

Abstract
Systems biology, with its associated technologies of proteomics, genomics and metabolomics, is driving the evolution of our understanding of cardiovascular physiology. Rather than studying individual molecules or even single reactions, a systems approach allows integration of orthogonal datasets from distinct tiers of biological data, including gene, RNA, protein, metabolite and other component networks. Together these networks give rise to emergent properties of cellular function and it is their reprogramming that causes disease. We present five observations regarding how systems biology is guiding a revisiting of the central dogma: (i) de-emphasizing the unidirectional flow of information from genes to proteins; (ii) revealing the role of modules of molecules as opposed to individual proteins acting in isolation; (iii) enabling discovery of novel emergent properties; (iv) demonstrating the importance of networks in biology; and (v) adding new dimensionality to the study of biological systems.
Introduction
Arguably the greatest post-modern coup for reductionism in biology was the articulation of the central dogma.1 Not since `humors' were discarded from medical practice and logic and experiment instituted as the cornerstones of physiology (which they remain today) had such a revolutionary idea transformed biology and enabled scientific inquiry. Because of its simplicity, the central dogma has the tantalizing allure of deduction—if one accepts the premises (that DNA encodes mRNA, and mRNA, protein), it seems one cannot deny the conclusions (that genes are the blueprint for life). As a result, the central dogma has guided research into causes of disease and phenotype, as well as constituted the basis for the tools used in the laboratory to interrogate these causes for the past half century.
The past decade, however, has witnessed a rapid accumulation of evidence challenging the linear logic of the central dogma. Four previously unassailable beliefs about the genome—that it is static throughout the life of the organism; that it is invariant between cell type and individual;2–4 that changes occurring in somatic cells cannot be inherited (also known as Lamarckian Evolution;5); and that necessary and sufficient information for cellular function is contained in the gene sequence—have all been called into question in the last few years. Revelations of similar scale have occurred in the transcriptome, with the discovery of the ubiquity (and variety) of mRNA splicing.6 And so too with the proteome, which has undergone perhaps the most dramatic shift in understanding as a result of the aforementioned changes to the transcriptome and the genome, as well as by the explosion of technology development that has enabled quantitative and qualitative analysis of large groups of proteins and their modifications in a single experiment. It is now clear that information flows multi-directionally between different tiers of biological information, of which genes, transcripts and proteins constitute only the most obvious three.
The ostensible fourth step in the central dogma—how molecules `encode' cells—clearly lacks the crystalline formulae that relate DNA to protein. While molecular details have been revealed for thousands of cellular events, no model exists that can explain how, for example, the modest erythrocyte is formed without error ~2 million times per second in adult H. sapiens. In contrast to a blueprint that can perfectly describe how to assemble a motorcycle or to build a city, we lack the knowledge to explain how a cell forms—with correct processes operational, cellular structures formed, and signaling mechanisms in place. It is in attempting to extend the logic of the central dogma beyond proteins where one realizes that the logic of biological systems and engineered ones are fundamentally different.7 Like the central dogma did for the investigation of basic and medical biological problems, a new synthesis for how cells form and function will result in philosophical shifts in research, as well as technological breakthroughs to enable it.
The purpose of the present work is to emphasize the contribution of proteomics and systems biology to extending the central dogma (Figure 1). Whenever possible, studies from the cardiovascular literature are used to highlight conceptual and technical breakthroughs. Excellent reviews exist on novel means of quantifying the proteome,8, 9 methods to analyze post-translational modifications,10, 11 cardiovascular diagnosis,12–14 and organelle proteomics;15–17 however, these areas are not the purview of the present work.
Figure 1. Role of proteomics in systems biology.

The advent of reproducible and high accuracy genomic and proteomic instrumentation has enabled characterization of distinct types of gene, RNA and protein networks. In many cases, quantitative data collected at fixed intervals after a stimulus (or, for example, during the development of disease) allow for dynamics of processes to be determined, and functional relationships in networks revealed. These technologies are fundamentally dependent on rigorously characterized models of disease (or human samples with extensive clinical data) to allow bioinformatic analyses to extract statistically significant relationships within and between large proteomic and genomic datasets. Mathematical modeling allows these observations to make predictions about behaviors underlying the system. The ultimate goal is to understand the relationships between different cellular networks (here the genome and proteome are represented, see also Figure 2) during health, such that we can engineer strategies to reprogram these network-network relationships therapeutically.
Proteomics as a Tool in Systems Biology
Whether systems biology as a discipline differs other than in semantics from physiology is worthy of the briefest clarification. It is our contention that the principle difference is the greater abundance, and higher rate of accumulation, of data in the former. While both concern themselves with the physical and chemical parts of a biological system and focus on revealing the oft-referenced mechanisms underlying phenotype, systems biology has been enabled by the development of ‴omics” technologies over the past 15 years. The sheer magnitude of data accumulated in studies of the genome, transcriptome, metabolome, proteome and so forth not only demanded a greater computational infrastructure for interpretation, but simultaneously revealed new dimensionality within tiers of biological information (witness: genome-wide association studies and unsupervised clustering of gene and protein expression data from high-throughput studies). Physiologists long ago appreciated the utility of simple engineering principles to model biological systems—the giant squid axon studies of Hodgkin and Huxley as well as Guyton's stunning mathematical (control theory) description of the brain and cardiovascular system come to mind—but the importance of mathematical biology has become pervasive in systems biology for interpretation (that is, bioinformatics) as well as hypothesis generation by modeling (wherein mathematical models become stand-alone entities that make predictions about future experiments). A hallmark of systems biology is the integration of large sets of data from measurements made on different tiers of the central dogma in the same experiment, to reveal emergent properties of the biological entity (Figure 2). Systems biology relies on the interplay between hypothesis- and discovery-driven research, and proteomics drives both these approaches.
Figure 2. Evolution of the central dogma.
