

Abstract
Helicobacter pylori (H. pylori) is the causative pathogen underlying gastric diseases such as chronic gastritis and gastric cancer. Previously, the authors revealed that α1,4-linked N-acetylglucosamine-capped O-glycan (αGlcNAc) found in gland mucin suppresses H. pylori growth and motility by inhibiting catalytic activity of cholesterol α-glucosyltransferase (CHLαGcT), the enzyme responsible for biosynthesis of the major cell wall component cholesteryl-α-d-glucopyranoside (CGL). Here, the authors developed a polyclonal antibody specific for CHLαGcT and then undertook quantitative ultrastructural analysis of the enzyme’s localization in H. pylori. They show that 66.3% of CHLαGcT is detected in the cytoplasm beneath the H. pylori inner membrane, whereas 24.7% is present on the inner membrane. In addition, 2.6%, 5.0%, and 1.4% of the protein were detected in the periplasm, on the outer membrane, and outside microbes, respectively. By using an in vitro CHLαGcT assay with fractionated H. pylori proteins, which were used as an enzyme source for CHLαGcT, the authors demonstrated that the membrane fraction formed CGL, whereas other fractions did not. These data combined together indicate that CHLαGcT is originally synthesized in the cytoplasm of H. pylori as an inactive form and then activated when it is associated with the cell membrane. This article contains online supplemental material at http://www.jhc.org. Please visit this article online to view these materials.
Keywords: cell wall, glycosyltransferase, O-glycan, Helicobacter pylori, immunocytochemistry
In the two decades since Marshall and Warren (1984) discovered Helicobacter pylori in the human stomach, it has been accepted that H. pylori is associated with pathogenesis of gastritis, peptic ulcer, gastric carcinoma, and gastric lymphoma of mucosa-associated lymphoid tissues (Peek and Blaser 2002). Although H. pylori infects more than half of the world’s population (Bruce and Maaroos 2008), most infections are asymptomatic, and only a fraction of individuals infected develop serious gastric diseases such as gastric ulcer and cancer, suggesting that the stomach harbors protective mechanisms.
H. pylori colonizes the surface mucin by binding blood group Lewis b and type H carbohydrates via the blood group antigen-binding adhesin (BabA; Ilver et al. 1998; van de Bovenkamp et al. 2003). By contrast, H. pylori is barely detectable in gland mucin secreted from gland mucous cells such as mucous neck cells and pyloric gland cells, which contain unique O-glycans having terminal α1,4-linked N-acetylglucosamine (αGlcNAc; Hidaka et al. 2001). Previously, we revealed that cholesteryl-α-d-glucopyranoside (CGL), a major cell wall lipid component of H. pylori, plays a critical role in bacterial survival and that αGlcNAc in gland mucin serves as a naturally occurring antibiotic against H. pylori by suppressing CGL biosynthesis, thus protecting gastric mucosa from H. pylori infection (Kawakubo et al. 2004).
Cholesterol α-glucosyltransferase (CHLαGcT), which we and others molecularly cloned from H. pylori, is the glycosyltransferase that transfers glucose from UDP-glucose to a carbon atom at the third position of cholesterol with an α1,3-linkage (Lee et al. 2006; Lebrun et al. 2006). High conservation of CHLαGcT amino acid sequence among Helicobacter species suggests that this enzyme plays an important role in Helicobacter growth (Lee et al. 2008). Recently, we have also shown that the suppressive effect of αGlcNAc on CGL biosynthesis is due to its competition with CHLαGcT in vitro (Lee et al. 2006). These results suggest that O-glycans exhibiting αGlcNAc in the gland mucin interact with CHLαGcT expressed by H. pylori, thus protecting the gastric mucosa from infection. However, the exact localization of CHLαGcT in H. pylori remains to be determined.
In the present study, we developed polyclonal antibodies specific to H. pylori CHLαGcT and used them to demonstrate the subcellular localization of this enzyme in H. pylori using immunoelectron microscopy. Then, we carried out an in vitro CHLαGcT assay using fractionated proteins isolated from H. pylori to test which fraction contained an active form of the enzyme.
Materials and Methods
Bacterial Strain and Culture Conditions
Three standard H. pylori strains—26695 (ATCC 700392), NCTC 11637 (ATCC 43504), and J99 (ATCC 700824)—obtained from American Type Culture Collection (Manassas, VA) were cultured on trypticase soy agar with 5% sheep blood (Becton Dickinson; Franklin Lakes, NJ) for 3 to 4 days at 35C in 15% CO2. Bacteria were then inoculated in Brucella broth (Becton Dickinson) supplemented with 10% horse serum (Invitrogen; Carlsbad, CA) and cultured at 35C in 15% CO2. Bacteria were subsequently cultured in Mueller Hinton broth (Eiken Chemical; Tokyo, Japan) supplemented with 5.5% horse serum.
