

Abstract
Immunofluorescent staining is central to nearly all cell-based research, yet only a few fluorescent signal amplification approaches for cell staining exist, each with distinct limitations. Here, the authors present a novel, fluorescent polymerization-based amplification (FPBA) method that is shown to enable similar signal intensities as the highly sensitive, enzyme-based tyramide signal amplification (TSA) approach. Being non-enzymatic, FPBA is not expected to suffer from nonspecific staining of endogenous enzymes, as occurs with enzyme-based approaches. FPBA employs probes labeled with photopolymerization initiators, which lead to the controlled formation of fluorescent polymer films only at targeted biorecognition sites. Nuclear pore complex proteins (NPCs; in membranes), vimentin (in filaments), and von Willebrand factor (in granules) were all successfully immunostained by FPBA. Also, FPBA was demonstrated to be capable of multicolor immunostaining of multiple antigens. To assess relative sensitivity, decreasing concentrations of anti-NPC antibody were used, indicating that both FPBA and TSA stained NPC down to a 1:100,000 dilution. Nonspecific, cytoplasmic signal resulting from NPC staining was found to be reduced up to 5.5-fold in FPBA as compared to TSA, demonstrating better signal localization with FPBA. FPBA’s unique approach affords a combination of preferred attributes, including high sensitivity and specificity not otherwise available with current techniques.
Keywords: immunofluorescent cell staining, immunocytochemistry, signal amplification, polymerization, non-enzymatic
Immunofluorescent staining of cells is a central technology in cell biology that is invaluable for localization of cellular proteins, elucidating cellular functions and aiding in pathology-based disease diagnosis. To localize fluorescent signals at antigenic biorecognition sites, an organic fluorophore is typically coupled to the primary or secondary antibody. In a similar approach, a biotinylated antibody probe is used, followed by fluorescently labeled avidin, which specifically binds biotin. Although straightforward to implement, direct labeling of protein probes is particularly inadequate for detection of low-abundance antigens (Van Heusden et al. 1997). Enzymatic fluorescent signal amplification techniques that use an enzyme to deposit fluors near biorecognition sites enable significantly improved detection limits and are commercially available, yet they also have limitations. One popular enzymatic approach is tyramide signal amplification (TSA), also referred to as catalyzed reporter deposition (CARD), in which probes are coupled to horseradish peroxidase (HRP), which catalyzes the formation of short-lived fluorescently labeled tyramide radicals that rapidly react with chemical groups near the enzyme, thereby immobilizing the fluorophores. Although enabling highly sensitive detection, TSA is often hindered by loss of signal localization as the tyramide radicals diffuse away from the biorecognition site prior to immobilization (Kerstens et al. 1995; Wiedorn et al. 1999). In addition, nonspecific staining from endogenous cellular peroxidases is often difficult or impractical to eliminate completely (Hunyady et al. 1996; Ishii et al. 2004; Pavlekovic et al. 2009). Another enzymatic method, enzyme-linked fluorescence (ELF), generates a precipitating fluorescent substrate but is only available with a single fluorophore (Larison et al. 1995). As an alternative to enzymatic approaches, probe-functionalized quantum dots (QDs) have also been employed for cellular immunostaining (Wu et al. 2003). The QD approach yields an exceptionally photostable stain and also provides especially narrow emission spectra, reducing cross-talk in multiplexed staining of multiple antigens. Although QDs are brighter than organic fluorophores, they typically do not generally achieve the same increase in fluorescence intensity as achieved by enzyme-based methods (Speel et al. 1999; Wu et al. 2003).
To improve on the currently available methodologies for fluorescent cellular immunostaining, there is a need to identify new signal amplification approaches that achieve the high sensitivity and strong fluorescence intensities required for low-abundance protein detection, without suffering from diffusion-related loss of signal localization and nonspecific staining of endogenous cellular enzymes that occur with enzymatic amplification methods. Here, we present a novel non-enzymatic, polymerization-based signal amplification method for immunofluorescent staining of cells that combines the attributes of high sensitivity, good signal localization, photostable fluorescence, and multiplexed staining ability, without interference from endogenous enzymes or the need for enzymatic processes.
Harnessing the amplification inherent in radical polymerization reactions, photoinitiator molecules are coupled to biological probes such that light exposure initiates the conversion of monomer and fluorescent moieties into a highly fluorescent polymer film localized at the biorecognition site (Figure 1). Specifically, this fluorescent polymerization-based signal amplification (FPBA) method uses protein probes labeled with eosin, a photosensitizing initiator. After the unbound eosin-labeled probe is removed, a monomer solution composed of poly(ethylene glycol) diacrylate (PEGDA) monomer, fluorescent nanoparticles (NPs), vinyl pyrrolidone as a comonomer, and N-methyldiethanolamine (MDEA) as a coinitiator is applied to the sample. The surface is then irradiated with visible light, which causes eosin on the biological probe to undergo energy, charge, and electron transfer with MDEA to generate initiating MDEA radicals that propagate through the monomer to yield a crosslinked polymer network (Avens and Bowman 2009). As the crosslinked polymer network forms, the fluorescent NPs in the monomer solution become entrapped wherever polymerization has occurred. After polymerization, the surface is rinsed to remove unreacted monomer and NPs that did not become entrapped in the polymer film. In this manner, FPBA yields highly fluorescent polymer films formed only at the biorecognition sites.
Figure 1.
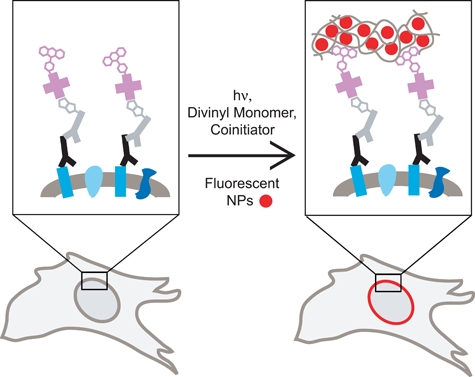
Conceptual depiction of fluorescent polymerization-based amplification (FPBA) method for immunostaining of a nuclear envelope membrane protein. Primary antibody (black) binds its target protein (blue rectangles), and a biotinylated secondary antibody probe (gray) binds primary antibody. Streptavidin coupled to eosin photoinitiators (pink) binds to biotin on the secondary antibody. Divinyl monomer, coinitiator, and fluorescent nanoparticles (NPs) are applied, and the sample is exposed to visible light to initiate polymerization. The growing polymer film entraps the fluorescent NPs, anchoring them to the biorecognition site and enabling detection.