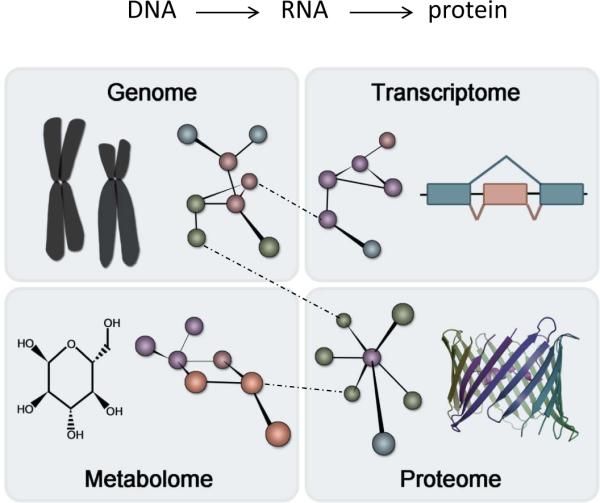
As described in the text, a new synthesis of the central dogma is emerging, in which different networks comprise each tier and information moves within and between the networks without strict directionality (see Table).
Proteomics has traditionally concerned itself with cataloging proteins in different cells and tissues, with an increasing focus of late on organelles. While this process has also been reviewed in greater depth elsewhere, it is important to note the conceptual advancement enabled by proteome maps. The first is the ability to navigate these maps, as proposed by Aebersold and colleagues, for which the current state of the art is directed mass spectrometry approaches (selected/multiple reaction monitoring, SRM/MRM experiments;9). This is now possible across the full dynamic range of expressed proteins in non-mammalian eukaryotes.18 Fortunately, generation of maps to guide this type of navigation is well underway for multiple organelles in the mammalian heart, most notably the mitochondria19–23 but also the nucleus20, 24 and proteasome.25 In virtually all cases, these maps have been generated by semi-quantitative (or in some cases, non-quantitative) tandem mass spectrometry, in which proteins extracted from purified organelles are separated intact, digested (usually by trypsin) to peptides which are then separated by liquid chromatography and introduced into the mass spectrometer by electrospray. The tandem mass spectra resulting from these experiments are then searched against a protein database to make identifications. These studies constitute the maps with which SRM/MRM experiments may now explore and quantify mammalian cardiac proteomes.
Second, conceptualizing proteomes as functional units of cellular phenotype has enabled researchers to analyze them as protagonists of cellular function as well as readouts for the actions of other tiers.
Measurement of proteomes as readouts of cardiac function is now widely utilized and rigorous standards have been established for the evaluation of mass spectrometry data. Such a framework has been essential to allow proteomic investigations to yield biologically and clinically relevant insights. In the realm of signaling, expression profiling proteomics have been used in the heart to study targets of type 5 adenylyl cyclase,26 the role of the mitochondria27, 28 or matrix metalloproteinases29 in cardiac ischemia and protection, as well as to study direct kinase targets important for cardiac phenotype,30, 31 to name only a few examples in what is now a rapidly growing body of literature.
Where proteomes as readouts have the greatest potential to enhance our understanding of cardiac function is when simultaneous measurements are made on other tiers of biological information and/or when proteomic data is integrated with rigorous gain and loss of function approaches. Both are discussed in greater detail below; the former is still quite a nascent field in the heart although some progress has been made with coupled transcriptomic and proteomic analysis of cardiac protection.32 In one example of the latter approach, Ago and colleagues used targeted proteomics to identify novel regulatory mechanisms that would have been virtually impossible to discover with traditional laboratory biochemistry approaches.33 The investigators used mass spectrometry to identify specific oxidized residues on HDAC4 that alter its nuclear-cytoplasmic partitioning in the setting of hypertrophy and revealed that thioredoxin counteracts this process by reducing these residues. In this study, the use of proteomics to identify novel modified residues facilitated the use of reductionist approaches (genetic and pharmacologic) to test causality, highlighting the powerful interplay of discovery-based and hypothesis-driven approaches.
The challenge is to shift the focus from proteomes as readouts into measuring them as the active agents of biological function. To use proteomics to reveal basic mechanisms of disease, one is, in essence, trying to develop a molecular EKG: an emergent property of the system that will allow us to diagnose the health of the cell. All of the proteins that influence the EKG are not known. However, we need not have a perfect understanding of all the factors that contribute to the network of molecules as long as we can quantify its existence and demonstrate its linkage to phenotype in a reproducible manner. The objective of a complete static wiring diagram for a biological system is incomplete, as biological systems defy the linear logic of those engineered by humans.
Network Biology: Insights from Proteomics
Types of Biological Networks
There has been an explosion in the last decade in our understanding of the role of networks in all aspects of biology. Several excellent reviews on the properties of networks and how they relate to cell biology exist;34–36 herein we will only highlight what we see as a few critical considerations for network logic in the interpretation of proteomic and genomic data, and to highlight how this data in turn is affecting the study of networks.
While active debate continues about the most accurate mathematical representation of biological networks (whether, for instance, their structures are scale-free or exponential, and the importance of local versus global features), there is almost universal agreement that network structure is non-random and plays a fundamental role in how information is processed and decisions made in the cell. How nodes (or individual features) are linked (also known as edges) to each other reveals core features about the network, including: how a signal is transmitted, whether it is amplified or dampened, the relative importance of individual nodes versus that of groups of nodes, and the dynamical processes that the network can perform. Topology, or how the network is organized, determines the local and global structural features, which, when dynamics are incorporated, include the emergence of feedback and feedforward motifs (both excitatory and inhibitory) as well as threshold behavior and biological memory.37–39 Indeed it is important to note that most `omics studies to date have used only static data representations to produce networks. The limitation of this approach is that cellular behavior results from dynamical properties of the network that cannot be appreciate from the static topology.