Antibody Production
Polyclonal antibodies directed to CHLαGcT of the H. pylori 26695 strain were generated using synthetic peptides as immunogens based on published sequences (Lee et al. 2006). Peptide sequences with high antigenicity were analyzed by database searches, and three peptides unique to H. pylori CHLαGcT, designated E17S, V17F, and P17K, were selected and synthesized (Table 1). The combined peptides were mixed with Freund’s complete adjuvant and intradermally injected into two Japanese white rabbits, followed by eight additional injections with Freund’s incomplete adjuvant. Sera were separately purified using each synthetic peptide conjugated to agarose gel (Pierce; Rockford, IL) for Western blot analysis to evaluate specificity as well as suitability for immunocytochemical staining. The experiment protocol was approved by the Institutional Animal Care and Use Committee at Medical and Biological Laboratories, Co., Ltd. (Nagoya, Japan).
Table 1.
Synthetic Peptides Used to Develop Anti-CHLαGcT Antibodies
| Peptide Name | Amino Acid Sequence | Amino Acid Residues |
|---|---|---|
| E17S | CEAIRDFKNNPHLFKTLSz | 373-389 |
| V17F | CVVDSFKDTSNGTSMTAF | 6-22 |
| P17K | CPHVDNLGSEEEGYYNLK | 40-56 |
C indicates cysteine residue at the N-terminus for coupling with keyhole limpet hemocyanin.
Western Blot Analysis
Histidine (His)–tagged CHLαGcT recombinant protein expressed by Escherichia coli (E. coli) HB101 competent cells was purified using a HisTrap HP column (GE Healthcare; Buckinghamshire, UK) as described (Lee et al. 2006). In parallel, H. pylori 26695, NCTC 11637, and J99 strains harvested in Brucella broth with 10% horse serum as well as E. coli transformed with pTKNd6xH-CHLαGcT or pTKNd6xH harvested in LB medium were centrifuged and sonicated in 50 mM Tris-HCl (pH 7.4) containing 300 mM NaCl and 0.5% Triton X-100. After centrifugation at 16 000 × g for 10 min, the supernatant served as a source for whole protein. To evaluate specificity of antibodies, Western blot analysis was carried out on His-tagged CHLαGcT recombinant protein and whole proteins from the three H. pylori strains. Equal amounts of protein were separated on 14% SDS-PAGE and transferred to a PVDF membrane (Millipore; Billerica, MA). Nonspecific binding was blocked with 5% skim milk in TBS, and the membrane was incubated with the three anti-CHLαGcT antibodies (anti-E17S, -V17F, and -P17K; 1:1000 dilution each) or anti-His tag (1:500 dilution; Santa Cruz Biotechnnology, Santa Cruz, CA) as a primary antibody. After rinsing with TBS, membranes were incubated with horseradish peroxidase (HRP)–conjugated goat anti-rabbit antibody (Jackson Immunoresearch; West Grove, PA), and immunoreactions were detected using the ECL Western blotting detection system (GE Healthcare). Specificity of the anti-E17S antibody was further evaluated by an antigen absorption test using various peptides: specifically, antibody was preincubated with 10 µg/ml of synthetic E17S peptide or the irrelevant peptides V17F and P17K and then immunoblotted using absorbed anti-E17S antibodies. Prestained protein ladder BenchMark (Invitrogen) was used as a size marker.
Immunocytochemistry
H. pylori 26695 strain was obtained from a 3-day culture in trypticase soy agar supplemented with 5% sheep blood (Beckton Dickinson). Bacteria were placed on MAS-coated glass (Matsunami glass; Osaka, Japan), air-dried, and fixed with 20% formalin neutral buffer solution, pH 7.4 (Wako; Osaka, Japan). Slides were incubated with 0.3% H2O2 in absolute methanol for 30 min and then permeabilized with 1% saponin dissolved in TBS containing 0.02% BSA. Because Western blot analysis described above revealed that the anti-E17S antibody was the most specific in recognizing recombinant and native CHLαGcT proteins, we used it for subsequent immunocytochemical analysis. After incubation with anti-E17S antibody (1:200 dilution) overnight at 4C, anti-rabbit immunoglobulins conjugated with HRP-labeled polymer Envision (DakoCytomation; Carpinteria, CA) were used as secondary antibody. HRP activity was visualized using 3,3′-diaminobenzidine (DAB; Dojindo, Kumamoto, Japan) with H2O2. As a control, anti-E17S antibody was omitted from the procedure, and no specific staining was found. Gram stain was carried out to observe the morphology of H. pylori.