A PEGDA-based monomer system was chosen because it is water soluble, it was found to generate films that entrap a higher density of fluorescent NPs than other formulations investigated, and it yields films that are appropriately thin so as to favor localized staining (Avens et al. 2010). Previous studies with this polymerization technique have demonstrated that although most of the initiating radicals are not surface tethered, anchoring of the polymer films to the biorecognition site occurs through a variety of methods, including the rapid, localized formation of a crosslinked polymer that is physically entangled with the surface from which it is initiated (Cruise et al. 1998), radical termination reactions that occur by combination of propagating radicals with eosin radicals (Kizilel et al. 2004), and chain transfer of propagating radicals to nearby functional groups followed by reinitiation (Avens et al. 2008). The films are not displaced during aqueous wash steps (Hansen, Sikes, Bowman 2008). Moreover, the polymer film is covalently crosslinked, rendering it resistant to chemical and physical perturbations and practically eliminating diffusion of the fluorescent NPs out of the films (Avens et al. 2010). The rate of film growth depends on the radical initiation rate, which is determined by the light intensity and the surface density of eosin initiators (Hansen, Sikes, Bowman 2008). As there is minimal attenuation of visible light through cells, the PEGDA films are expected to demonstrate uniform three-dimensional growth from cellular structures where the eosin-labeled probe has been selectively bound (Cruise et al. 1998).
Compared to individual fluorophores, the fluorescent NPs employed here for FPBA exhibit enhanced photostability (Mayr et al. 2009) and undergo fewer unwanted side reactions during the polymerization (Avens and Bowman 2010). Specifically, FPBA was performed using FluoSpheres (commercially available from Invitrogen; Carlsbad, CA), which are polystyrene NPs with carboxylate surface functionalities that have fluorophores embedded in the interior. Because the fluorophores are embedded within the polystyrene environment, they are relatively shielded from problematic side reactions such as photobleaching and nonspecific photoinitiation (Avens and Bowman 2010).
In previously published reports, polymerization-based signal amplification (PBA) in a variety of configurations has been demonstrated to be a sensitive and valuable signal amplification approach for surface-based biosensing (Lou et al. 2005; Hansen, Sikes, Bowman 2008; Hansen, Avens, et al. 2008; He et al. 2008; Sikes et al. 2008; Hansen et al. 2009; Sikes et al. 2009; Avens and Bowman 2010), although its application to biodetection and immunostaining of cells has never before been reported. Non-fluorescent PBA has been shown to enable instrument-free detection of as few as ~1000 surface-bound biomarkers (Sikes et al. 2008). In addition, in an antibody microarray format, FPBA has been demonstrated to yield a 100-fold improvement in sensitivity compared to the use of fluor-labeled streptavidin (Avens and Bowman 2010). These dramatic improvements in sensitivity associated with PBA are well suited for extension to immunofluorescent staining where, for reasons of reducing costs, improving sensitivity, decreasing nonspecific staining, and simplifying procedures, there is a strong desire for improved methodologies.
Hypothesizing that PBA would be an advantageous signal amplification approach for visualization of biorecognition events in cells, we have developed and evaluated here FPBA for immunofluorescent staining of cellular antigens in cultured cells. In the work presented here, eosin was coupled to streptavidin to create a generalized approach for detecting a variety of biotinylated targets; however, coupling eosin to a primary or secondary antibody probe is readily achievable and would enable fewer immunostaining steps. FPBA was compared to Alexa 488–labeled streptavidin (SA–Alexa 488) for the detection of vimentin, von Willebrand factor (vWF), and nuclear pore complex proteins (NPCs). The staining pattern, specificity, and resolution were evaluated and compared for each of these targets. In addition, the relative sensitivities of FPBA, TSA, and SA–Alexa 488 were evaluated in the context of NPC staining by using decreasing concentrations of primary antibody. Use of low primary antibody concentrations typically results in fewer biorecognition events, thereby facilitating a comparison of the relative sensitivities of the three staining methods. Because FPBA and SA–Alexa 488 both employed the streptavidin-biotin approach, a TSA kit was selected as a control that used HRP coupled to streptavidin (SA-HRP) in conjunction with Alexa 488–labeled tyramide. FPBA’s capability for multicolor detection of two antigens was evaluated using two sequential FPBA steps, each employing differently colored fluorescent NPs. Finally, the photostability of FPBA was evaluated and compared to the photostability of SA–Alexa 488 and FITC-labeled streptavidin (SA-FITC) staining.
Materials and Methods
Materials
Monoclonal mouse IgG1 anti-NPC (MAb414) was purchased from Covance (Princeton, NJ; catalogue #MMS-120P). Polyclonal rabbit anti-vWF was purchased from Dako (Glostrup, Denmark; catalogue #A0082). Biotinylated polyclonal goat IgG anti-mouse IgG (H+L; catalogue #BA-9200), biotinylated polyclonal goat IgG anti-rabbit IgG (H+L; catalogue #BA-1000), streptavidin fluorescein (SA-FITC), normal horse serum, and Vectashield hard set mounting medium for fluorescence were purchased from Vector Laboratories (Burlingame, CA). Denhardt’s solution (50×), SA–Alexa 488, and TSA kit with HRP-streptavidin (SA-HRP) and Alexa Fluor488 tyramide were purchased from Invitrogen (Carlsbad, CA). Hydromount was purchased from Life Science Products (Frederick, CO). Bovine serum albumin (BSA) was purchased from Fisher Scientific (Waltham, MA). Paraformaldehyde was purchased from Electron Microscopy Sciences (Hatfield, PA). Monoclonal mouse IgG1 anti-vimentin (V9; catalogue #V6389), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), 10× phosphate-buffered saline (PBS), 50× Denhardt’s solution, Triton-X, PEGDA (Mn = 575), MDEA, and 1-vinyl-2-pyrrolidinone were purchased from Sigma Aldrich (St Louis, MO). Streptavidin-eosin (SA-eosin) was prepared as described previously (Hansen, Sikes, Bowman 2008). Water was purified using a Milli-Q system. The isolation of the human endothelial colony-forming cells used in these experiments has been described previously (Baker et al. 2009). The fibroblasts were isolated from human umbilical cord outgrowth cells. The endothelial cells were positive for eNOS (610297; BD Biosciences, Franklin Lakes, NJ), CD31 (M0823; Dako), vWF-8 (A0082; Dako), VE-Cadherin (160840; Cayman, Ann Arbor, MI), and VEGFR-2 (SC504; Santa Cruz Biotechnology, Santa Cruz, CA) and displayed a cobblestone morphology. Fibroblasts were negative for these markers and displayed a spindle-like appearance.
Nanoparticles
The NPs used in this study are yellow/green FluoSpheres (20 nm diameter) and Nile red FluoSpheres (20 nm diameter) purchased from Invitrogen. They are composed of polystyrene with fluorophores embedded in the interior, and they have carboxylate surface functionalization. The yellow/green NPs have a maximum absorbance at 505 nm and a maximum emission at 515 nm, which is well matched to SA–Alexa 488 (495/519) and SA-FITC (494/518). The Nile red NPs have a maximum absorbance at 535 nm and a maximum emission at 575 nm. Both the yellow/green NPs and the Nile red NPs have diameters of 24 ± 4 nm, as indicated by Invitrogen.