A fundamental defining feature of biological networks is whether they are physical or functional. In a physical network, the nodes are connected to each other directly or indirectly in a physical manner—an obvious example being a protein interaction network, in which the proteins are the nodes and the links exist between proteins that bind to each other. Information flowing through such a network is also physically defined (and physically relayed in a direct manner). In the same example, this could include one protein post-translationally modifying another, or otherwise directly altering its activity. Physical networks can also exist between different types of biological molecules, for instance the network defined by the genes whose promoters are bound by a transcription factor. Again the network is physical so long as the given protein directly binds to an individual gene; information flows through this network in the form of altering gene expression (although the actual process of altering gene expression likely does not arise from the solitary act of a protein binding a DNA sequence, and thus this example is instructive of how a physical network then becomes a functional one). Functional, or influence networks, are ones in which nodes are connected by virtue of their ability solely to influence each other, without implication (or at least without direct evidence) of physical interaction between the nodes. Simply put, a link in an influence network means that the two nodes affect each other in some manner and/or participate in some shared process in the cell. Moving from the previous gene expression example, the protein transcription factor is unlikely to be sufficient to express a gene: it must bind to DNA and/or histones and recruit other factors, notably RNA polymerase II, to initiate transcription. Some of these nodes interact directly where as others clearly are connected only through influence. The most extensively studied influence networks are gene co-expression networks, in which genes (nodes) are connected if their expression behavior is similar before or after an intervention. Clearly there could be scores of intermediate physical links, not directly measured in a microarray or RNA sequencing study, that influence the connection between two nodes in such a network. In the cardiac realm, several proteomic and genomic investigations have revealed both physical31, 40 and functional41, 42 networks relevant for heart function (to cite just a few examples).
Network-Network Interactions
Like proteins and genes, molecular networks are interconnected and interdependent. Information flows between networks and one must integrate distinct datasets to represent a complete picture of how the cell functions. Several recent studies are illustrative of this point.
Lage and colleagues used multiple forms of data including protein interaction studies and phenotype-linked mutational studies on individual genes to construct networks controlling specific processes in different anatomical locations of the heart.43 The result is a systems-level view of the networks underlying spatial (in terms of within the organ) and temporal development of the prenatal myocardium. The multidimensionality of the data analysis allowed the investigators to extract features of these networks, such as the relationship between protein module complexity and protein/transcript abundance, which would escape appreciation if intuition alone were used to evaluate the data. Furthermore, the networks examined in this study included physical ones based on published protein interactions, as well as functional ones, based on shared involvement in different stages or features of embryonic heart formation.
An intuitive example of obligatory symbiosis between tiers of biological molecules is the eukaryotic genome, defined herein as the DNA molecules and the chromatin structural proteins that constitutively bind to them. The packaging of DNA in the nucleus can be understood on the basis of the following structural hierarchy: a segment (~147 nucleotides in length) of the double helical DNA molecule wraps around a protein complex containing two copies each of four core histones (H2A, H2B, H3 and H4), constituting a nucleosome; this octomeric, DNA-bound, protein complex in turn forms higher ordered structures of less well-defined architecture through interactions with linker histones (like H1) and other chromatin structural proteins (such as high mobility group proteins). These chromatin domains determine the overall shape and presentation of each individual chromosome. While the organization of these DNA-protein complexes—the chromosomes—during cell division is well established, and extensive work has been done to characterize changes in nucleosome positioning, there exists no dogma articulating how chromatin structure regulates gene expression. To address this issue, an adaptation of conventional 3C (chromosomal conformation capture), a technique that allows determination of physical proximity of specific genetic loci in three dimensions, was used to map physical arrangement of entire genomes in intact interphase nuclei. This approach was applied separately to human44 and yeast45 genomes to reveal that the 3D structure of the genome resembles a fractal globule, a structure that has several desirable features such as non-overlapping segments and modular architecture, both of which would facilitate differential accessibility by transcription factors.46 While elegant, this model is incomplete in that it cannot account for differential modification of this genome structure in different cell types and within the same cell under varied transcriptional states. Furthermore, little known about how these interesting observations from non-cardiac systems will be examined in the setting of the heart, with its inherent challenge of heterogeneity in terms of cell type. Extensive work has been done, including in the heart, to show how various classes of proteins modulate chromatin accessibility and thereby gene expression (including but not limited to HDACs/HATs, HMG proteins, lysine and arginine methyltransferases/demethylases, kinases/phosphatases and chromatin remodeling proteins47,48). Importantly recent reviews have highlighted critical considerations for analyzing these types of networks (i.e. those involving DNA and protein), including how to capture discrete molecular details of genetic circuits, and considerations for distinguishing specificity and affinity in studying regulation.49 However, these studies have yet to reveal an invariant relationship between structure and function. There is no code that can account for how the hierarchical structure of DNA and proteins establishes the complex genomic regulatory programs that exist in distinct differentiated cells, as there is to explain, for example, how DNA encodes RNA and RNA, protein.
Other work has recently advanced our view of the landscape of genomic regulation by networks of proteins in the eukaryotic nucleus. One recent study demonstrated the power of combining proteomic and genomic approaches to reveal insights into both physical and influence networks on a large-scale. Using a modified histone-derived peptide as bait, potential chromatin-modifiers were identified by subsequent mass spectrometry analysis.50 The genome-wide localization of select members of this group was then mapped by chromatin immunoprecipitation followed by DNA sequencing. The core insight from this study was at the level of two types of networks and their integration in the setting of the activation or inhibition of gene expression, as dictated by the respective histone post-translational modifications. An innovative approach to use protein influence networks to define genomic influence networks also comes from the realm of chromatin regulation.51 A sizable group (53) of chromatin binding proteins with known genome occupancy profiles but with no preconceived relationship to each other was selected. The occupancy profiles were used to discriminate 5 domains of chromatin based on principal component analyses (in brief, regions where the proteins had similar occupancy across the genome were defined as functional domains). As a result, all loci were assigned to one of these 5 exclusive chromatin types, thus constituting a previously unrecognized influence network. Importantly, these genomic features reveal themselves based not on a single DNA sequence feature, post-translational modification or bound protein; it is only through the combination of proteins bound to a given region that the feature of this genome-proteome interaction emerges. A third study undertook a massive analysis combining published data and RNAi screens to identify a list of target proteins which were subsequently engineered with GFP or tandem affinity tags and utilized for microscopy-based localization or affinity purification and mass spectrometry analysis, respectively.52 This is a noteworthy example of unbiased, discovery-based analysis of a system. In addition, the combination of approaches gives physical information on protein interactions and anatomical reference within the context of an intact cell. The result was a phenotype and function-linked catalog of chromosomal segregation proteins that regulate mitosis in mammalian cells.
