

Abstract
Congenital muscular dystrophy type 1A, a severe neuromuscular disease characterized by early-onset muscle weakness and degeneration, is caused by insufficient levels of laminin α2 (LAMA2) in the basal lamina surrounding muscle fibers and other cells. A better understanding of the molecular mechanisms leading to muscle loss is needed to develop therapeutic interventions for this disease. Here, the authors show that inflammation is an early feature of pathogenesis in Lama2-deficient mouse muscle, indicated by elevated expression of tenascin C in the endomysium around muscle fibers, infiltration of macrophages, and induction of the inflammatory cytokines tumor necrosis factor α (TNFα) and IL-1β. In addition, the expression of lymphatic vessel endothelial hyaluronan receptor-1 (LYVE-1), a specific marker for lymphatic vessel endothelial cells, is dramatically reduced early in Lama2-deficient muscle pathogenesis. LYVE-1 expression, which is inhibited by TNFα, is also decreased in muscles undergoing degeneration due to dystrophin deficiency and cardiotoxin damage. LYVE-1 expression thus provides a useful biomarker to monitor the onset of muscle pathogenesis, likely serving as an indicator of inflammatory signals present in muscles. Together, the data show that inflammatory pathways are activated in the earliest stages of Lama2-deficient disease progression and could play a role in early muscle degeneration.
Keywords: congenital muscular dystrophy, laminin alpha2, LYVE-1, inflammation, tenascin C, muscle
Congenital muscular dystrophy type IA (MDC1A) is a severe early-onset disease for which there is currently no cure. The mechanisms that lead to early loss of muscle function and muscle degeneration in this disease are not known. We have investigated early features of laminin α2 (Lama2)-deficient muscle pathology to identify molecular pathways that could contribute to muscle degeneration. An understanding of such pathways would allow the development of new strategies for therapeutic intervention at the earliest stages of this devastating disease.
At least 30 different types of muscular dystrophy have been identified, which are characterized by loss of muscle function due to muscle fiber degeneration, fibrosis, fat deposition, and often neurological impairment. The underlying genetic abnormalities responsible for many muscular dystrophies are known (Bushby et al. 2009). MDC1A is caused by a deficiency in the expression of LAMA2 in the basal lamina surrounding muscle fibers (Xu et al. 1994; Helbling-Leclerc et al. 1995). Laminin is a heterotrimeric protein consisting of α, β, and γ chains folded into a cross-shape configuration and is one of the more abundant proteins found in the basal lamina (Durbeej 2010). The predominant laminin isoform surrounding muscle fibers is laminin 211 (α2β1γ1, also called laminin 2 or merosin). Absence of the LAMA2 chain causes loss of muscle along with impaired motor nerve function because Schwann cells, which are responsible for nerve myelination, also require LAMA2. Key receptors for laminin 211 on muscle fibers include α-dystroglycan and α7β1 integrin. Interactions between these receptors and laminin 211 in the basal lamina are critical for postnatal muscle function and survival.
We have been studying the early pathogenesis of LAMA2-deficient muscular dystrophy using a Lama2-/- mouse model for this disease (Lama2dy-W) to gain insight into the early events leading to muscle loss. Lama2-/- muscles appear normal at birth, but apoptosis and necrosis occur between 1 and 2 weeks of age, leading to significant muscle death. Muscle regeneration occurs but is inefficient because newly regenerated fibers are unstable in the laminin-deficient environment and die through mechanisms that involve apoptosis (Vachon et al. 1996; Miyagoe et al. 1997; Kuang et al. 1999; Mukasa et al. 1999). Fibrosis becomes a prominent feature of Lama2-/- muscle. Poor regeneration leads to significant lethality within 4 weeks after birth and little survival beyond 4 months.