Immunostaining
Cells in eight-well culture slides were washed with 1× PBS, fixed with 4% paraformaldehyde in PBS for 20 to 30 min, and then stored refrigerated in PBS. Staining was conducted 1 to 30 days postfixation with no observed difference in staining intensity or loss of antigens. Immediately prior to staining, the cells were washed with PBS, permeabilized for 5 min with 0.1% Triton-X in PBS, and then washed with PBS. Next, the cells were blocked with 2% horse serum in PBSA (0.1% BSA in PBS) for 1 hr, followed by washing with PBSA. Then, primary antibody in PBSA was applied at the specified dilution for 1 hr, followed by washing with PBSA. Next, secondary antibody in PBSA was applied at the specified dilution for 1 hr, followed by washing with PBSA. Finally, the SA species (SA-eosin, SA-FITC, or SA-HRP) was prepared at 10 µg/ml in 1× PBS and 5× Denhardt’s solution and applied for 30 min, followed by washing with PBS and finally with water. An orbital shaker was used during the washing, permeabilization, blocking, binding, and rinsing steps. All PBSA and PBS wash steps consisted of three washes of 2 min each. The water rinse consisted of a single 2-min rinse. For each staining method and target investigated, negative controls were performed in which the primary antibody was omitted, whereas all other steps were performed as previously described.
FPBA Signal Amplification
Cells treated with SA-eosin were contacted with 420 mM PEGDA, 210 mM MDEA, 35 mM 1-vinyl-2-pyrrolidinone, and 0.05 wt% fluorescent NPs in water. For the eight-well culture well slides, 100 µl monomer solution was used per well. Polymerization was initiated by a 20-min exposure to 30 mW/cm2 of light at wavelengths greater than 480 nm, followed by three water washes to remove unreacted monomer and NPs that had not been entrapped in polymer films. Argon flow was employed during polymerization to reduce ambient oxygen, which inhibits polymerization. Specifically, the slides were placed in a sealed, clear plastic bag, and a needle was used to introduce argon to the bag, with the tank pressure set to approximately 3 psi. Argon was introduced for 5 min before the light was turned on, and argon flow continued throughout the photopolymerization reaction. Investigation of other polymerization reaction times revealed that times shorter than 20 min yielded less intense staining, whereas longer polymerization times often resulted in excessive evaporation of the monomer solution, leading to nonspecific binding of the NPs to the surface. An orbital shaker was used for the washes but not during the polymerization reaction. The polymer films formed were stable to the wash steps. The light source was an Acticure (Exfo, Quebec, Canada) high-pressure mercury lamp with an in-house internal bandpass filter (350-650 nm) and an external 490-nm longpass filter (Edmund Optics; Barrington, NJ) positioned at the end of a light guide and a collimating lens. The light intensity was measured using an International Light radiometer (International Light, Peabody, MA).
TSA Signal Amplification
Cells treated with SA-HRP underwent a 10-min Alexa 488–tyramide labeling reaction following the manufacturer’s instructions. Specifically, an Alexa 488–tyramide working solution was prepared by diluting the stock solution (provided in the TSA kit) 1:100. For the eight-well culture slides, 100 µl of working solution was used per well. After the 10-min reaction, three PBS rinses of 2 min each were performed, followed by one 2-min water rinse. An orbital shaker was employed during the rinse steps but not during the labeling reaction with Alexa 488–tyramide.
Imaging
Counterstaining of the nucleus was achieved by applying 2 mg/ml DAPI in water for 3 min, followed by two rinses with water. Cells stained by SA–Alexa 488 or by TSA were mounted with Vectashield or Hydromount mounting mediums. Cells stained by FPBA were imaged without mounting medium (simply placing a cover glass on top of the dry slide). Vectashield and Hydromount were investigated for use with FPBA and were found to be incompatible, resulting in almost complete signal loss, likely as a result of swelling the polystyrene NPs, which subsequently extracted the encapsulated fluorophores. Nonetheless, appropriate images were achieved, even in the absence of any mounting medium. It was determined that water is an appropriate mounting medium for FPBA, and it was used to take the image in Figure 2C. Epifluorescence microscopy was performed with a Nikon TE 2000 using a 20× air objective (Nikon, Tokyo, Japan) and an X-Cite 120 lamp (Exfo). Confocal scanning laser microscopy (CLSM) was performed with a Zeiss LSM 510 instrument (Carl Zeiss; Oberkochen, Germany) with a 40× oil objective or 63× oil objective, as indicated.
Figure 2.
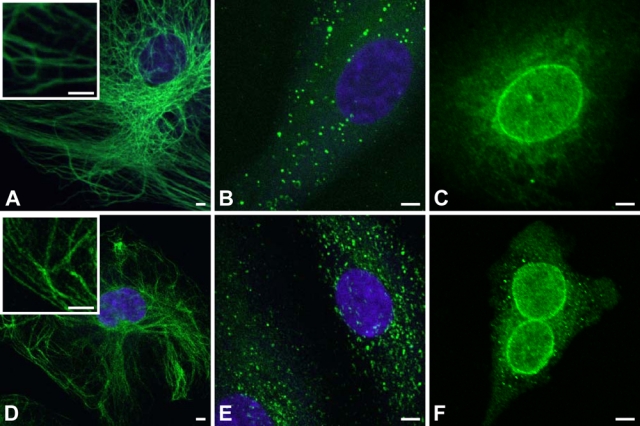
Comparison of fluorescent polymerization-based amplification (FPBA) versus SA–Alexa 488 for immunofluorescent imaging of various antigens. (A) Human endothelial cells were immunostained with mouse anti-vimentin (1:50,000), biotinylated anti-mouse secondary (1:400), and fluorescently labeled by FPBA. (B) Human endothelial cells were stained for von Willebrand factor (vWF) with rabbit anti-vWF (1:250,000), biotinylated anti-rabbit secondary (1:500), and fluorescently labeled by FPBA. (C) Human endothelial cells were immunostained with mouse anti–nuclear pore complex protein (NPC; 1:1000), biotinylated anti-mouse secondary (1:400), and fluorescently labeled by FPBA. (D-F) Same as A through C, respectively, except that the fluorescent labeling was achieved using SA–Alexa 488 rather than FPBA. Scale bar is 5 µm. Images were taken on a confocal laser scanning microscope (CLSM) using a 40× oil objective for A, B, D, and E and a 63× oil objective for C and F. The nucleus is counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; purple). DAPI staining in C and F confirmed that immunostaining is localized around the nucleus, but the DAPI stain is not depicted so as to more clearly indicate the NPC staining.