Literature on protein interaction networks is extensive and this remains an area of active technology development in proteomics. An important and potentially obfuscated consideration for these studies is that virtually all protein interaction data is recovered in a binary manner, in which knowledge on levels of interactions, if present, is lost, along with insights into whether interactions are persistent or fleeting. This issue was addressed in a novel way by a recent paper that focused on structural features of the individual proteins53 such as disordered regions, protein interaction domains and states of activation. High-throughput heterologous systems like yeast two-hybrid assays and tandem affinity purification tagging in E. coli are not amenable to fine-tuning experimental conditions to capture distinct type of protein interactions, and so much of this information needs to be recovered by repeat experimentation in different systems (example: reciprocal purification) or by alternative method, such as in situ co-localization.
As a final example for this section, consider a study in which numerous types of experiments were carried out to first generate and then explore hippocampal neuronal networks.54 In this study, the nodes were primarily proteins and the edges included direct interactions (as in protein complexes), signal transduction (indirect or imperfectly defined interactions), ligand-receptor binding, enzymatic activity, and common use of cofactors. This analysis revealed signaling motifs that conferred distinct behavior and could account for many of the higher-level phenotypic properties of neurons that individual proteins, or simple `pathways' could not. Furthermore, the use of graph theory to investigate the role of local and global connectivity in the flow of information through networks allowed the large datasets to yield meaningful insights into specific cellular processes. Similar integrated models have been developed and explored for the cardiac myocyte,55–57 which like the neuron offers itself to representation based on electrical engineering principles.
How do biological networks form?
We would like to briefly address the following paradox with regard to the formation of cellular networks: unlike any other networks (including human interactions, the internet, transportation, power grids, computers, predator-prey interactions and so forth) cellular networks appear to inherently require the absence of flexibility in their formation. This statement requires some clarification. On the one hand, cellular networks are infinitely flexible after they are formed (that is, in a normally functioning cell); this is the observation we are all well aware of and which is commonly written off as the large degree of redundancy in cellular networks. However, inflexibility appears to be a requirement during the formation of the network. We must refute the hypothesis that formation of networks during the birth of a cell is a deterministic event if we wish to claim variability exists in the formation of these networks. While the response to a given stimulus in an existent network may be enacted by more than one mechanism (illustrating the so-called redundancy), the network allowing for this occurrence must, if we reason the response was non-random and reproducible, harbor properties that specifically produce this behavior. The presence of such properties suggests emergent control in the formation of the network in addition to emergent control of its function once formed. How can this emergence be measured rather than just observed? An enigma in biology is how a cell with (at least) thousands of proteins can reproducibly form and behave in the same manner without central control. This argument means one of two explanations must be true: either there is an as yet unknown process that governs invariant formation of networks or biological networks are not invariant (e.g., the precise structure and/or means of formation of a protein interaction network in two cells can differ). Clearly we must return to the laboratory to resolve this issue. An experiment to resolve this issue would be one in which a given network (e.g. gene expression or protein expression) were mapped multiple times in distinct sets of the same cells without pooling the data between the individual technical replicates. If sufficient resolution was achieved (which is now possible due to next generation nucleotide sequencing58 and high mass accuracy mass spectrometers9 and other techniques59), algorithms could be devised to distinguish noise from true variation in the system. A simulation of how much noise should result from the known limits of the instrumentation when compared to the data resultant from said experiment would reveal whether there is detectable variation in the architecture of the networks in multiple copies of the same cell. If sufficient variation existed between distinct samples of the same network, this observation would refute the hypothesis that architecture of molecular networks is invariant.
Specific Challenges for Cardiac Proteomics
As with any cell or organ, the challenges with proteomic and genomic dissection mirror the unique physiology of the system. Four prescient challenges for cardiovascular proteomics are: the contribution of distinct cell types; distinguishing between primary and secondary effects; determining what aspects of endogenous remodeling are beneficial versus harmful; and the availability of healthy human samples.
The heart's reason for existence is to pump blood—hence, the cells of primary interest in this objective are the cardiac myocytes. Experimental harnessing of cardiac-specific genetic manipulation has enabled extensive analysis of in vivo roles for individual proteins in cardiac myocytes.60, 61 The result has been an astounding advancement in our understanding of cardiac cellular networks62, 63 from the standpoint of adding or removing individual nodes. What we currently lack is the ability to manipulate the roles of individual proteins in the many other cell types that constitutively populate the heart (like vascular smooth muscle, endothelial cells, and fibroblasts64) along with those that are transiently recruited there in the setting of stress (macrophages, for example). Furthermore, the specialized cells in numerous vascular beds no doubt establish distinct networks that carry out their unique physiology—compare cardiac microvascular endothelial cells to those from the gut or kidney, and then to those commonly used for large scale analyses, which arise from the aorta. These challenges inherently impact proteomic and genomic interrogation of the heart, in that one must examine the heart en toto, accepting that information on cell type is lost, or fractionate the cell populations ex vivo prior to analyses, accepting in this scenario the unpredictable modifications to gene and protein abundance/modification that will occur during the time-consuming isolation process. The reality, in our opinion, is that both approaches must be pursued and that this will remain the case for the foreseeable future. Many groups are making progress in identifying cell-type specific markers which may enable comprehensive cell sorting in the future, but even in this case the concern of time between in vivo and measurement will remain.
How does one distinguish primary and secondary effects when the organ systems—and ergo the proteomes—of heart and vasculature are linked in health and disease (as is true of all vascularized organ systems)? Conditions of atherosclerosis, myocardial ischemia, cardiac hypertrophy and heart failure are interrelated in the clinical setting and are amenable to individual dissection only in experimental models. As with cell type specificity, the roles of heart and vascular proteomes in distinct disease can only be revealed from a divide and conquer strategy at the present time—one that uses and experimental model that allows disease to develop with heart and vasculature in normal apposition, dissects out (literally) the roles of different cells, and then uses the method of systems biology (Figure 1) to reassemble the insights into a coherent model. In the future, real-time and in vivo imaging techniques (such as PET and MRI), along with specific probes (whose design must be guided by the insights from proteomics), will allow for non-destructive analysis of the cardiovascular system, but at present these techniques are in their infancy with regard to molecular level resolution.
A third major challenge is decoding the natural remodeling that occurs following stress to the heart and vasculature to separate the nefarious processes (to be targeted for inhibition) from those that are protective (to be engineered for enhancement). This challenge too is not unique to proteomic and genomic studies, but takes on an acute problem of scale when one is faced with a mountain of experimental data. The more sensitive the instruments become, the greater this challenge will be. Again relying on the method for systems analyses, the genetic models created in the field, many of which have been characterized to have ostensibly normal physiology until presented with cardiovascular stress, can be used to distinguish (in the proteomic/genomic data) true drivers of the pathological phenotypes from general stress response molecules. This knowledge in turn allows our maps of proteomic and genomic networks to encapsulate more details and to become more perfect representations of biological processes.