In one study of early Lama2-deficient mouse pathology using the 129ReJ dy/dy mouse strain, elevated expression of tenascin C (TN-C) was observed in the extracellular matrix around some muscle fibers a week after birth and was attributed to a possible delay in muscle maturation (Ringelmann et al. 1999). TN-C plays a role in regulating cell migration, tissue morphogenesis, and wound healing, and it could be important for cell migration within the diseased muscle tissue. TN-C is expressed throughout the interstitium of many tissues during embryonic development and postnatally is normally expressed in regions of mechanical stress such as tendons, ligaments, and myotendinous or osteotendinous junctions (Jarvinen et al. 2000; Chiquet-Ehrismann and Chiquet 2003; Tucker and Chiquet-Ehrismann 2009). In perinatal muscle, TN-C is expressed in the endomysium near myotendinous or osteotendinous junctions (Chiquet and Fambrough 1984). Shortly after birth, TN-C becomes restricted to the epimysium around muscle bundles throughout the length of the muscle but is not in the perimysium or endomysium surrounding individual fibers. In pathological conditions, TN-C becomes highly expressed during inflammation and tissue regeneration and serves as a good indicator of inflammation and pathological stress (Jarvinen et al. 2000; Chiquet-Ehrismann and Chiquet 2003; Tucker and Chiquet-Ehrismann 2009). Its expression in Lama2-deficient muscles thus suggests inflammatory signals are present very early in disease progression.
During inflammation, the blood and the lymphatic vascular systems become altered to promote cell migration from the blood into affected tissues and to allow cells, fluids, and degraded proteins to be removed through the lymphatic system to the lymph nodes where an immune response can be elicited. The lymphatic vasculature plays a critical role in normal tissue fluid homeostasis and in the inflammatory process and development of immunity. As reviewed recently (Cueni and Detmar 2008; Maby-El Hajjami and Petrova 2008; Tammela and Alitalo 2010), the development of a functional lymphatic vascular system is essential for survival. Mice lacking key lymphangiogenesis regulators such as VEGFR-3 and Prox1 die during embryogenesis. Enhanced sprouting of lymphatic vessels is a prominent feature of chronic inflammation and tumor pathology, promoted by the lymphangiogenic growth factor VEGF-C, which, along with other lymphangiogenic regulators such as Prox1 and the VEGF-C receptor VEGFR-3, is induced by proinflammatory cytokines tumor necrosis factor α (TNFα) and IL-1β (Ristimaki et al. 1998; Flister et al. 2010). Lymphatic vessels have not been investigated significantly in muscles undergoing degeneration or remodeling due to disease or trauma; thus, it is not known whether changes in lymphatic vessel patterning or gene expression are features of such muscle pathology.
To investigate early events in Lama2-/- muscle pathogenesis, we have used the Lama2-/- mouse model to examine the expression of TN-C along with the lymphatic vessel marker LYVE-1 (lymphatic vessel endothelial hyaluronan receptor-1) in both Lama2-/- and normal muscles. LYVE-1 is a hyaluronan (HA) receptor homologous to CD44 that is expressed only by lymphatic endothelial cells (LECs) (Jackson 2004) and by a unique population of hepatic sinusoidal vessel endothelial cells restricted to the liver (Mouta Carreira et al. 2001) and thus is a very specific marker for lymphatic vessels within muscle. LYVE-1 is rapidly degraded in LECs exposed to TNFα (Johnson et al. 2007), demonstrating its responsiveness to a proinflammatory environment.
In the current work, we show that one of the earliest pathological features of Lama2-/- mouse muscle is the dramatic reduction in expression of LYVE-1 in lymphatic capillary vessels of Lama2-/- skeletal muscles, occurring within 1 week after birth and before evidence of significant muscle degeneration. Strong expression of TN-C around some affected muscle fibers also occurs during this time, correlating with areas of macrophage infiltration. In addition, we show that the expression of the proinflammatory cytokines TNFα and IL-1β is significantly elevated during this early period of muscle pathology, which could contribute to the changes in both LYVE-1 and TN-C expression and macrophage migration. A decrease in LYVE-1 expression also occurs in muscles degenerating due to dystrophin deficiency and cardiotoxin damage. Thus, the altered pattern of LYVE-1 expression provides a useful biomarker to monitor the onset of pathogenesis during diseased-related muscle degeneration. Our data show that inflammation is a predominant early feature of Lama2-deficient mouse muscle pathogenesis and could contribute to the initial loss of muscle function in this disease.