Immunoelectron Microscopy
The H. pylori 26695 strain was washed with phosphate buffer and plated onto poly-l-lysine-coated specimen carrier (Leica; Vienna, Austria) (Sawaguchi et al. 2003). Samples were quick frozen using a high-pressure freezing system EM PACT (Leica) and fixed with 1% glutaraldehyde in acetone, followed by gradual warming from −85C to −3C by auto-freeze substitution using EM AFS (Leica). After washing with cold acetone, fixed cells were embedded in LR White Resin (London Resin Company; Berkshire, UK) and polymerized with UV at −20C for 24 hr. Immunogold staining was carried out as described (Kasai et al. 2003). Briefly, ultrathin sections with 0.1-µm thickness cut by ultramicrotome were mounted on nickel grids coated with formvar (Nacalai Tesque; Kyoto, Japan). To prevent nonspecific reaction, sections were treated with 1% BSA in PBS and then incubated with anti-E17S antibody (1:50 dilution). After washing with PBS, sections were incubated with goat anti-rabbit IgG conjugated with 15 nm colloidal gold (1:50 dilution) purchased from BBInternational (Cardiff, UK). After washing with PBS and distilled water (DW), sections were poststained with 2% uranyl acetate in DW and coated with carbon and observed using conventional transmission electron microscopy (TEM), JEM-1200EX (JEOL; Tokyo, Japan), at an accelerating voltage of 80 kV. Control samples omitting primary antibody showed no specific signal.
Fractionation of Bacterial Proteins
The H. pylori 26695 strain harvested in Brucella broth with 10% horse serum was washed with 10 mM Tris-HCl (pH 7.5) containing 30 mM NaCl and suspended in 30 mM Tris-HCl (pH 7.5) containing 20% sucrose. EDTA was added to 1 mM and the sample incubated at room temperature with agitation. The pellet obtained by centrifugation was immediately suspended in 0.5 mM MgCl2 and incubated at 4C with gentle agitation. After centrifugation at 8000 × g, the supernatant was carefully removed and used as the periplasmic fraction. The pellet was resuspended in 10 mM HEPES buffer (pH 7.0) and sonicated. To remove unbroken cells, the cell suspension was centrifuged at 3000 × g for 20 min and supernatant ultracentrifuged at 100 000 × g for 1 hr. The supernatant was collected as the cytoplasmic fraction, and the pellet was dissolved in 10 mM HEPES buffer containing 0.5% Triton X-100 and served as the membrane fraction. All samples contained a protease inhibitor cocktail (Roche; Mannheim, Germany). To validate the purity of fractionated proteins, the enzymatic activity of a cytoplasmic protein, malate dehydrogenase, was measured as described (Pitson et al. 1999; Appels and Haaker 1988). Briefly, mixtures of each fractionated protein with 0.1 M potassium phosphate buffer (pH 7.5) and 0.33 mM oxaloacetate were incubated at 25C for 5 min. After adding 143 µM NADH, the tube was mixed immediately and then incubated at 25C for 60 min. The decrease in absorbance at 340 nm was monitored. The molecular sizes of each fractionated protein were determined by Western blot analysis with anti-E17S antibody as described before.
In Vitro CHLαGcT Assay
The enzyme activity of CHLαGcT was measured using an in vitro CHLαGcT assay as described (Lee et al. 2006). Briefly, recombinant CHLαGcT was prepared by transforming E. coli JM109 cells with pTKNd6xH-CHLαGcT, followed by purification with a ProtinoR Ni-TED 2000 packed column (Macherey-Nagel; Duren, Germany) according to the manufacturer’s protocol. The eluted protein was dialyzed against 50 mM HEPES buffer (pH 7.5) containing 15% glycerol and 5 mM dithiothreitol (DTT) by using an ultrafiltration unit (Millipore) at 4C and then used as an enzyme source for positive control. In parallel, the fractionated proteins isolated from H. pylori as described before were concentrated to 20 times, and 10 µl of the solution was incubated with 25 mM Tris-HCl buffer (pH 8.0) containing 3.7 µM UDP-[14C]glucose (111 000 dpm/µl), 500 µM cholesterol, and 0.1% Triton X-100. In addition, 0.5 µl of the recombinant CHLαGcT was used as well. After an 18-hr incubation at 30C, the reaction was stopped by adding 3 µl of 1 N HCl, and then 10 volumes of ethyl acetate were added and vortexed. After brief centrifugation, radioactivity in the ethyl acetate (upper) layer, which contained α-glucosyl cholesterol, was measured using a liquid scintillation counter (Beckman Coulter; Brea, CA). In addition, these samples were also subjected to TLC with ethyl acetate layer. Briefly, the ethyl acetate layer was laid on preparative TLC (HPTLC LiChrospher Silica gel 60 F254s; Merck, Darmstadt, Germany) and separated in chroloform/methanol (85:15), and radioactive materials were visualized by fluorography (Typhoon 8600; GE Healthcare).