Lastly, there is, for obvious reasons, a lack of healthy human cardiac and specialized vascular tissue. There are no grounds to posit that fundamental processes do not differ in the human heart as compared to the model systems commonly employed experimentally, most frequently being the mouse. Thus we ultimately must develop the means to characterize via proteomics and genomics both the healthy and diseased adult human myocardium, the way we can today with echocardiography, so as to track the deterioration or recovery of cardiac proteomes. A promising avenue to overcome this challenge is guided differentiation of embryonic or adult stem cells into a cardiac lineage, and some genomic studies on these cells have already emerged.65, 66 Much work clearly remains before these cells can be labeled bona fide cardiac cells, including sorting out to what extent an apparently reprogrammed phenotype completely recapitulates the appropriate epigenetic state,67 but rapid progress is being made in this area and may be combined in the future with proteomics and in vivo labeling techniques (as described above) to develop a signature for cardiac health and disease in humans.
Conclusion: Impact of Recent Advances in Systems Biology on the Central Dogma
The aforementioned advances in proteomics and network biology are driving the evolution of our understanding of the central dogma of molecular biology. In addition to the increasingly appreciated role of non-protein-coding RNA in biological function, next generation sequencing is ushering in a new generation of proteomics, in which we will now have the ability to measure whether (and if so how) variation propagates from genome to proteome, affecting function. Because proteomic experiments are, in the majority of cases, still dependent on DNA/mRNA sequence, the availability of multiple genomes from a given species will dramatically increase the search space and dimensionality of proteomics. In a recent study, Pelak and colleagues sequenced 20 human genomes, identifying on average 165 unique protein-truncating variants in each genome;2 likewise, the preliminary report of the 1000 Genomes Project suggests variation on a similar scale, with ~250–300 genes per individual being different from the reference genome.4 It is likely that we will have the sequences of 100,000 to 1 million human genomes in the next few years, and if these estimates of interpersonal variation are demonstrated in larger populations, this would mean potentially hundreds of millions of additional protein variants solely on the basis of genome variation. These data analysis challenges will make today's bioinformatic loads seem paltry by comparison.
In closing, we identify five ways in which “'omics” technologies are changing basic and clinical research and contributing to a revisiting of the central dogma (Table): First, de-emphasizing the unidirectional flow of information (i.e. DNA to RNA to protein; Figure 2, top); second, placing an emphasis on modules68–70 of genes/proteins/molecules rather than individual factors; third, enabling the discovery and quantification of emergent properties present at different scales of information;71 fourth, revealing the role of networks in biological function; and fifth, allowing for new dimensionality in the analysis of all biological molecules (Figure 2, bottom).
Table.
Contribution of Systems Biology to Evolution of the Central Dogma
| Multidirectional Flow of Information |
Demonstrated
|
| Role of micro-RNAs in controlling phenotype72 | |
| Use of slicing to expand the proteome6 | |
| Contribution of proteins to genomic structure and regulation73,74 | |
|
Predicted
| |
| Global regulation of genomic structure by proteins is a key feature of cellular plasticity | |
|
| |
| Modules |
Demonstrated
|
| Evolutionary conservation of molecular signaling units,75 e.g. MAPK76 | |
| Few molecules act alone or have single functions70 | |
| Cells are hierarchically organized according to functional groups of molecules77,78 | |
|
Predicted
| |
| Modules of proteins are themselves direct substrates for evolution | |
|
| |
| Emergent Properties |
Demonstrated
|
| Resistance of cells to noise and variation79 | |
| Motifs are formed within networks, enabling complex behaviors and dynamics39 | |
| Pattern formation underlies many biological processes71,74 | |
|
Predicted
| |
| Protein modules and networks are diagnostic for disease and targets for therapy | |
|
| |
| Networks |
Demonstrated
|
| Topology of molecular interactions determines cellular function80 | |
| Cells have built-in redundancies and information is processed according to nonlinear principles81 | |
| Stimuli are integrated, and responses coordinated, in an ordered manner82 | |
|
Predicted
| |
| Network formation is a biological attractor–the lowest energy state of the cell | |
|
| |
| New Dimensionality |
Demonstrated
|
| Epigenetic inheritance of `acquired' traits83 | |
| Comparative analyses of individual genomes84,85 and non-coding regions86 | |
| Large-scale correlation between molecular networks and phenotype75,87 | |
|
Predicted
| |
| The definition of `gene' and `protein' will significantly change with results from massive sequencing experiments | |
The greatest present challenge for biology is the limit of reductionism. The term systems biology itself underscores our linguistic circumscription of this problem. A system, no matter how complex, is a defined entity; it is a human creation. We conceptualize an evolvable central information unit that describes (and orchestrates) the piecewise assembly of the machine that is a cell. We conceptualize watches, even if we shun the watchmaker.
Acknowledgments
The authors thank Peipei Ping, PhD and James N. Weiss, MD for support and inspiration and Alan Garfinkel, PhD for critical feedback on the manuscript (all from UCLA). The authors express their gratitude to the many investigators whose work contributed to our “five ways the central dogma is evolving” but was not formally acknowledged because of space limitations.
Funding Sources Work in the Vondriska laboratory is supported in part by the National Heart, Lung and Blood Institute at the NIH and the Laubisch Endowment at UCLA.