Fluorescence Quantification
Quantification of fluorescence intensities was done using ImageJ 1.40g (National Institutes of Health; Bethesda, MD). Error bars represent standard error. p values were determined for one-tailed Student’s t-test. For each staining condition quantified, measurements were made for all cells in four images obtained from at least two separate staining sessions. Background signal is defined as an average intensity from an area the size of the nucleus and far away from any cellular material. Specific NPC signal is defined as the signal that is co-localized with the DAPI nuclear staining, whereas the specific fluorescence intensity is defined as the background signal subtracted from the specific NPC signal. Nonspecific signal in cells imaged for NPC is quantified by measuring the average fluorescence in an area equal to the size of the nucleus but immediately to the left of the nucleus, and the nonspecific fluorescence intensity is defined as the background signal subtracted from the nonspecific signal. Signal-to-noise ratios are calculated as the specific fluorescence intensity divided by the nonspecific fluorescence intensity.
Double Immunostaining with FPBA
Binding reactions to stain NPC were performed as described previously, except that blocking and antibody binding steps were only 45 min rather than 1 hr, and SA-eosin was applied to the surface for only 20 min rather than 30 min. The first round of polymerization employed Nile red NPs. Immediately following the first polymerization step, the cells were blocked again, and binding reactions were performed to stain vimentin, using 45-min reaction times for the blocking and antibody binding steps and 20 min for the SA-eosin binding step. The second polymerization used yellow/green NPs. The two polymerization steps incorporated NPs of different colors to enable facile discrimination of the independent responses. Two negative controls were performed: 1) the NPC primary antibody was omitted, whereas all other steps were performed the same, and 2) alternatively, vimentin primary antibody was omitted, whereas all other steps were performed as usual. Each of the negative controls was imaged for detection of both Nile red NPs and yellow/green NPs.
Photostability
Epifluorescence microscopy was performed as above, except the excitation source was an Acticure (Exfo) high-pressure mercury lamp with an in-house internal bandpass filter (350-650 nm). This lamp is designed to achieve an exceptionally stable light intensity. The slides were continuously illuminated while images were taken at the indicated times. All images were taken without mounting medium present to ensure a valid comparison, as mounting medium can alter the photostability of the dye (Wu et al. 2003). A cover glass was placed over the dry slide.
Results
Comparison of FPBA and SA–Alexa 488 for Staining a Variety of Cellular Antigens
By generating a fluorescent film in response to biorecognition, FPBA immobilizes a significantly greater number of fluors to the surface as compared to staining with probes that are directly labeled with fluorophores; however, the generation of a polymer film with a finite thickness brings into question the spatial resolution of the stain and the types of structures that may be imaged. To verify that FPBA achieves similar staining patterns as fluor-labeled probes, staining of various antigens was performed using biotinylated secondary antibodies and either FPBA or SA–Alexa 488 to generate a fluorescent signal. SA–Alexa 488 was selected for comparison because Alexa 488 absorbance is well matched to the photoluminescent properties of the yellow/green NPs used for FPBA. Moreover, because SA-eosin is used for FPBA, SA–Alexa 488, as opposed to a fluorescent antibody, was chosen for comparisons such that both methods employ a similar streptavidin-biotin approach. Figure 2 demonstrates that the two staining methods yielded similar staining patterns and resolution for a variety of fine cellular structures, including filamentous vimentin in the cytoplasm of fibroblasts, the NPC located in the nuclear envelope, and vWF, which is present in the cytoplasm of endothelial cells, often concentrated in granules. In all targets tested, the presence of polymer did not obscure or alter any subcellular feature. For both FPBA and SA–Alexa 488, staining of vimentin yielded images in which many of the filaments were measured to be 500 nm wide. Because the fluorescent polymer is layered on top of the cellular feature that is being stained, and because film formation occurs uniformly as it moves away from the filament surface, the polymer film itself must be less than 250 nm thick. This result is consistent with previously published microarray format experiments in which films formed from surface immobilized SA-eosin under similar monomer and polymerization conditions were found to be 5 to 200 nm thick (Avens et al. 2008). In the case of NPC immunostaining, both FPBA and SA–Alexa 488 yielded a distinct ring structure around the nucleus, in which the ring was measured to be approximately 500 nm thick.
In NPC imaging, both FPBA and SA–Alexa 488 resulted in nonspecific staining around the nucleus, which is attributable to nonspecific binding of the primary antibody. For each of these staining targets, negative controls were performed in which the primary antibody was omitted and all other steps were performed as usual. Under matched imaging conditions, the negative controls showed no visible staining. In addition, it is of note that the FPBA CSLM images in Figure 2A,B were obtained without mounting medium, simply setting a cover glass on the dry slide, whereas Figure 2C used water as a mounting medium. The SA–Alexa 488 CLSM images (Figure 2D-F), on the other hand, were obtained using Hydromount mounting medium. The use of no mounting medium or water as a mounting medium is anticipated to negatively affect the fluorescence intensity and the resolution compared to the use of Hydromount. It is expected that development or identification of a more suitable mounting medium for FPBA will yield higher resolution FPBA images. Overall, these results verify that FPBA achieves similar staining patterns to those observed when using a fluor-labeled probe.
Comparison of the Relative Sensitivity of FPBA, SA–Alexa 488, and TSA
To assess the magnitude of the signal amplification afforded by FPBA, this method was directly compared to the use of a fluor-labeled probe (SA–Alexa 488) for the immunofluorescent imaging of NPC in fibroblast cells at decreasing primary antibody concentrations. Reduced primary antibody concentrations generally result in fewer biorecognition events, thereby enabling a systematic comparison of the relative sensitivities of each of the staining methodologies. Furthermore, to benchmark FPBA relative to a commercially available enzymatic signal amplification approach, NPC staining was also performed by TSA, using SA-HRP and Alexa 488–tyramide. All three staining approaches employed fluorophores that have similar excitation and emission spectra as discussed in the Methods section, such that each staining approach was equally well suited for the epifluorescence filter set that was used. In addition, all three methods employed the streptavidin- biotin approach.