Statistical Analysis
All statistical data are presented as means ± SD. Statistical analysis of enzyme activity in multiple comparisons was determined by one-way ANOVA with a Bonferroni multiple comparisons test using InStat 3 software (GraphPad Software; San Diego, CA). p<0.05 was considered significant.
Results
CHLαGcT Antibody Specificity
The reactivity of anti-CHLαGcT antibodies raised against three CHLαGcT-specific peptides, E17S, V17F, and P17K, was verified by Western blot analysis using whole-protein lysates of E. coli transformed with CHLαGcT vector or vehicle alone. Both anti-E17S and anti-P17K antibodies reacted with recombinant CHLαGcT protein, whereas the immunoreaction of the anti-V17F antibody was less potent (Fig. 1A).
Figure 1.
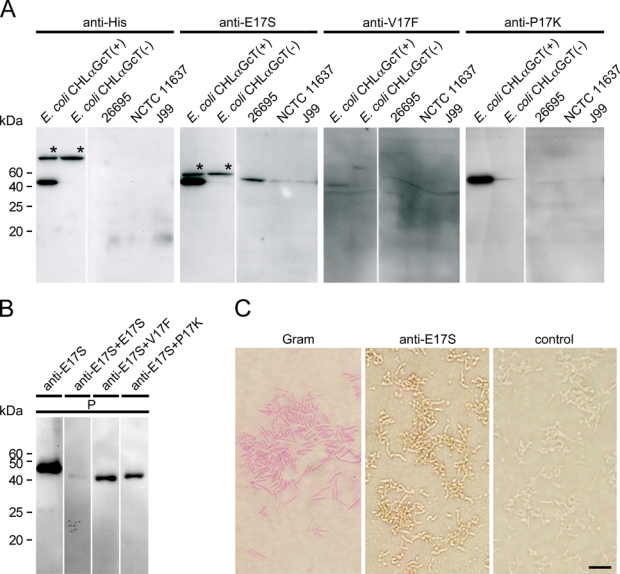
Characterization of three anti-CHLαGcT antibodies. (A) Immunoreactivity against various Helicobacter pylori strains (26695, NCTC 11637, J99) and Escherichia coli transformed by pTKNd6xH-CHLαGcT (CHLαGcT (+)) or pTKNd6xH (CHLαGcT (−)) reacted with anti-His (anti-His) and anti-CHLαGcT antibodies (anti-E17S, anti-V17F, and anti-P17K). Asterisks indicate extra bands for proteins other than CHLαGcT. (B) Inhibition of anti-E17S antibody using E17S peptide. Recombinant His-tagged CHLαGcT protein, which was preincubated with 10 µg/ml of the synthetic peptides E17S, V17F, or P17K, was reacted with anti-E17S antibody. P, His-purified cell lysate prepared from E. coli transformed by pTKNd6xH-CHLαGcT used for a positive control. (C) Gram staining and immunocytochemistry using anti-E17S antibody against H. pylori strain 26695. Control indicates secondary antibody alone. Bar = 5 µm.
To determine whether anti-E17S and anti-P17K antibodies react with native CHLαGcT protein extracted from H. pylori, Western blot analysis using whole-protein lysates from 3 strains of H. pylori (26695, NCTC 11637, and J99) was carried out. The anti-E17S antibody clearly reacted with native CHLαGcT isolated from all H. pylori strains, but its immunoreactivity was less potent against NCTC 11637 and J99 strains than against the 26695 strain (Fig. 1A). By contrast, anti-P17K antibody did not react with native CHLαGcT proteins extracted from any H. pylori strain tested, although it reacted strongly with recombinant CHLαGcT protein (Fig. 1A).