Footnotes
Conflict of Interest Disclosure Sarah Franklin – none Thomas M. Vondriska - none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Crick F. Central dogma of molecular biology. Nature. 1970;227:561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- 2.Pelak K, Shianna KV, Ge D, Maia JM, Zhu M, Smith JP, Cirulli ET, Fellay J, Dickson SP, Gumbs CE, Heinzen EL, Need AC, Ruzzo EK, Singh A, Campbell CR, Hong LK, Lornsen KA, McKenzie AM, Sobreira NL, Hoover-Fong JE, Milner JD, Ottman R, Haynes BF, Goedert JJ, Goldstein DB. The characterization of twenty sequenced human genomes. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, Shendure J, Drmanac R, Jorde LB, Hood L, Galas DJ. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDaniell R, Lee BK, Song L, Liu Z, Boyle AP, Erdos MR, Scott LJ, Morken MA, Kucera KS, Battenhouse A, Keefe D, Collins FS, Willard HF, Lieb JD, Furey TS, Crawford GE, Iyer VR, Birney E. Heritable individual-specific and allele-specific chromatin signatures in humans. Science. 328:235–239. doi: 10.1126/science.1184655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly K. Out of Control: The New Biology of Machines, Social Systems and the Economic World. Perseus; New York: 1994. [Google Scholar]
- 8.Park SK, Venable JD, Xu T, Yates JR., 3rd A quantitative analysis software tool for mass spectrometry-based proteomics. Nat Methods. 2008;5:319–322. doi: 10.1038/nmeth.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahrens CH, Brunner E, Qeli E, Basler K, Aebersold R. Generating and navigating proteome maps using mass spectrometry. Nat Rev Mol Cell Biol. 2010;11:789–801. doi: 10.1038/nrm2973. [DOI] [PubMed] [Google Scholar]
- 10.Siuti N, Kelleher NL. Decoding protein modifications using top-down mass spectrometry. Nat Methods. 2007;4:817–821. doi: 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choudhary C, Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nat Rev Mol Cell Biol. 11:427–439. doi: 10.1038/nrm2900. [DOI] [PubMed] [Google Scholar]
- 12.Gerszten RE, Wang TJ. The search for new cardiovascular biomarkers. Nature. 2008;451(7181):949–952. doi: 10.1038/nature06802. [DOI] [PubMed] [Google Scholar]
- 13.Matt P, Fu Z, Fu Q, Van Eyk JE. Biomarker discovery: proteome fractionation and separation in biological samples. Physiol Genomics. 2008;33:12–17. doi: 10.1152/physiolgenomics.00282.2007. [DOI] [PubMed] [Google Scholar]
- 14.Eagle KA, Ginsburg GS, Musunuru K, Aird WC, Balaban RS, Bennett SK, Blumenthal RS, Coughlin SR, Davidson KW, Frohlich ED, Greenland P, Jarvik GP, Libby P, Pepine CJ, Ruskin JN, Stillman AE, Van Eyk JE, Tolunay HE, McDonald CL, Smith SC., Jr. Identifying patients at high risk of a cardiovascular event in the near future: current status and future directions: report of a national heart, lung, and blood institute working group. Circulation. 121:1447–1454. doi: 10.1161/CIRCULATIONAHA.109.904029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agnetti G, Husberg C, Van Eyk JE. Divide and conquer: the application of organelle proteomics to heart failure. Circ Res. 2011;108:512–526. doi: 10.1161/CIRCRESAHA.110.226910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huber LA, Pfaller K, Vietor I. Organelle proteomics: implications for subcellular fractionation in proteomics. Circ Res. 2003;92:962–968. doi: 10.1161/01.RES.0000071748.48338.25. [DOI] [PubMed] [Google Scholar]
- 17.Yates JR, 3rd, Gilchrist A, Howell KE, Bergeron JJ. Proteomics of organelles and large cellular structures. Nat Rev Mol Cell Biol. 2005;6:702–714. doi: 10.1038/nrm1711. [DOI] [PubMed] [Google Scholar]
- 18.Picotti P, Rinner O, Stallmach R, Dautel F, Farrah T, Domon B, Wenschuh H, Aebersold R. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat Methods. 7:43–46. doi: 10.1038/nmeth.1408. [DOI] [PubMed] [Google Scholar]
- 19.Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, Scott MS, Gramolini AO, Morris Q, Hallett MT, Rossant J, Hughes TR, Frey B, Emili A. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell. 2006;125:173–186. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 20.Foster LJ, de Hoog CL, Zhang Y, Zhang Y, Xie X, Mootha VK, Mann M. A mammalian organelle map by protein correlation profiling. Cell. 2006;125:187–199. doi: 10.1016/j.cell.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 21.Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, Murphy AN, Gaucher SP, Capaldi RA, Gibson BW, Ghosh SS. Characterization of the human heart mitochondrial proteome. Nat Biotechnol. 2003;21:281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Li X, Mueller M, Wang Y, Zong C, Deng N, Vondriska TM, Liem DA, Yang JI, Korge P, Honda H, Weiss JN, Apweiler R, Ping P. Systematic characterization of the murine mitochondrial proteome using functionally validated cardiac mitochondria. Proteomics. 2008;8:1564–1575. doi: 10.1002/pmic.200700851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, Patterson N, Lander ES, Mann M. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 24.Franklin S, Zhang MJ, Chen H, Paulsson AK, Mitchell-Jordan SA, Li Y, Ping P, Vondriska TM. Specialized compartments of cardiac nuclei exhibit distinct proteomic anatomy. Mol Cell Proteomics. 2011 doi: 10.1074/mcp.M110.000703. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomes AV, Zong C, Edmondson RD, Li X, Stefani E, Zhang J, Jones RC, Thyparambil S, Wang GW, Qiao X, Bardag-Gorce F, Ping P. Mapping the murine cardiac 26S proteasome complexes. Circ Res. 2006;99:362–371. doi: 10.1161/01.RES.0000237386.98506.f7. [DOI] [PubMed] [Google Scholar]
- 26.Yan L, Vatner DE, O'Connor JP, Ivessa A, Ge H, Chen W, Hirotani S, Ishikawa Y, Sadoshima J, Vatner SF. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007;130:247–258. doi: 10.1016/j.cell.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 27.Arrell DK, Elliott ST, Kane LA, Guo Y, Ko YH, Pedersen PL, Robinson J, Murata M, Murphy AM, Marban E, Van Eyk JE. Proteomic analysis of pharmacological preconditioning: novel protein targets converge to mitochondrial metabolism pathways. Circ Res. 2006;99:706–714. doi: 10.1161/01.RES.0000243995.74395.f8. [DOI] [PubMed] [Google Scholar]