Figure 3 shows the imaging results for FPBA, SA–Alexa 488, and TSA at four different anti-NPC primary antibody concentrations, using matched camera and image settings (4-sec exposure, no binning of pixels). In addition, the fluorescence intensity of NPC staining was quantified and is depicted in Figure 4A. Although a 4-sec exposure resulted in saturated fluorescence in the cells that were treated with 1:100 primary antibody dilution and stained by FPBA, for the purpose of making quantitative comparisons, the same exposure time was used in all conditions. Also, epifluorescence microscopy, as opposed to CLSM, was used for Figures 3 and 4 because of its ease of signal quantification. Epifluorescence microscopy captures fluorescence emitted from all planes of the image, readily enabling quantification of an overall fluorescence emission. On the other hand, CLSM images only a single slice of the specimen at a time, making quantification more complicated as it depends on the depth of the slices, the number of slices, and the space between the slices. Although CLSM provides optimal discrimination of in-plane versus out-of-plane fluorescence, it also leads to a greater challenge in defining a specific and nonspecific signal in three dimensions. In epifluorescent staining, it is trivial to use DAPI staining and assume the immunostaining will be co-localized. The use of co-localized immunostaining was ruled out due to concerns over potential interference with any of the three staining approaches. We believe epifluorescent imaging is the simplest approach to minimize any analytical bias.
Figure 3.
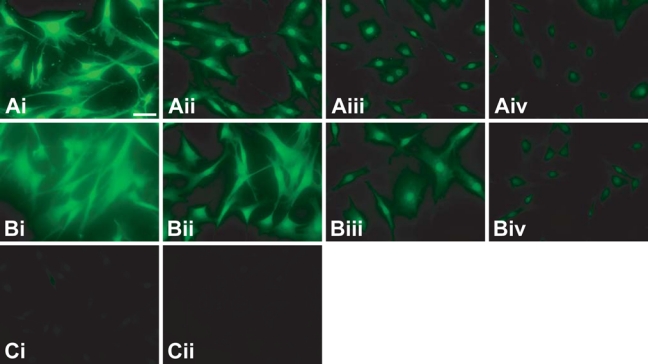
Comparison of fluorescent polymerization-based amplification (FPBA) to other detection methods for the fluorescent imaging of nuclear pore complex proteins (NPCs) in human fibroblast cells. (A) FPBA is compared to (B) tyramide signal amplification (TSA) and (C) SA–Alexa 488. Cells were stained using (i) 1:102, (ii) 1:103, (iii) 1:104, or (iv) 1:105 dilution of primary antibody. In all cases, the secondary antibody was biotinylated anti-mouse at a 1:400 dilution. These images were obtained by epifluorescence microscopy, using a 20× objective lens, a 4-sec exposure time, and no binning of pixels. The scale bar is 200 µm. DAPI (4′,6-diamidino-2-phenylindole dihydrochloride) nuclear counterstaining verified that staining is centered on the nuclei, but the DAPI stain is not depicted so as to more clearly indicate the NPC staining.
Figure 4.
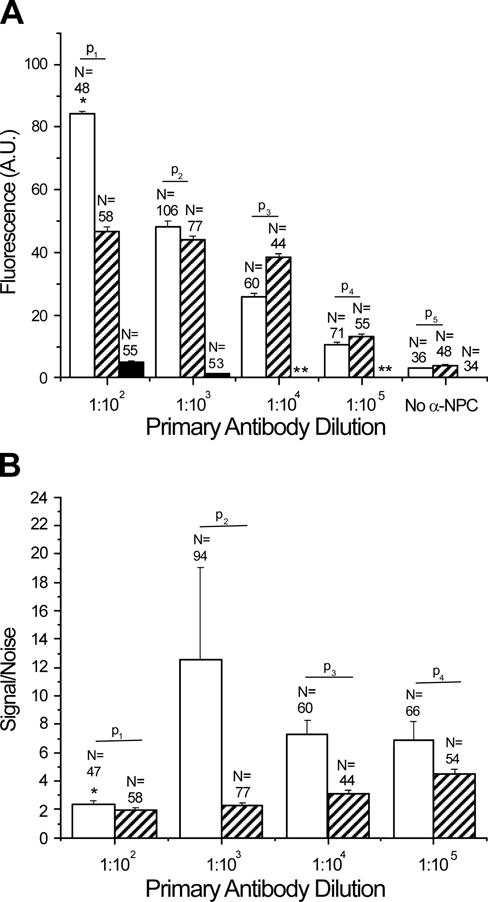
(A) Comparison of fluorescence intensity of nuclear pore complex protein (NPC) staining in human fibroblast cells using fluorescent polymerization-based amplification (FPBA; light gray), tyramide signal amplification (TSA; dark gray), or SA–Alexa 488 (black). The p-values for a one-tailed Student’s t-test between FPBA and TSA were p1 = 1.1 × 10−3, p2 = 3.4 × 10−2, p3 = 2.7 × 10−11, p4 = 1.1 × 10−3, and p5 = 1.9 × 10−1. Although p-values were determined for all data sets, it is of note that the data sets for the no-primary antibody condition were not normally distributed, nor were the FPBA data for the 1:100 antibody dilution. *Indicates that most of these signal intensities were saturated. **Indicates that these measurements were not taken. (B) Nonspecific staining (“noise”) was assessed in each cell by measuring the fluorescence in an area equal to the size of the nucleus but immediately adjacent to the nucleus. For each cell, the intensity of staining in the nucleus (“signal”) was divided by the noise value. The ratio of signal/noise was compared between FPBA (light gray) and TSA (dark gray). Although these data sets were not normally distributed, p-values for a one-tailed t-test were determined: p1 = 8.5 × 10−2, p2 = 6.0 × 10−2, p3 = 1.0 × 10−4, p4 = 3.6 × 10−3. For both A and B, measurements were taken for human fibroblast cells stained under the same staining and imaging conditions as used for the images in Figure 3. Also, for each primary antibody dilution in A and B, measurements were made for all cells in each of four images obtained from at least two separate staining sessions; however, when it was not possible to take a noise measurement immediately adjacent to the nucleus, that cell was not included in the signal/noise measurements. For A and B, intercellular background signal measured far from the cells was subtracted from the measured signal and noise intensities. DAPI (4′,6-diamidino-2-phenylindole dihydrochloride) staining was used to delineate areas that were expected to be stained positive for NPC.
At the highest NPC antibody concentration (1:100 dilution), FPBA signal was saturated, whereas SA–Alexa 488 showed no visually detectable staining, although quantification did reveal a small amount of fluorescence signal (Figures 3A,C and 4A). At 1:1000 anti-NPC dilution, the FPBA signal was 48, whereas the SA–Alexa 488 signal was only 1.2, below the camera’s linear detection range. On the basis of these measurements, it is concluded that FPBA enables a greater than 40-fold improvement in fluorescence intensity compared to SA–Alexa 488.
