphates of nucleoside analogues: role of the cellular membrane in drug release. Mol Pharm. 2005;2:449–461. doi: 10.1021/mp0500364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galmarini CM, Warren G, Kohli E, Zeman A, Mitin A, Vinogradov SV. Polymeric nanogels containing the triphosphate form of cytotoxic nucleoside analogues show antitumor activity against breast and colorectal cancer cell lines. Mol Cancer Ther. 2008;7:3373–3380. doi: 10.1158/1535-7163.MCT-08-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aryal S, Hu CM, Zhang L. Combinatorial drug conjugation enables nanoparticle dual-drug delivery. Small. 2010;6:1442–1448. doi: 10.1002/smll.201000631. [DOI] [PubMed] [Google Scholar]
- 21.Ferey G. Hybrid porous solids: past, present, future. Chem Soc Rev. 2008;37:191–214. doi: 10.1039/b618320b. [DOI] [PubMed] [Google Scholar]
- 22.Ulbrich K, Strohalm J, Kopecek J. Polymers containing enzymatically degradable bonds. VI. Hydrophilic gels cleavable by chymotrypsin. Biomaterials. 1982;3:150–154. doi: 10.1016/0142-9612(82)90004-7. [DOI] [PubMed] [Google Scholar]
- 23.Galmarini CM, Clarke ML, Jordheim L, Santos CL, Cros E, Mackey JR, Dumontet C. Resistance to gemcitabine in a human follicular lymphoma cell line is due to partial deletion of the deoxycytidine kinase gene. BMC Pharmacol. 2004;4:8. doi: 10.1186/1471-2210-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ullman B. Dideoxycytidine metabolism in wild type and mutant CEM cells deficient in nucleoside transport or deoxycytidine kinase. Adv Exp Med Biol. 1989;253B:415–420. doi: 10.1007/978-1-4684-5676-9_61. [DOI] [PubMed] [Google Scholar]
- 25.Sinha ND, Biernat J, McManus J, Koster H. Polymer support oligonucleotide synthesis XVIII: use of beta-cyanoethyl-N,N-dialkylamino-/N-morpholino phosphoramidite of deoxynucleosides for the synthesis of DNA fragments simplifying deprotection and isolation of the final product. Nucleic Acids Res. 1984;12:4539–4557. doi: 10.1093/nar/12.11.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vinogradov SV, Suzdaltseva Y, Alakhov V, Kabanov AV. Inhibition of herpes simplex virus 1 reproduction with hydrophobized antisense oligonucleotides. Biochem Biophys Res Commun. 1994;203:959–966. doi: 10.1006/bbrc.1994.2275. [DOI] [PubMed] [Google Scholar]
- 27.Nochi T, Yuki Y, Takahashi H, Sawada S, Mejima M, Kohda T, Harada N, Kong IG, Sato A, Kataoka N, Tokuhara D, Kurokawa S, Takahashi Y, Tsukada H, Kozaki S, Akiyoshi K, Kiyono H. Nanogel antigenic protein-delivery system for adjuvant-free intranasal vaccines. Nat Mater. 2010;9:572–578. doi: 10.1038/nmat2784. [DOI] [PubMed] [Google Scholar]
- 28.Yusa S, Kamachi M, Morishima Y. Hydrophobic self-association of cholesterol moieties covalently linked to polyelectrolytes: Effect of spacer bond. Langmuir. 1998;14:6059–6067. [Google Scholar]
- 29.Gimenez V, Reina JA, Mantecon A, Cadiz V. Unsaturated modified poly(vinyl alcohol). Crosslinking through double bonds. Polymer. 1999;40:2759–2767. [Google Scholar]
- 30.Moreno B, Urbina JA, Oldfield E, Bailey BN, Rodrigues CO, Docampo R. 31P NMR spectroscopy of Trypanosoma brucei, Trypanosoma cruzi, and Leishmania major. Evidence for high levels of condensed inorganic phosphates. J Biol Chem. 2000;275:28356–28362. doi: 10.1074/jbc.M003893200. [DOI] [PubMed] [Google Scholar]
- 31.Warnecke S, Meier C. Synthesis of nucleoside Di- and triphosphates and dinucleoside polyphosphates with cycloSal-nucleotides. J Org Chem. 2009;74:3024–3030. doi: 10.1021/jo802348h. [DOI] [PubMed] [Google Scholar]
- 32.Chen N, Murata S, Yoshikawa K. Dramatic change in the tertiary structure of giant DNA without distortion of the secondary structure caused by pteridine-polyamine conjugates. Chemistry. 2005;11:4835–4840. doi: 10.1002/chem.200500135. [DOI] [PubMed] [Google Scholar]
- 33.Razzell WE, Khorana HG. Studies on polynucleotides. III. Enzymic degradation; substrate specificity and properties of snake venom phosphodiesterase. J Biol Chem. 1959;234:2105–2113. [PubMed] [Google Scholar]
- 34.Garcia-Diaz M, Avalos M, Cameselle JC. Methanol esterification reactions catalyzed by snake venom and bovine intestinal 5′-nucleotide phosphodiesterases. Formation of nucleoside 5′-monophosphate methyl esters from guanosine 5′-triphosphate and other nucleoside 5′-polyphosphates. Eur J Biochem. 1991;196:451–457. doi: 10.1111/j.1432-1033.1991.tb15836.x. [DOI] [PubMed] [Google Scholar]
- 35.Mikkola S. Hydrolytic reactions of diadenosine 5′,5′-triphosphate. Org Biomol Chem. 2004;2:770–776. doi: 10.1039/b313200e. [DOI] [PubMed] [Google Scholar]
- 36.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 37.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 38.Garcia-Diaz M, Avalos M, Cameselle JC. Methanol esterification reaction catalyzed by snake venom and bovine intestinal 5′-nucleotide phosphodiesterases. Eur J Biochem. 2006;196:451–457. doi: 10.1111/j.1432-1033.1991.tb15836.x. [DOI] [PubMed] [Google Scholar]
- 39.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.