Material and Methods
Animals
All procedures involving mice followed guidelines of the US Public Health Service Policy on Humane Care and Use of Laboratory Animals and the USDA Animal Welfare Act. Mice were maintained in a pathogen-free, temperature-controlled environment. Wild-type C57BL/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME). Lama2-deficient mice (Lama2dy-W), which have a targeted insertion of a lacZ cassette that disrupts normal Lama2 expression, were obtained from Dr Eva Engvall (Kuang et al. 1998). They have been maintained in our laboratory for more than 5 years by interbreeding heterozygous Lama2+/- animals because of early lethality of homozygous-null animals. Some mice were additionally derived from breeding Lama2+/- animals with MyoD-hBcl-2 transgenic mice (in a C57BL/6J background) that overexpress human Bcl-2 specifically in skeletal muscles (Dominov et al. 2005), but none of the mice used in this study carried the transgene. Dystrophin-deficient mdx (C57/10ScSn-Dmdmdx/J) mice or unaffected dystrophin(+) siblings, some carrying the MyoD-hBCl-2 transgene (MyoD-hBcl-2 tg(+), as noted in figures*), were generated as previously described (Girgenrath et al. 2004; Dominov et al. 2005). Of note is that the MyoD-hBcl-2 transgene does not affect muscle morphology (Dominov et al. 2005) or expression of the markers shown (Dominov, manuscript in preparation). All mice were genotyped as previously described (Dominov et al. 2005). Muscle samples were collected at various ages and immediately snap frozen on dry ice for RNA analysis. For immunohistological analyses, additional muscle samples or intact lower hindlimbs of young mice (≤2 weeks old) were embedded in OCT freezing compound (Tissue-Tek, Sakura-Finetek, Torrance, CA) then quickly frozen in isopentane chilled with liquid nitrogen.
Cardiotoxin Injury
To induce acute muscle damage, 100 µl of cardiotoxin (Calbiochem 217504, 10 µM in PBS, filter sterilized), was injected into the left tibialis anterior (TA) muscles of adult mice anesthetized with isoflurane. The right TA muscles served as uninjured controls. Mice were euthanized 5 days after injury, and the TA muscles were frozen in OCT for immunohistological analysis.
Immunostaining
Frozen muscle or hindlimb samples were cryosectioned (10-µm sections) then immunostained to analyze protein expression. For immunostaining with rat or rabbit primary antibodies, sections were fixed with ice-cold acetone or methanol for 5 min or with 2% paraformaldehyde for 10 min at room temperature, flushed briefly and then washed 5 min. with PBS + 0.1% Triton X-100 (PBS/Triton). All subsequent incubations were done at room temperature. Samples were incubated for 1 hr in blocking solution (2% bovine serum albumin fractionV, 2% horse serum, 2% goat serum, 0.1% Triton X-100 in PBS), then incubated overnight in primary antibodies. For immunostains using a mouse anti-embryonic myosin heavy chain (EmbMyHC) antibody, additional processing steps were included to reduce the level of background caused by secondary antibody reactivity with endogenous mouse immunoglobulins. Sections were fixed with 2% paraformaldehyde for 10 min at room temperature and then washed three times, 5 min each, with PBS/Triton. They were then dehydrated then rehydrated through a series of ethanol and xylene rinses (3 min each: 50%, 70%, 80%, 95%, and 100% [twice] ethanol; xylene [twice]; 95%, 70% ethanol) then PBS rinsed. Samples were then steamed in 0.01M Na citrate pH 6.0 20 min, rinsed in PBS + 0.5% Triton, then incubated for 30 min in blocking solution followed by 30 min in M.O.M. mouse Ig blocking solution (Vector Labs, Burlingame, CA), and then overnight in primary antibody. After all primary antibody incubations, samples were washed 4 times, 5 min each, in PBS/Triton, incubated with fluorescently labeled secondary antibodies along with the nuclear stain bisbenzimide (Hoechst 33258, 1 µg/ml, Invitrogen, Carlsbad, CA) for 1 hr, then washed (4 × 5 min) with PBS/Triton.