Comparison of FPBA with TSA revealed that both methods generated detectable NPC staining across all four primary antibody concentrations investigated. Both methods enabled NPC staining down to 1:100,000 primary antibody dilution, with fluorescence intensities of 11 ± 1 for FPBA and 13 ± 1 for TSA (Figures 3A,B and 4A). Likewise, both methods displayed a small amount of nonspecific nuclear staining in the absence of primary antibody, possibly due to nonspecific binding of secondary antibody. At the highest antibody concentration (1:100 dilution), FPBA generated saturated signals, indicating that the true average signal was actually higher than 84; in contrast, the TSA signal was only 47. Indeed, the TSA signal appeared to plateau as the primary antibody concentration was increased, whereas FPBA continued to increase with increasing primary antibody concentration. Notably, decreasing the exposure time from 4 sec to 500 msec prevented signal saturation of the FPBA-treated cells and enabled more clear visualization of the NPC staining; however, for the purpose of this comparative quantitative study, exposure times were not varied.
The TSA NPC stain in Figure 3 is observed to be less localized than the FPBA stain obtained with the same primary antibody dilution. Signal/noise calculations, as well as definitions of specific and nonspecific signals, are provided in the Materials and Methods section. FPBA was found to have signal/noise values ranging from 6.9 to 13 for dilutions greater than 1:100, whereas for the 1:100 primary antibody dilution, the positive signal was truncated due to saturation of the camera’s detector (Figure 4B). In contrast, TSA displayed signal-to-noise values ranging from 2.0 to 4.5, with the best signal/noise occurring at 1:100,000 primary antibody dilution, where specific signal was the lowest. Some of the nonspecific signal adjacent to the nucleus likely arose from nonspecific binding of the primary antibody; however, differences observed between signal/noise in FPBA and TSA were likely attributable to differences in signal localization between the two methods. The fact that the FPBA tended to have higher signal/noise suggests that FPBA yielded better signal localization than TSA. In addition, it is of note that no mounting medium was used on the FPBA slides for the data used in Figures 3 and 4, whereas Vectashield mounting medium was employed for the TSA and SA–Alexa 488 samples. As mounting medium is intended to improve signal intensity and image resolution, it is expected that identification of a suitable mounting medium for FPBA would yield images with further increased signal intensities and possibly better signal/noise as a consequence of improved image resolution.
Multicolor Staining of Two Antigens by Sequential FPBA Reactions
It was of interest to determine whether multiple antigenic targets in the same cell could be stained by sequential rounds of FPBA. Figure 5 demonstrates successful sequential immunostaining of two targets by FPBA in endothelial cells. In the first round of staining, NPC was stained by FPBA using Nile red NPs. Immediately following this first staining reaction, the cells were stained for vimentin by a second round of FPBA using yellow/green NPs. Two negative controls were performed: 1) the primary antibody against NPC was omitted (Figure 5B), and 2) the primary antibody against vimentin was omitted (Figure 5C). Each negative control was imaged for detection of both Nile red and yellow/green NPs. The successful vimentin staining depicted in Figure 5A,B verifies that the polymerization reaction is mild enough that antigenic sites are still intact and available for antibody recognition and immunostaining after the first round of FPBA. Furthermore, Figure 5A,C reveals that the polymer films from the first round of staining are not displaced or rendered nonfluorescent during the second round of staining. An additional anticipated challenge of two-round FPBA is that eosin photoinitiators remaining from the first round of polymerization might initiate polymerization during the second round, yielding nonspecific staining. Figure 5C demonstrates that no nonspecific green staining occurred around the nucleus, verifying that eosin from the previous round of FPBA did not interfere with the second FPBA detection reaction.
Figure 5.
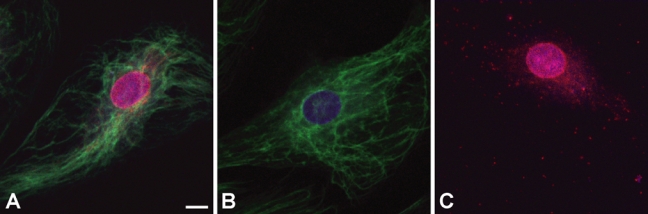
Dual-color immunostaining of two antigens was achieved by sequential fluorescent polymerization-based amplification (FPBA) reactions. (A) Human endothelial cells were immunostained for nuclear pore complex protein (NPC) using Nile red nanoparticles (NPs; red), followed by a second round of immunostaining for vimentin using yellow/green NPs (green). (B) As a negative control for NPC staining, the NPC primary antibody was omitted from the antibody dilution buffer during the first staining reaction. (C) As a negative control for vimentin staining, the vimentin primary antibody was omitted from the antibody dilution buffer during the second staining reaction. The nucleus was stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; purple). The scale bar is 10 microns. Images were taken with a 40× oil objective on confocal scanning laser microscope. Mouse anti-NPC antibody was used at 1:1000 dilution, mouse anti-vimentin antibody was used at 1:5000 dilution, and biotinylated anti-mouse antibody was used at 1:400 dilution.
Evaluation of the Photostability of Fluorescence Signals Generated by FPBA
Generally, photobleaching during composing images and adjusting acquisition settings causes the intensity of the fluorescent stain to decrease, resulting in irreproducible results and loss of weakly fluorescent signals. Because FPBA uses fluorescent NPs, which are reported to be more photostable than free fluors (Mayr et al. 2009), it is expected that FPBA will yield a more photostable fluorescent stain compared to other staining approaches. On the other hand, the fact that the photopolymerization step of FPBA requires the fluorescent NPs to experience a 20-min exposure to intense light calls into question whether the NP fluorescence will be as photostable as has been reported previously. To evaluate this question, the photostability of FPBA with yellow/green NPs was compared to staining with SA–Alexa 488 and SA-FITC in the context of imaging vimentin in fibroblast cells. All images were taken without mounting medium present to ensure a valid comparison, as mounting medium can alter the photostability of the dye (Wu et al. 2003). Alexa 488 is well known for being highly photostable, whereas FITC is generally acknowledged as being quite unstable. FPBA and SA–Alexa 488 showed similar rates of photobleaching, whereas staining with SA-FITC resulted in more rapid photobleaching (Figure 6). After 5 min of illumination, the fluorescence from FPBA and SA–Alexa 488 remained at greater than 80% of its initial value, whereas the fluorescence from SA-FITC had decreased to nearly half its original intensity. Although FPBA showed similar photobleaching rates as SA–Alexa 488, it should be noted that FPBA generated many-fold higher signal intensities than SA–Alexa 488 or SA-FITC, such that a nearly 10-fold longer exposure time was required for imaging the cells stained by the latter methods.
Figure 6.