Next, the specificity of anti-E17S antibody for CHLαGcT was confirmed using an antigen absorption test. Anti-E17S immunoreactivity was apparently decreased when antibody was preincubated with the synthetic E17S peptide used as its immunogen. On the other hand, a distinct CHLαGcT band was detected when anti-E17S antibody was preincubated with the irrelevant peptides V17F and P17K (Fig. 1B).
Last, the specificity of the anti-E17S antibody was confirmed by immunocytochemistry. H. pylori strain 26695 was positive for the anti-E17S antibody (Fig. 1C). These results collectively indicate that the anti-E17S antibody specifically recognized recombinant and native CHLαGcT proteins and was suitable for immunocytochemical analysis of H. pylori.
Subcellular Distribution of CHLαGcT in H. pylori as Determined by Immunoelectron Microscopy
To determine the subcellular localization of CHLαGcT in H. pylori, the 26695 strain labeled with immunogold was observed by TEM using the anti-E17S antibody. Immunoelectron microscopy revealed that CHLαGcT was largely cytoplasmic (Fig. 2A,B) and adjacent to the inner membrane (Fig. 2C,D) and that some CHLαGcT signals were also detected in cell wall components (Fig. 2E,F). To quantitate the number of gold particles representing CHLαGcT in H. pylori, cellular components were divided into five regions—cytoplasm, inner membrane, periplasm, outer membrane, and extracellular space—and gold particles were counted approximately up to 1000 in total. In this analysis, particles touching the inner or outer membranes were counted as residing in the inner membrane or outer membrane, respectively (Table 2). We found that 710 (66.3%) of 1070 gold particles indicative of CHLαGcT were located in the cytoplasm, and 264 (24.7%) particles were present on the inner membrane. Notably, 28 (2.6%) and 53 (5.0%) particles were also detected in the respective periplasm and on the outer membrane. Fifteen (1.4%) particles were present outside of bacteria.
Figure 2.
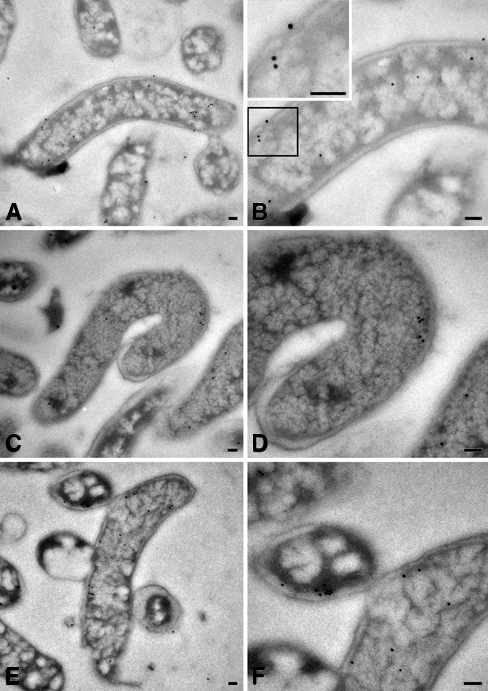
Ultrastructural localization of CHLαGcT in Helicobacter pylori strain 26695. Gold particles indicate CHLαGcT immunoreactivity. B, D, and F are enlarged images of A, C, and E, respectively. Bar = 0.1 µm.
Table 2.
Number (%) of Gold Particles Indicative of CHLαGcT in Helicobacter pylori
| Cytoplasm | Inner Membrane | Periplasm | Outer Membrane | Outside of Cell | Total |
|---|---|---|---|---|---|
| 710 (66.3) | 264 (24.7) | 28 (2.6) | 53 (5.0) | 15 (1.4) | 1070 (100) |
Detection of CHLαGcT Activity in the Fractionated Protein of H. pylori
To test which subcellular fractions contained the active form of CHLαGcT, we carried out an in vitro CHLαGcT assay using fractionated H. pylori compartments. To validate the method of fractionation, we monitored the activity of malate dehydrogenase, which is a marker for cytoplasmic protein, and confirmed that the malate dehydrogenase activity was detected only in the cytoplasmic fraction (data not shown).
Thus, we carried out an in vitro CHLαGcT assay using 20-fold concentrated proteins. Western blot analysis revealed that molecular weights of each fractionated protein were the same (Fig. 3A). However, the enzyme activity was detected only in the membrane fraction (Fig. 3B). By contrast, such enzyme activity for CHLαGcT was not detected from other fractions, including the cytoplasm and periplasm. Similar results were also obtained when the samples were tested by TLC, and the positive signal indicative of CGL was detected only in the membrane fraction (Fig. 3C).