The primary antibodies used, diluted in immunostain blocking solution, were rat anti–TN-C (clone MTn-12, Sigma T3413, diluted 1/500), rabbit anti–LYVE-1 (Abcam 14917, 1/500), rat anti-CD31 (PECAM-1) (BD Pharmingen 550274, 1/2000), mouse anti-EmbMyHC (clone F1.652, Developmental Studies Hybridoma Bank, 1/5 diluted hybridoma supernatant), and Alexa Fluor 488 labeled rat anti-mouse CD11b (M1/70, BD Pharmingen, 557672, 1/500). The secondary antibodies used were Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen A11070, 1/1000), Alexa Fluor 546 goat anti-mouse Ig (Invitrogen A11018, 1/1000), and Cy3 goat anti-rat IgG (Jackson Immunoresearch 112-165-006, 1/2000). Some samples were double immunostained using two antibodies as indicated in figure legends. When this was not possible (e.g., because the optimal fixation and tissue processing varied among antibodies), neighboring sections were individually stained and aligned for comparison.
Samples were imaged using a Leica DMR microscope and imaging system with accompanying Leica IM50 Image Manager software. Adobe Photoshop was used to make minor linear adjustments in brightness and contrast to images. These adjustments were applied to the entire image, and the same adjustments were applied to each sample within an experimental set.
RNA Expression
RNA was extracted from frozen muscle tissues using TRIzol reagent (Invitrogen) and the manufacturer’s protocol. RNA was DNAse treated (Ambion DNA-Free Kit), ethanol precipitated, and then reverse transcribed (BioRad iScript cDNA Synthesis Kit) using manufacturers’ protocols. Resulting cDNAs (from 50 ng input RNA) were used in quantitative PCR reactions (triplicate reactions/sample) with specific primer and probe sets (Table 1). We used Hotmaster Taq DNA polymerase and reagents (5 PRIME) and reaction conditions of 94°C 2 min, then 50 cycles of 94°C for 20 sec., 60°C for 1 min with output read at each cycle using an Opticon II (Biorad, Hercules, CA) quantitative PCR machine. Additional reactions performed without reverse transcriptase were used to confirm that RNA samples did not contain DNA that contributed to amplified PCR products. Relative mRNA expression levels were calculated after normalizing to glyceraldehyde-3-phosphate dehydrogenase expression in each sample. RNA and body mass data were analyzed for statistical significance using unpaired t-tests and Prism 4.0c statistical analysis software (GraphPad software).
Table 1.
Primers Used in Quantitative PCR Analyses
| Gene | Primers/Probes | Sequence |
|---|---|---|
| IL-1β (Overbergh et al. 1999) | Forward primer (5’- 3’) | CAACCAACAAGTGATATTCTCCATG |
| Reverse primer (5’ – 3’) | GATCCACACTCTCCAGCTGCA | |
| Probe (5’ Fam- 3’ Tamra) | CTGTGTAATGAAAGACGGCACACCCACC | |
| Tumor necrosis factor α (El-Karef et al. 2007) | Forward primer (5’- 3’) | CATCTTCTCAAAATTCGAGTGACAA |
| Reverse primer (5’ – 3’) | TGGGAGTAGACAAGGTACAACCC | |
| Probe (5’ Fam- 3’ Tamra) | CACGTCGTAGCAAACCACCAAGTGGA | |
| Tenascin C (El-Karef et al. 2007) | Forward primer (5’- 3’) | TCAAGGAAGTCATTGTGGGGC |
| Reverse primer (5’ – 3’) | CAGGAGTCCAATTGTTGTGAAG | |
| Probe (5’ Fam- 3’ Tamra) | CACCCACTACTCAGCAAGGATCCAGG | |
| Glyceraldehyde-3-phosphate dehydrogenase | Forward primer (5’- 3’) | TGTGTCCGTCGTGGATCTGA |
| Reverse primer (5’ – 3’) | CCTGCTTCACCACCTTCTTGA | |
| Probe (5’ Fam- 3’ Tamra) | CCTGGAGAAACCTGCCAAGTATGA |
Results
Early Pathology of Lama2-/- Mice
At birth, Lama2-/- mice appear normal, having the same body mass and relative activity level as that of wild-type or heterozygous Lama2+/- siblings (Figure 1A). Although some newborn death has been observed, the overall pup survival frequencies (number of Lama2 (+/+): (+/-): (-/-) pups) observed at 1 day (27: 37: 24) and 7 days (12: 35: 14) of age (separate mouse cohorts at each age group) indicate that survival of knockout mice is similar to that of wild-types as predicted for heterozygous breeding (1: 2: 1). By 7 days of age, Lama2-null mice can be distinguished from normal siblings by their smaller size, as evident by a statistically significant difference in body mass (Figure 1A). No obvious differences in motility or behavior are typically evident until after 2 to 3 weeks of age when symptoms of hindlimb weakness become apparent and mortality increases. The earliest features of abnormal muscle function are inward contraction and slight tremors of hindlimbs when animals are suspended, followed by hindlimb fatigue and dragging at later ages, and then finally paralysis and rigid contractures that typically occur in animals surviving beyond 8 weeks of age.