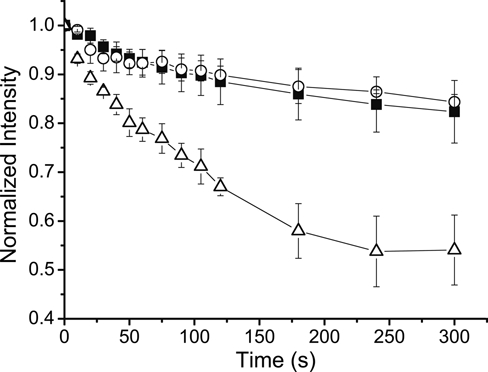
Fluorescent polymerization-based amplification (FPBA), SA–Alexa 488, and SA-FITC were compared for their photostability during imaging. Human fibroblast cells were stained for vimentin and are fluorescently labeled by either FPBA (yellow/green nanoparticles [NPs]; solid squares), SA–Alexa 488 (open circles), or SA-FITC (open triangles). The cells were continuously illuminated while images were taken at the indicated times after the illumination began (time = 0 sec). The images were obtained by epifluorescence microscopy, with binning set to 2. Exposure times: FPBA = 0.125 sec; SA–Alexa 488 and SA-FITC = 1 sec. Mouse anti-vimentin antibody was used at 1:500 dilution, and biotinylated anti-mouse antibody was used at 1:400 dilution.
Discussion
The results presented here highlight FPBA as uniquely advantageous in providing highly sensitive and specific detection accompanied by extremely strong fluorescent signals without suffering from the disadvantages associated with enzymatic amplification. FPBA was demonstrated to enable similar resolution and staining patterns as those achieved with SA–Alexa 488 (Figure 2). The observation that staining cellular features with FPBA generates films at least as thin as 250 nm is consistent with previously published microarray format experiments in which polymer films formed from surface-immobilized SA-eosin under similar conditions were found to be 5 to 200 nm thick (Avens et al. 2008).
The improvements in signal intensity and detection sensitivity afforded by FPBA relative to staining with SA–Alexa 488 are similar to those achieved by the enzymatic TSA approach (Figures 3 and 4). Both FPBA and TSA enabled use of reduced primary antibody concentrations, which are associated with a significant cost savings as primary antibodies are often rare and costly, frequently constituting the most expensive component of the staining reagents. In addition, as TSA is known to be a highly sensitive amplification technique capable of visualizing single binding events (Schmidt et al. 1997), these results suggest that FPBA will likewise prove to be suitable for detection of very low-abundance antigens. Finally, although SA–Alexa 488 staining of NPC was visualized easily using CLSM, only extremely faint staining was achieved using epifluorescence imaging. In contrast, FPBA and TSA both enabled facile epifluorescence imaging of NPC. As epifluorescence microscopy is much less expensive and more widely available than CSLM, signal amplification methods that permit the use of more affordable and accessible instrumentation are of value.
Although both FPBA and TSA are able to achieve similar fluorescence gains and sensitivities, Figures 3 and 4 indicate that FPBA tends to have better signal/noise and better signal localization. One explanation for the enhanced localization of FPBA relative to TSA is that FPBA signal amplification occurs by formation of a localized, crosslinked film at the biorecognition site, which hinders initiating radicals from diffusing far from the site of biorecognition. The TSA process, on the other hand, results in enzymatically activated tyramide–Alexa 488 molecules that are free to diffuse away from the biorecognition site before covalently attaching to cellular structures. Previously published studies have noted that TSA often requires prolonged meticulous adjustment of the primary antibody concentration or the enzyme reaction time to mitigate diffusion-related loss of signal localization; however, these measures also reduce the signal amplification, ultimately limiting the sensitivity of the TSA method (Kerstens et al. 1995; Wiedorn et al. 1999). Likewise, it is expected that shorter polymerization times may improve FPBA signal localization by generating thinner polymer films, although signal intensity would likely be reduced as well. A second advantage of FPBA is that it is non-enzymatic and therefore is not expected to be affected by endogenous enzymes. A significant problem with the TSA approach, which has been reported to be difficult and sometimes infeasible to completely eliminate, is endogenous peroxidase activity of some cell types that results in nonspecific staining (Hunyady et al. 1996; Ishii et al. 2004; Pavlekovic et al. 2009). Thus, FPBA generates similarly high levels of signal amplification as TSA yet tends to yield better stain localization and is not expected to be affected by endogenous enzymes.
The capability of FPBA to immunostain multiple targets was demonstrated by successfully staining NPC and vimentin via sequential immunostaining reactions. Negative controls confirmed that the polymerization conditions do not excessively damage antigens and that eosin from the first staining reaction does not initiate nonspecific polymerization in subsequent FPBA steps. The eosin from the first reaction was likely either consumed or trapped within the polymer film during the first photopolymerization reaction. The polymer film formed in the first round of FPBA acts as a diffusive barrier, reducing the transport of MDEA coinitiator and monomer to eosin, thereby hindering additional polymerization. It should also be noted that the capability of the fluorescent polymer film to act as a diffusion barrier may affect the second round of staining negatively. For example, after FPBA is used to stain NPC, it might become more difficult to stain antigens that are colocalized on the nuclear envelope or subnuclear antigens that require antibodies to pass through the nuclear envelope. Although this potential limitation has not yet been investigated experimentally, if it does indeed prove to be problematic, simply selecting an appropriate order of antigen staining would circumvent these complications in most instances. Combining FPBA with other fluorescent staining methods for multiantigen staining is also expected to be successful, provided that FPBA is the first method employed, as the illumination during the photopolymerization reaction may excessively photobleach fluors that are not embedded within NPs.
The photostability of FPBA staining was found to be better than SA-FITC and equal to SA–Alexa 488 while achieving dramatically brighter fluorescence intensities than either SA–Alexa 488 or SA-FITC. Although Alexa488 is well known for being an extremely photostable fluor, it is noteworthy that FPBA is not more photostable than SA–Alexa 488, as the former method uses fluorescent NPs that have been reported to be more photostable than Alexa 488 (Mayr et al. 2009). It is likely that some aspect of FPBA staining, such as the illumination during photopolymerization or a component of the monomer formulation, may negatively affect photostability during imaging, thereby counteracting any benefits associated with the fluorescent NPs. Nonetheless, it is observed that FPBA has photostability equivalent to Alexa 488 while enabling many-fold brighter fluorescence intensity. By providing both good photostability and intense fluorescence signals, FPBA facilitates reproducible imaging and helps prevent loss of weak signals during image acquisition.
In conclusion, FPBA was shown to be highly sensitive, generating a greater than 40-fold increase in fluorescent signal compared to SA–Alexa 488. Compared to the TSA enzymatic approach, FPBA appears to have similar sensitivity as both methods enable the use of similarly low primary antibody concentrations for detection of NPC. The results also suggest that FPBA improves upon TSA by achieving better signal localization. Furthermore, because FPBA is non-enzymatic, the method is not expected to be affected by endogenous enzymes as occurs with enzymatic amplification methods. Finally, FPBA provides photostability equivalent to Alexa 488 and the capability for multicolor staining of multiple cellular targets. The results presented here establish FPBA as providing a combination of advantageous attributes not currently available by any other immunofluorescent cell staining method.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This material is based on work supported by NIH R21 CA 127884 and a National Science Foundation Graduate Research Fellowship to HJA. Also, this work has been supported by the state of Colorado and the University of Colorado Technology Transfer Office.
