

Abstract
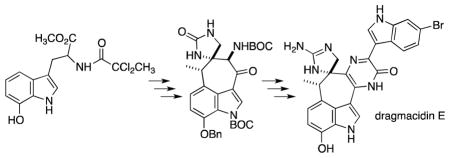
The bis indole sponge alkaloid dragmacidin E was synthesized in racemic form over 25 steps starting from 7-benzhydroxyindole. Key steps include (a) a Witkop cyclization to facilitate construction of the indole-spanning 7-membered ring and (b) a cyclodehydrative pyrazinone synthesis that unites the two indole-containing sectors.
The bis indole alkaloid dragmacidin E (1) along with congener dragmacidin D was isolated from a Spongosortes sp. collected in Australian waters.1 Both of these species are described as exhibiting potent serine-threonine protein phosphatase inhibitory activity. The challenges inherent in the unique molecular architecture of 1 and related species dragmacidins D and F has fueled much synthesis effort,2,3 culminating in total syntheses of dragmacidins D and F by Stoltz and colleagues.2 Our own efforts have focused on dragmacidin E, and in particular the assembly of the cycloheptannelated indole core via application of diradical cyclization chemistry to forge the key C(4″)–C(6‴) bond.4 Additionally, successful preparation of 1 will have to utilize chemistry that is not derailed by (1) severe steric congestion along the upper rim of the cycloheptanoid unit, (2) ring strain resulting from installation of five contiguous sp2 carbons in a seven-membered ring, and (3) the sensitive guanidine unit. Preliminary studies that probed these issues resulted in the preparation of the model system 2, lacking both the C(7″) hydroxyl and the C(6′) bromide.4b In this report, the application and extension of this chemistry to the total synthesis of the intact (±)-dragmacidin E target is described. The key transformative step involves Witkop cyclization5 of a tryptophan derivative to deliver the C(3″)-C(4″) bridged indole core of the target.
The synthesis plan commenced with preparation of the Witkop cyclization precursor 6 from the known 7-diphenylmethyl ether of indole, 3, itself available via Bartoli indole synthesis in 2 steps,6 Scheme 1. Installation of the tryptophan side chain proceeded via initial formylation of 3 at C(3) and then Emmons-Horner alkene extension with the anion of methyl (2,2-dichloropropionylamino)-(dimethylphosphoryl)acetate to furnish the (Z)-dehydrotryptophan derivative 5. Exposure of 5 to catalytic Wilkinson’s catalyst and high pressure hydrogen gas served to both saturate the side chain and cleave the diphenylmethyl ether function at C(7″) to deliver the Witkop substrate 6 following deprotection of the indole nitrogen.
Scheme 1.

Synthesis of the Witkop cyclization precursor.
The key Witkop cyclization proceeded smoothly upon irradiation of dilute solutions of 6 in acetonitrile in a quartz vessel with 254 nm light, Scheme 2. The isolated product, 13, represents one of the highest yielding Witkop cyclizations reported to date.8 The Witkop cyclization has been subject to numerous mechanistic studies7 and has been utilized as a pivotal step in many total syntheses of cognate indole alkaloids.8 Application of the consensus Witkop mechanistic paradigm to the chemistry of 6 is detailed below. This speculation features two consecutive photochemically mediated electron transfers that labilize both C–Cl bonds for further chemistry. In the first sequence, chloride loss promotes diradical 8 generation, and regioselective closure within 8 then constitutes the key C–C bond forming step of this Witkop cyclization. A subsequent photoelectron transfer within 9 provides a low-energy pathway for Cl–, and then H+, loss to deliver the enone product 13, although it is not unreasonable to imagine that HCl loss may proceed from 9 without intervention of excited state chemistry.
Scheme 2.

Witkop cyclization of 6, with mechanistic speculation.
Conversion of the 8-membered bridging lactam into a cycloheptanoid structure constituted the next synthesis objective, Scheme 3. This formal one-atom ring contraction could be effected by initial conjugate addition of hydride to the enone unit, which promoted a Dieckmann cyclization of the first-formed enolate 14. The strained tetracyclic product 15 was readily hydrolyzed to deliver the desired cycloheptane-bridged indole substructure of dragmacidin E, 16. Tricycle 16 was formed as an inconsequential mixture of stereoisomers at C(6‴); both diastereomers later converged to the same product.
Scheme 3.