- 28.Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res. 106:1681–1691. doi: 10.1161/CIRCRESAHA.109.213645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zamilpa R, Lopez EF, Chiao YA, Dai Q, Escobar GP, Hakala K, Weintraub ST, Lindsey ML. Proteomic analysis identifies in vivo candidate matrix metalloproteinase-9 substrates in the left ventricle post-myocardial infarction. Proteomics. 10:2214–2223. doi: 10.1002/pmic.200900587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ping P, Zhang J, Pierce WM, Jr., Bolli R. Functional proteomic analysis of protein kinase C epsilon signaling complexes in the normal heart and during cardioprotection. Circ Res. 2001;88:59–62. doi: 10.1161/01.res.88.1.59. [DOI] [PubMed] [Google Scholar]
- 32.McCully JD, Bhasin MK, Daly C, Guerrero MC, Dillon S, Liberman TA, Cowan DB, Mably JD, McGowan FX, Levitsky S. Transcriptomic and proteomic analysis of global ischemia and cardioprotection in the rabbit heart. Physiol Genomics. 2009;38:125–137. doi: 10.1152/physiolgenomics.00033.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, Vatner SF, Sadoshima J. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133:978–993. doi: 10.1016/j.cell.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 34.Strogatz SH. Exploring complex networks. Nature. 2001;410:268–276. doi: 10.1038/35065725. [DOI] [PubMed] [Google Scholar]
- 35.Lusis AJ, Weiss JN. Cardiovascular networks: systems-based approaches to cardiovascular disease. Circulation. 121:157–170. doi: 10.1161/CIRCULATIONAHA.108.847699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Genet. 2010;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qu Z, Vondriska TM. The effects of cascade length, kinetics and feedback loops on biological signal transduction dynamics in a simplified cascade model. Phys Biol. 2009;6:16007. doi: 10.1088/1478-3975/6/1/016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shoval O, Alon U. SnapShot: network motifs. Cell. 2010;143:326–e321. doi: 10.1016/j.cell.2010.09.050. [DOI] [PubMed] [Google Scholar]
- 39.Ferrell JE., Jr. Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr Opin Cell Biol. 2002;14:140–148. doi: 10.1016/s0955-0674(02)00314-9. [DOI] [PubMed] [Google Scholar]
- 40.Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, Yates JR, 3rd, Lefkowitz RJ. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U S A. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matkovich SJ, Zhang Y, Van Booven DJ, Dorn GW., 2nd Deep mRNA sequencing for in vivo functional analysis of cardiac transcriptional regulators: application to Galphaq. Circ Res. 2010;106:1459–1467. doi: 10.1161/CIRCRESAHA.110.217513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blaxall BC, Spang R, Rockman HA, Koch WJ. Differential myocardial gene expression in the development and rescue of murine heart failure. Physiol Genomics. 2003;15:105–114. doi: 10.1152/physiolgenomics.00087.2003. [DOI] [PubMed] [Google Scholar]
- 43.Lage K, Mollgard K, Greenway S, Wakimoto H, Gorham JM, Workman CT, Bendsen E, Hansen NT, Rigina O, Roque FS, Wiese C, Christoffels VM, Roberts AE, Smoot LB, Pu WT, Donahoe PK, Tommerup N, Brunak S, Seidman CE, Seidman JG, Larsen LA. Dissecting spatio-temporal protein networks driving human heart development and related disorders. Mol Syst Biol. 2010;6:381. doi: 10.1038/msb.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, Noble WS. A three-dimensional model of the yeast genome. Nature. 465:363–367. doi: 10.1038/nature08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Steensel B, Dekker J. Genomics tools for unraveling chromosome architecture. Nat Biotechnol. 2010;28:1089–1095. doi: 10.1038/nbt.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Schreiber SL, Bernstein BE. Signaling network model of chromatin. Cell. 2002;111(6):771–778. doi: 10.1016/s0092-8674(02)01196-0. [DOI] [PubMed] [Google Scholar]
- 49.Stormo GD, Zhao Y. Determining the specificity of protein-DNA interactions. Nat Rev Genet. 11:751–760. doi: 10.1038/nrg2845. [DOI] [PubMed] [Google Scholar]
- 50.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, Mann M. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 51.Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, van Steensel B. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 143:212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hutchins JR, Toyoda Y, Hegemann B, Poser I, Heriche JK, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J, Conrad C, Comartin D, Schleiffer A, Sarov M, Pozniakovsky A, Slabicki MM, Schloissnig S, Steinmacher I, Leuschner M, Ssykor A, Lawo S, Pelletier L, Stark H, Nasmyth K, Ellenberg J, Durbin R, Buchholz F, Mechtler K, Hyman AA, Peters JM. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science. 328:593–599. doi: 10.1126/science.1181348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Transient protein-protein interactions: structural, functional, and network properties. Structure. 18:1233–1243. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 54.Ma'ayan A, Jenkins SL, Neves S, Hasseldine A, Grace E, Dubin-Thaler B, Eungdamrong NJ, Weng G, Ram PT, Rice JJ, Kershenbaum A, Stolovitzky GA, Blitzer RD, Iyengar R. Formation of regulatory patterns during signal propagation in a Mammalian cellular network. Science. 2005;309:1078–1083. doi: 10.1126/science.1108876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang JH, Saucerman JJ. Computational models reduce complexity and accelerate insight into cardiac signaling networks. Circ Res. 2011;108:85–97. doi: 10.1161/CIRCRESAHA.110.223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cortassa S, Aon MA, O'Rourke B, Jacques R, Tseng HJ, Marban E, Winslow RL. A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys J. 2006;91:1564–1589. doi: 10.1529/biophysj.105.076174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weiss JN, Nivala M, Garfinkel A, Qu Z. Alternans and arrhythmias: from cell to heart. Circ Res. 2011;108:98–112. doi: 10.1161/CIRCRESAHA.110.223586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shachaf CM, Elchuri SV, Koh AL, Zhu J, Nguyen LN, Mitchell DJ, Zhang J, Swartz KB, Sun L, Chan S, Sinclair R, Nolan GP. A novel method for detection of phosphorylation in single cells by surface enhanced Raman scattering (SERS) using composite organic-inorganic nanoparticles (COINs) PLoS One. 2009;4:e5206. doi: 10.1371/journal.pone.0005206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J Biol Chem. 1991;266:24613–24620. [PubMed] [Google Scholar]