References
- Avens HJ, Bowman CN. 2009. Mechanism of cyclic dye regeneration during eosin-sensitized photoinitiation in the presence of polymerization inhibitors. J Polym Sci A Polym Chem. 47:6083-6094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avens HJ, Bowman CN. 2010. Development of fluorescent polymerization-based amplification for sensitive and non-enzymatic biodetection in antibody microarrays. Acta Biomater. 6:83-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avens HJ, Chang EL, May AM, Berron BJ, Seedorf GJ, Balasubramaniam V, Bowman CN. 2010. Fluorescent polymeric nanocomposite films generated by surface-mediated photoinitiation of polymerization. J Nanopart Res. doi: 10.1007/s11051-010-0034-z [DOI] [Google Scholar]
- Avens HJ, Randle TJ, Bowman CN. 2008. Polymerization behavior and polymer properties of eosin-mediated surface modification reactions. Polymer. 49:4762-4768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CD, Ryan SL, Ingram DA, Seedorf GJ, Abman SH, Balasubramaniam V. 2009. Endothelial colony-forming cells from preterm infants are increased and more susceptible to hypoxia. Am J Respir Crit Care Med. 180:454-461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruise GM, Hegre OD, Scharp DS, Hubbell JA. 1998. A sensitivity study of the key: parameters in the interfacial photopolymerization of poly(ethylene glycol) diacrylate upon porcine islets. Biotechnol Bioeng. 57:655-665 [DOI] [PubMed] [Google Scholar]
- Hansen RR, Avens HJ, Shenoy R, Bowman CN. 2008. Quantitative evaluation of oligonucleotide surface concentrations using polymerization-based amplification. Anal Bioanal Chem. 392:167-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RR, Johnson LM, Bowman CN. 2009. Visual, base-specific detection of nucleic acid hybridization using polymerization-based amplification. Anal Biochem. 386:285-287 [DOI] [PubMed] [Google Scholar]
- Hansen RR, Sikes HD, Bowman CN. 2008. Visual detection of labeled oligonucleotides using visible-light-polymerization-based amplification. Biomacromolecules. 9:355-362 [DOI] [PubMed] [Google Scholar]
- He P, Zheng W, Tucker EZ, Gorman CB, He L. 2008. Reversible addition—fragmentation chain transfer polymerization in DNA biosensing. Anal Chem. 80:3633-3639 [DOI] [PubMed] [Google Scholar]
- Hunyady B, Mezey E, Pacak K, Palkovits M. 1996. Identification of endogenous peroxidase-containing cells as eosinophils in the gastrointestinal system. Histochem Cell Biol. 106:447-456 [DOI] [PubMed] [Google Scholar]
- Ishii K, Mußmann M, MacGregor BJ, Amann R. 2004. An improved fluorescence in situ hybridization protocol for the identification of bacteria and archaea in marine sediments. FEMS Microbiol Ecol. 50:203-212 [DOI] [PubMed] [Google Scholar]
- Kerstens HMJ, Poddighe PJ, Hanselaar AGJM. 1995. A novel in situ hybridization signal amplification method based on the deposition of biotinylated tyramine. J Histochem Cytochem. 43:347-352 [DOI] [PubMed] [Google Scholar]
- Kizilel S, Pérez-Luna VH, Teymour F. 2004. Photopolymerization of poly(ethylene glycol) diacrylate on eosin-functionalized surfaces. Langmuir. 20:8652-8658 [DOI] [PubMed] [Google Scholar]
- Larison KD, BreMiller R, Wells KS, Clements I, Haugland RP. 1995. Use of a new fluorogenic phosphatase substrate in immunohistochemical applications. J Histochem Cytochem. 43:77-83 [DOI] [PubMed] [Google Scholar]
- Lou X, Lewis MS, Gorman CB, He L. 2005. Detection of DNA point mutation by atom transfer radical polymerization. Anal Chem. 77:4698-4705 [DOI] [PubMed] [Google Scholar]
- Mayr T, Moser C, Klimant I. 2009. Performance of fluorescent labels in sedimentation bead arrays: a comparison study. J Fluoresc. 19:303-310 [DOI] [PubMed] [Google Scholar]
- Pavlekovic M, Schmid MC, Schmider-Poignee N, Spring S, Pilhofer M, Gaul T, Fiandaca M, Löffler FE, Jetten M, Schleifer K-H, et al. 2009. Optimization of three FISH procedures for in situ detection of anaerobic ammonium oxidizing bacteria in biological wastewater treatment. J Microbiol Methods. 78:119-126 [DOI] [PubMed] [Google Scholar]
- Schmidt BF, Choa J, Zhu Z, DeBiasio RL, Fisher G. 1997. Signal amplification in the detection of single-copy DNA and RNA by enzyme-catalyzed deposition (CARD) of the novel fluorescent reporter substrate Cy3.29-tyrmaide. J Histochem Cytochem. 45:365-373 [DOI] [PubMed] [Google Scholar]
- Sikes HD, Hansen RR, Johnson LM, Jenison R, Birks JW, Rowlen KL, Bowman CN. 2008. Using polymeric materials to generate an amplified response to molecular recognition events. Nat Mat. 7:52-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikes HD, Jenison R, Bowman CN. 2009. Antigen detection using polymerization-based amplification. Lab Chip. 9:653-656 [DOI] [PubMed] [Google Scholar]
- Speel EJM, Hopman AHN, Komminoth P. 1999. Amplification methods to increase the sensitivity of in situ hybridization: play CARD(s). J Histochem Cytochem. 47:281-288 [DOI] [PubMed] [Google Scholar]
- Van Heusden J, de Jong P, Ramaekers F, Bruwiere H, Borgers M, Smets G. 1997. Fluorescein-labeled tyramide strongly enhances the detection of low bromodeoxyuridine incorporation levels. J Histochem Cytochem. 45:315-319 [DOI] [PubMed] [Google Scholar]
- Wiedorn KH, Kühl H, Galle J, Caselitz J, Vollmer E. 1999. Comparison of in-situ hybridization, direct and indirect in-situ PCR as well as tyramide signal amplification for the detection of HPV. Histochem Cell Biol. 111:89-95 [DOI] [PubMed] [Google Scholar]
- Wu X, Liu H, Liu J, Haley KN, Treadway JA, Larson JP, Ge N, Peale F, Bruchez MP. 2003. Immunofluorescent labeling of cancer marker Her2 and other cellular targets with semiconductor quantum dots. Nat Biotechnol. 21:41-46 [DOI] [PubMed] [Google Scholar]