Synthesis of the cycloheptannelated indole core
Conversion of the C(5‴) carbonyl into the spiroimidazolone unit with correct relative stereochemistry defined the next task, Scheme 4. Functionalizing this ketone proved quite challenging, and, curiously, related difficulties did not plague the similar des-C(7″)-oxy model system preceding 2; the long-range reactivity-altering effects of the C(7″) OBn unit on the chemistry of the C(5‴) carbonyl were not anticipated. Through much trial-and-error, an effective and reproducible Strecker reaction-based experimental procedure utilizing methanolic ammonia at elevated temperature (75°C) in a sealed tube, followed by rapid TMSCl addition to the crude reaction mixture, was developed. Diastereomeric cyanoamines 17a and 17b were formed in a ~ 5:1 ratio favoring the desired isomer 17a; in the model system preceding 2, only the desired diastereomer was produced. Epimerization at C(6‴) during the reaction ensured that only the most thermodynamically favorable cis methyl/NHBOC stereochemical arrangement emerged from these Strecker reaction conditions. Diastereomers 17a and 17b could be separated by chromatography, and the free amine of major isomer 17a was immediately protected as the methyl carbamate. Without this protection, attempted transformations on 17a/17b tended to promote retro-Strecker reaction and deliver either an imine of 16 or 16 itself following imine hydrolysis. This methyl carbamate was subjected to the nitrile reduction conditions of Satoh.9 The crude amino carbamate product was cyclized to furnish the imidazolone unit within 18 under base mediation, which removed the indole N-BOC group as well. The relative stereochemistry of 18 was established by single crystal X-ray analysis, as indicated in Scheme 4.10 Functionalization of 18 at C(6) was required to install the pyrazinone ring of dragmacidin E, and that operation was accomplished by DDQ-mediated oxidation of this indolic position. Subsequent reduction of the C(6) carbonyl to a pair of labile alcohols, and then conversion of those alcohols to the diastereomeric azides 20, proceeded through standard transformations. The relative stereochemistry at C(6) was not germane to the synthesis effort.
Scheme 4.

Installation of the spiro 2-imidazolone; structural determination by X-ray analysis.
Coupling the tetracyclic indole core unit 20 with the second indole component, in the form of acid chloride 21,11 was preceded by azide reduction to the primary amines, Scheme 5. The product amide 22 was formed as a ~ 2:1 mixture of diastereomers at C(6); both species were useful. Pyrazinone formation proceeded through a two-stage protocol involving first cyclization of the primary amine liberated by TFA-mediated deprotection of the C(5) NHBOC moiety with the indolic ketone of 22 to give a dihydropyrazinone product. This pale yellow species immediately was subjected to DDQ-based oxidation to deliver the stable pyrazinone-containing product 23 as a bright yellow solid.
Scheme 5.

Merging the two indole cores; synthesis of the pyrazinone 23.
Completion of the dragmacidin E synthesis required successful execution of two transformations; (1) conversion of the spiroimidazolone’s urea unit into a guanidine, and (2) removal of the C(7‴) benzyl protecting group. A priori, it was not clear which maneuver should be approached first. As it turned out, initial work on the imidazolone unit proved satisfactory, Scheme 6. O-methylation of the nucleophilic carbonyl oxygen within the urea appeared to be slightly more facile that methylation at other potentially nucleophilic sites in 23, and so moderate yields of the 2-methoxyamidine derivation of 23 could be isolated under carefully controlled reaction conditions. Replacement of the MeO function with NH2 was accomlished by heating a methanolic solution of this 2-methoxyamidine with ammonia in a sealed tube, and the sensitive guanidine-contaning product was subjected to the benzyl ether deprotection conditions reported by Stoltz in his dragmacidin D synthesis2a to give, following SiO2 chromatography, (±)-dragmacidin E (1) in modest yield. The final structural identity of synthetic 1 was derived from a comparison of the 1H NMR and 13C NMR spectral data with those described by Capon, and by co-injection HPLC with an authentic sample of natural dragmacidin E. (see Supporting Information for details).
Scheme 6.

Completion of dragmacidin E synthesis via guanidine introduction.
Supplementary Material
Figure 1.