Figure 3.
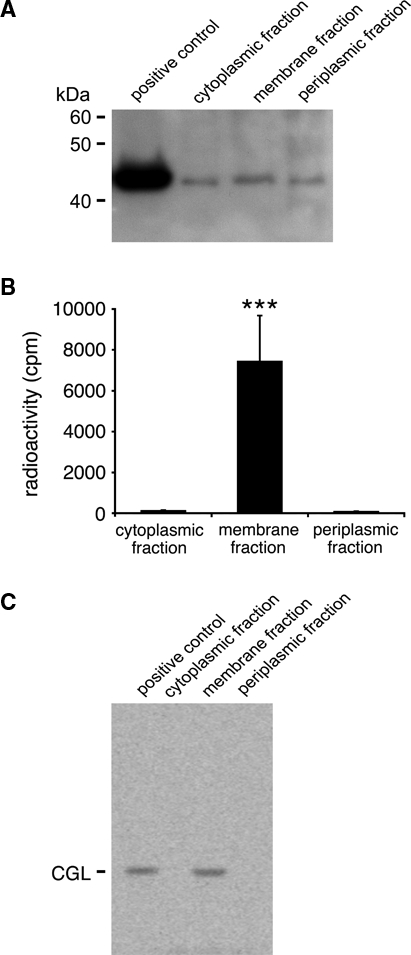
In vitro CHLαGcT assay using fractionated proteins of Helicobacter pylori. (A) Molecular weights of each fractionated protein were examined using Western blot analysis with anti-E17S antibody. (B) Radioactivity in each fractionated protein of H. pylori incubated with a solution containing UDP-[14C]glucose and cholesterol was measured by a liquid scintillation counter. Radioactivity of the membrane fraction is significantly higher than cytoplasmic and membrane fractions (***p<0.001). Data are presented as means ± SD. (C) In parallel, these samples were analyzed by TLC. The product indicative of cholesteryl-α-d-glucopyranoside (CGL) was formed only when the membrane fraction was used as an enzyme source.
Discussion
CHLαGcT is the glycosyltransferase that forms CGL, a major cell wall glycolipid of Helicobacter species, including H. pylori. CGL was originally discovered as one of three major cholesteryl glucosides (CGs) that account for 25% of total cell wall lipids of Helicobacter species, including H. pylori, whereas other CGs include cholesteryl-6-O-tetradecanoyl-α-d-glucopyranoside (CAG) and cholesteryl-6-O-phosphatidyl-α-d-glucopyranoside (CPG; Hirai et al. 1995). Recently, Wunder et al. (2006) showed that CGL is required for H. pylori to evade the host’s immune response. In addition, we have shown that CGL is critical for H. pylori survival, and CGL biosynthesis by H. pylori is significantly suppressed when microbes are cultured with αGlcNAc (Kawakubo et al. 2004). We have also demonstrated that CHLαGcT activity is inhibited by αGlcNAc in vitro (Lee et al. 2006; Lee et al. 2008). Despite its important roles of CGL on H. pylori survival, the expression and activation of CHLαGcT in H. pylori have not been demonstrated so far.
In this study, we first developed a polyclonal antibody against CHLαGcT by immunizing the same rabbit with a cocktail of three different peptides specific for CHLαGcT. One technical benefit of our antibody production strategy is to reduce the number of animals used for immunization. Sera were then purified with each of the three peptides using affinity columns, and immunoreactivity to recombinant CHLαGcT protein and CHLαGcT isolated from H. pylori strains was evaluated. Anti-E17S antibody reacted with both recombinant and native CHLαGcT proteins (see Fig. 1A). An extra band with anti-E17S antibody found in both protein lysates of E. coli CHLαGcT (+) and E. coli CHLαGcT (−) was most possibly caused by the cross-reactivity of the anti-E17S antibody with unidentified protein endogenously expressed by E. coli, because an antigen absorption test using 10 µg/ml and 100 µg/m of the synthetic E17S peptide revealed that this extra band and the band indicative for CHLαGcT were diminished in a dose-dependent manner (unpublished data). Interestingly, the reactivity to CHLαGcT protein from H. pylori NCTC 11637 and J99 strains was much weaker than that from the 26695 strain. We thus compared the amino acid sequence of CHLαGcT of the H. pylori 26695 strain with those of the NCTC 11637 and J99 strains. No amino acid substitution was found in the E17S sequence of NCTC 11637, whereas two amino acid substitutions were found in the E17S sequence of J99 (Suppl. Fig. S1). Thus, weak reactivity for the anti-E17S antibody to J99 and NCTC 11637 strains might be caused by the difference of the primary structure of CHLαGcT for J99 and the weak expression level of the CHLαGcT protein for NCTC 11637, respectively. On the other hand, anti-P17K antibody reacted with recombinant CHLαGcT produced in E. coli but not with native protein from H. pylori. It is not obvious why anti-P17K antibody could recognize only recombinant protein. It might be possible that the P17K sequence of the native CHLαGcT was cryptic due to its secondary structure. By contrast, anti-V17F antibody reacted with neither recombinant nor native CHLαGcT (see Fig. 1A). It is not clear why anti-V17F antibody was less potent compared with other antibodies, despite that the V17F sequence of recombinant and native proteins was the same (Suppl. Fig. S1). The binding site for anti-V17F antibody on both proteins might be masked due to the secondary structure of CHLαGcT. Further study will be necessary to address these problems.