- 61.Robbins J. Genetic modification of the heart: exploring necessity and sufficiency in the past 10 years. J Mol Cell Cardiol. 2004;36:643–652. doi: 10.1016/j.yjmcc.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 62.Molkentin JD, Robbins J. With great power comes great responsibility: using mouse genetics to study cardiac hypertrophy and failure. J Mol Cell Cardiol. 2009;46:130–136. doi: 10.1016/j.yjmcc.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olson EN. A decade of discoveries in cardiac biology. Nat Med. 2004;10:467–474. doi: 10.1038/nm0504-467. [DOI] [PubMed] [Google Scholar]
- 64.Bowers SL, Borg TK, Baudino TA. The dynamics of fibroblast-myocyte-capillary interactions in the heart. Ann N Y Acad Sci. 2010;1188:143–152. doi: 10.1111/j.1749-6632.2009.05094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takeuchi JK, Bruneau BG. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature. 2009;459:708–711. doi: 10.1038/nature08039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu H, Yi BA, Chien KR. Shortcuts to making cardiomyocytes. Nat Cell Biol. 2011;13:191–193. doi: 10.1038/ncb0311-191. [DOI] [PubMed] [Google Scholar]
- 67.Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol. 2010;28:1079–1088. doi: 10.1038/nbt.1684. [DOI] [PubMed] [Google Scholar]
- 68.Bhalla US, Iyengar R. Functional modules in biological signalling networks. Novartis Found Symp. 2001;239:4–13. doi: 10.1002/0470846674.ch2. discussion 13–15, 45–51. [DOI] [PubMed] [Google Scholar]
- 69.Vondriska TM, Klein JB, Ping P. Use of functional proteomics to investigate PKC epsilon-mediated cardioprotection: the signaling module hypothesis. Am J Physiol Heart Circ Physiol. 2001;280:H1434–1441. doi: 10.1152/ajpheart.2001.280.4.H1434. [DOI] [PubMed] [Google Scholar]
- 70.Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402:C47–52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 71.Weiss JN, Qu Z, Garfinkel A. Understanding biological complexity: lessons from the past. Faseb J. 2003;17:1–6. doi: 10.1096/fj.02-0408rev. [DOI] [PubMed] [Google Scholar]
- 72.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 73.Segal E, Widom J. What controls nucleosome positions? Trends Genet. 2009;25:335–343. doi: 10.1016/j.tig.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Z, Pugh BF. High-resolution genome-wide mapping of the primary structure of chromatin. Cell. 2011;144:175–186. doi: 10.1016/j.cell.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McGary KL, Park TJ, Woods JO, Cha HJ, Wallingford JB, Marcotte EM. Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proc Natl Acad Sci U S A. 2010;107:6544–6549. doi: 10.1073/pnas.0910200107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010;90:1507–1546. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rives AW, Galitski T. Modular organization of cellular networks. Proc Natl Acad Sci U S A. 2003;100:1128–1133. doi: 10.1073/pnas.0237338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- 79.Barkai N, Shilo BZ. Variability and robustness in biomolecular systems. Mol Cell. 2007;28:755–760. doi: 10.1016/j.molcel.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 80.Venkatesan K, Rual JF, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, Hao T, Zenkner M, Xin X, Goh KI, Yildirim MA, Simonis N, Heinzmann K, Gebreab F, Sahalie JM, Cevik S, Simon C, de Smet AS, Dann E, Smolyar A, Vinayagam A, Yu H, Szeto D, Borick H, Dricot A, Klitgord N, Murray RR, Lin C, Lalowski M, Timm J, Rau K, Boone C, Braun P, Cusick ME, Roth FP, Hill DE, Tavernier J, Wanker EE, Barabasi AL, Vidal M. An empirical framework for binary interactome mapping. Nat Methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 82.Jordan JD, Landau EM, Iyengar R. Signaling networks: the origins of cellular multitasking. Cell. 2000;103:193–200. doi: 10.1016/s0092-8674(00)00112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Turner BM. Environmental sensing by chromatin: An epigenetic contribution to evolutionary change. FEBS Lett. 2011 doi: 10.1016/j.febslet.2010.11.041. In Press. [DOI] [PubMed] [Google Scholar]
- 84.Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, Chinwalla A, Conrad DF, Fu Y, Grubert F, Hajirasouliha I, Hormozdiari F, Iakoucheva LM, Iqbal Z, Kang S, Kidd JM, Konkel MK, Korn J, Khurana E, Kural D, Lam HY, Leng J, Li R, Li Y, Lin CY, Luo R, Mu XJ, Nemesh J, Peckham HE, Rausch T, Scally A, Shi X, Stromberg MP, Stutz AM, Urban AE, Walker JA, Wu J, Zhang Y, Zhang ZD, Batzer MA, Ding L, Marth GT, McVean G, Sebat J, Snyder M, Wang J, Eichler EE, Gerstein MB, Hurles ME, Lee C, McCarroll SA, Korbel JO. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raney BJ, Cline MS, Rosenbloom KR, Dreszer TR, Learned K, Barber GP, Meyer LR, Sloan CA, Malladi VS, Roskin KM, Suh BB, Hinrichs AS, Clawson H, Zweig AS, Kirkup V, Fujita PA, Rhead B, Smith KE, Pohl A, Kuhn RM, Karolchik D, Haussler D, Kent WJ. ENCODE whole-genome data in the UCSC genome browser (2011 update) Nucleic Acids Res. 2011;39(Database issue):D871–875. doi: 10.1093/nar/gkq1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alexander RP, Fang G, Rozowsky J, Snyder M, Gerstein MB. Annotating non-coding regions of the genome. Nat Rev Genet. 2010;11:559–571. doi: 10.1038/nrg2814. [DOI] [PubMed] [Google Scholar]
- 87.Bennett BJ, Farber CR, Orozco L, Kang HM, Ghazalpour A, Siemers N, Neubauer M, Neuhaus I, Yordanova R, Guan B, Truong A, Yang WP, He A, Kayne P, Gargalovic P, Kirchgessner T, Pan C, Castellani LW, Kostem E, Furlotte N, Drake TA, Eskin E, Lusis AJ. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 2010;20:281–290. doi: 10.1101/gr.099234.109. [DOI] [PMC free article] [PubMed] [Google Scholar]