Dragmacidin E (1) and a simpler model compound 2.
Acknowledgments
We thank the National Institutes of Health, General Medical Sciences (GM 72572) for financial support of this work; the X-Ray facility is supported by NSF CHE 0131112. In addition, we thank Dr. R. Capon (Institute for Molecular Bioscience, The University of Queensland, St. Lucia, QLD 4072, Australia) for providing copies of the 1H NMR and 13C NMR spectra of authentic dragmacidin E in CD3OD, and Andrew Piggott (The University of Queensland) for conducting the HPLC-based and UV-based comparisons of 1 with authentic dragmacidin E.
Footnotes
Supporting Information Available. General experimental, and detailed experimental descriptions along with copies of 1H and 13C NMR spectra for 4–6, 13, 15–23, and 1; X-ray data for 18; HPLC traces and UV traces of synthetic and authentic dragmacidin E. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Capon RJ, Rooney F, Murray LM, Collins E, Sim ATR, Rostas JAP, Butler MS, Carrol AR. J Nat Prod. 1998;61:660–662. doi: 10.1021/np970483t. [DOI] [PubMed] [Google Scholar]
- 2.(a) Garg NK, Sarpong R, Stoltz BM. J Am Chem Soc. 2002;124:13179–13184. doi: 10.1021/ja027822b. [DOI] [PubMed] [Google Scholar]; (b) Garg NK, Caspi DD, Stoltz BM. J Am Chem Soc. 2004;126:9552–9553. doi: 10.1021/ja046695b. [DOI] [PubMed] [Google Scholar]; (c) Garg NK, Caspi DD, Stoltz BM. J Am Chem Soc. 2005;127:5970–5978. doi: 10.1021/ja050586v. [DOI] [PubMed] [Google Scholar]; (d) Garg NK, Stoltz BM. J Chem Soc, Chem Commun. 2006:3769–3779. doi: 10.1039/b605929e. [DOI] [PubMed] [Google Scholar]; (e) Garg NK, Caspi DD, Stoltz BM. Synlett. 2006:3081–3087. [Google Scholar]
- 3.(a) Huntley RJ, Funk RL. Org Lett. 2006;8:4775–4778. doi: 10.1021/ol0617547. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huntley RJ. PhD Thesis. Pennsylvania State University; 2008. [Google Scholar]
- 4.(a) Feldman KS, Ngernmeesri P. Org Lett. 2005;7:5449–5452. doi: 10.1021/ol0522081. [DOI] [PubMed] [Google Scholar]; (b) Feldman KS, Ngernmeesri P. Org Lett. 2010;12:4502–4505. doi: 10.1021/ol1018008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yonemitsu O, Cerutti P, Witkop B. J Am Chem Soc. 1966;88:3941–3945. doi: 10.1021/ja00969a010. [DOI] [PubMed] [Google Scholar]
- 6.Dobson D, Todd A, Gilmore J. Synth Commun. 1991;21:611–617. [Google Scholar]
- 7.(a) McCall MT, Hammond GS, Yonemitsu O, Witkop B. J Am Chem Soc. 1970;92:6991–6993. [Google Scholar]; (b) Naruto S, Yonemitsu O. Tetrahedron Lett. 1975;16:3399–3402. [Google Scholar]; (c) Hamada T, Ohmori M, Yonemitsu O. Tetrahedron Lett. 1977;18:1519–1522. [Google Scholar]; (d) Naruto S, Yonemitsu O. Chem Pharm Bull. 1980;28:900–909. [Google Scholar]
- 8.(a) Kobayashi T, Spande TF, Aoyagi H, Witkop B. J Org Chem. 1969;12:636–638. doi: 10.1021/jm00304a017. [DOI] [PubMed] [Google Scholar]; (b) Anderson NG, Lawton RG. Tetrahedron Lett. 1977:1843–1846. [Google Scholar]; (c) Bosch J, Amat M, Sanfeliu E, Miranda MA. Tetrahedron. 1985;41:2557–2566. [Google Scholar]; (d) Klohr SE, Cassady JM. Synth Commun. 1988;18:671–674. [Google Scholar]; (e) Beck AL, Mascal M, Moody CJ, Slawin AMZ, Williams DJ, Coates WJ. J Chem Soc Perkin Trans. 1992;1:797–811. [Google Scholar]; (f) Beck AL, Mascal M, Moody CJ, Coates WJ. J Chem Soc Perkin Trans. 1992;1:813–822. [Google Scholar]; (g) Mascal M, Moody CJ, Slawin AMZ, Williams DJ. J Chem Soc Perkin Trans. 1992;1:823–830. [Google Scholar]; (h) Nagata R, Endo Y, Shudo K. Chem Pharm Bull. 1993;41:369–372. [Google Scholar]; (i) Mascal M, Moody CJ, Morrell AI, Slawin AMZ, Williams DJ. J Am Chem Soc. 1993;115:813–814. [Google Scholar]; (j) Mascal M, Wood IG, Begley MJ, Batsanov AS, Walsgrove T, Slawin AMZ, Williams DJ, Drake AF, Siligardi G. J Chem Soc Perkin Trans. 1996;1:2427–2433. [Google Scholar]; (k) Bennasar ML, Zulaica E, Ramirez A, Bosch J. Heterocycles. 1996;43:1959–1966. [Google Scholar]; (l) Ruchkina EL, Blake AJ, Mascal M. Tetrahedron Lett. 1999;40:8443–8445. [Google Scholar]; (m) Bremner JB, Russell HF, Skelton BW, White AH. Heterocycles. 2000;53:277–296. [Google Scholar]; (n) Mascal M, Modes KV, Durmus A. Angew Chem Int Ed. 2011;50:4445–4446. doi: 10.1002/anie.201006423. [DOI] [PubMed] [Google Scholar]; (o) Qin H, Xu Z, Cui Y, Jia Y. Angew Chem Int Ed. 2011;50:4447–4449. doi: 10.1002/anie.201100495. [DOI] [PubMed] [Google Scholar]
- 9.Satoh T, Suzuki S, Suzuki Y, Miyaji Y, Imai Z. Tetrahedron Lett. 1969;10:4555–4588. [Google Scholar]
- 10.Cambridge Crystallographic Data Centre deposition number for 18: CCDC 826169. The data can be obtained free from Cambridge Crystallographic Data Centre via. http://www.ccdc.cam.ac.uk/data_request/cif.
- 11.Guinchard X, Vallee Y, Denis JN. J Org Chem. 2007;72:3972–3975. doi: 10.1021/jo070286r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.