After confirming the specificity for anti-E17S antibody by a peptide inhibition test (see Fig. 1B), we carried out immunocytochemistry and showed that the CHLαGcT protein was most abundant in the cytoplasm, particularly beneath the inner cell membrane (see Fig. 2). Moderate and small amounts of enzyme were also expressed in the inner cell membrane and the outer membrane/periplasm, respectively. Then, we measured the enzyme activity using fractionated proteins and revealed that the active form of CHLαGcT was present only in membrane fraction, even though the molecular sizes of each fractionated protein were the same (see Fig. 3). Although this result is consistent with the data reported before (Lebrun et al. 2006), it seems to be paradoxical because the majority of CHLαGcT found in the cytoplasm lacked its activity, whereas the membrane fraction contained the active form of CHLαGcT despite a lack of transmembrane domain. At this moment, it is unknown why CHLαGcT is inactive in the cytoplasm and how this enzyme is converted to an active form in the membrane. These questions will be important future issues to be addressed.
It is noteworthy that such an expression pattern of CHLαGcT is similar to that of H. pylori α1,3/4-fucosyltransferase (α1,3/4-FucT), the enzyme responsible for the biosynthesis of Lewis antigens attached to lipopolysaccharide on the cell wall (Ge et al. 1997). Interestingly, both α1,3/4-FucT and CHLαGcT lack a transmembrane domain (Ge et al. 1997; Lee et al. 2006), suggesting that both are soluble proteins. Recently, Ma et al. (2003) suggested that α1,3/4-FucT could associate with the inner membrane through C-terminal regions rich in positively charged and hydrophobic residues functioning as a membrane anchor. These data are consistent with our present observation that the active form of CHLαGcT is expressed in the cell membrane. Recently, we found that the N-terminal region of CHLαGcT, which contains hydrophobic residues, is critical for its function because the truncated form of CHLαGcT lacking the N-terminal 18 amino acid resides was functionally inactive (YI and MFukuda, personal communication, 2008). It might be possible that CHLαGcT was associated with the cell membrane via the N-terminal hydrophobic residues.
Thus, these combined results for the subcellular localization of CHLαGcT, as demonstrated by immunoelectron microscopy, and its enzyme activity, as revealed by the in vitro CHLαGcT assay, indicated that CHLαGcT was originally synthesized in the cytoplasm of H. pylori as an inactive form and then activated when it was associated with the cell membrane. It is also noted that the membrane fraction thus obtained in the present study could contain protein fractions derived from both the inner and outer membranes. Although the gold particles indicative for CHLαGcT were more frequently found on the inner membrane compared with the outer membrane, it remains to be addressed which membrane is the major one associated with the active form of CHLαGcT. Future study should address this problem.
In summary, the present study developed a CHLαGcT-specific polyclonal antibody and then demonstrated that active form of CHLαGcT was associated with the cell membrane. Because CGL is critical for H. pylori’s survival, the present study provides useful information for developing novel and potentially safe therapeutic agents targeting the active form of CHLαGcT to treat H. pylori infection in humans.
Acknowledgments
We thank Dr. Elise Lamar for editing the manuscript and Dr. Masatomo Kawakubo for helpful discussion.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by the Program for Creating University Ventures from the Japan Science and Technology Agency (to JN) and in part by NCI grants CA33000 and CA71932 from the National Institutes of Health (to MFukuda).
References
- Appels MA, Haaker H. 1988. Identification of cytoplasmic nodule-associated forms of malate dehydrogenase involved in the symbiosis between Rhizobium leguminosarum and Pisum sativum. Eur J Biochem. 171:515-522 [DOI] [PubMed] [Google Scholar]
- Bruce MG, Maaroos HI. 2008. Epidemiology of Helicobacter pylori infection. Helicobacter. 13:1-6 [DOI] [PubMed] [Google Scholar]
- Ge Z, Chan NW, Palcic MM, Taylor DE. 1997. Cloning and heterologous expression of an α1,3-fucosyltransferase gene from the gastric pathogen Helicobacter pylori. J Biol Chem. 272:21357-21363 [DOI] [PubMed] [Google Scholar]
- Hidaka E, Ota H, Hidaka H, Hayama M, Matsuzawa K, Akamatsu T, Nakayama J, Katsuyama T. 2001. Helicobacter pylori and two ultrastructurally distinct layers of gastric mucous cell mucins in the surface mucous gel layer. Gut. 49:474-480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai Y, Haque M, Yoshida T, Yokota K, Yasuda T, Oguma K. 1995. Unique cholesteryl glucosides in Helicobacter pylori: composition and structural analysis. J Bacteriol. 177:5327-5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Borén T. 1998. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 279:373-377 [DOI] [PubMed] [Google Scholar]
- Kasai H, Nadano D, Hidaka E, Higuchi K, Kawakubo M, Sato T, Nakayama J. 2003. Differential expression of ribosomal proteins in human normal and neoplastic colorectum. J Histochem Cytochem. 51:567-573 [DOI] [PubMed] [Google Scholar]
- Kawakubo M, Ito Y, Okimura Y, Kobayashi M, Sakura K, Kasama S, Fukuda M, Katsuyama T, Nakayama J. 2004. Natural antibiotic function of a human gastric mucin against Helicobacter pylori infection. Science. 305:1003-1006 [DOI] [PubMed] [Google Scholar]
- Lebrun AH, Wunder C, Hildebrand J, Churin Y, Zähringer U, Lindner B, Meyer TF, Heinz E, Warnecke D. 2006. Cloning of a cholesterol-α-glucosyltransferase from Helicobacter pylori. J Biol Chem. 281:27765-27772 [DOI] [PubMed] [Google Scholar]
- Lee H, Kobayashi M, Wang P, Nakayama J, Seeberger PH, Fukuda M. 2006. Expression cloning of cholesterol α-glucosyltransferase, a unique enzyme that can be inhibited by natural antibiotic gastric mucin O-glycans, from Helicobacter pylori. Biochem Biophys Res Commun. 349:1235-1241 [DOI] [PubMed] [Google Scholar]
- Lee H, Wang P, Hoshino H, Ito Y, Kobayashi M, Nakayama J, Seeberger PH, Fukuda M. 2008. α1,4GlcNAc-capped mucin-type O-glycan inhibits cholesterol α-glucosyltransferase from Helicobacter pylori and suppresses H. pylori growth. Glycobiology. 18:549-558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Wang G, Palcic MM, Hazes B, Taylor DE. 2003. C-terminal amino acids of Helicobacter pylori α1,3/4 fucosyltransferases determine type I and type II transfer. J Biol Chem. 278:21893-21900 [DOI] [PubMed] [Google Scholar]
- Marshall BJ, Warren JR. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1:1311-1315 [DOI] [PubMed] [Google Scholar]
- Peek RM, Jr, Blaser MJ. 2002. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2:28-37 [DOI] [PubMed] [Google Scholar]
- Pitson SM, Mendz GL, Srinivasan S, Hazell SL. 1999. The tricarboxylic acid cycle of Helicobacter pylori. Eur J Biochem. 260:258-267 [DOI] [PubMed] [Google Scholar]
- Sawaguchi A, Yao X, Forte JG, McDonald KL. 2003. Direct attachment of cell suspensions to high-pressure freezing specimen planchettes. J Microsc. 212:13-20 [DOI] [PubMed] [Google Scholar]
- van de Bovenkamp JH, Mahdavi J, Korteland-Van Male AM, Buller HA, Einerhand AW, Boren T, Dekker J. 2003. The MUC5AC glycoprotein is the primary receptor for Helicobacter pylori in the human stomach. Helicobacter. 8:521-532 [DOI] [PubMed] [Google Scholar]
- Wunder C, Churin Y, Winau F, Warnecke D, Vieth M, Lindner B, Zähringer U, Mollenkopf HJ, Heinz E, Meyer TF. 2006. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat Med. 12:1030-1038 [DOI] [PubMed] [Google Scholar]