Figure 1.
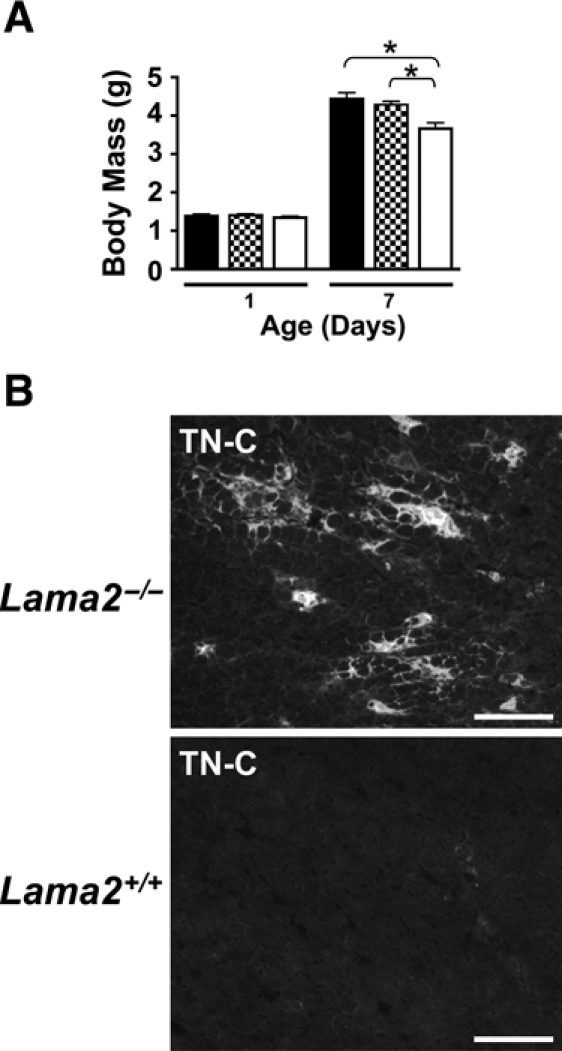
Pathological features are evident in Lama2-deficient mice within 7 days after birth. (A) Lama2-/- mice are of normal size at birth but are smaller than are Lama2-expressing siblings by 7 days of age. Body mass at 1 and 7 days of age is shown for wild-type Lama2+/+ (solid bars), Lama2+/- (shaded bars), and Lama2-/- (white bars) mice. The two age groups represent two separate cohorts of mice, n≥12 mice for each data point (see text). Data shown as mean ± SE, *p<0.002. (B) Tenascin-C (TN-C), a marker of inflammation, is dramatically upregulated in Lama2-/- quadriceps muscle at 7 days of age. Muscles immunostained for TN-C expression show deposition of TN-C in the endomysium surrounding muscle fibers in patches or foci within the muscle, including regions where loss of muscle integrity is not apparent morphologically. There is no TN-C in endomysium of wild-type muscle fibers at this age (scale bars=100 µm).
At the histological level, muscle pathology in Lama2-/- mice is evident by the expression of TN-C in the endomysium that surrounds muscle fibers, as observed in 7-day-old Lama2-/- muscles (Figure 1B). Consistent with others who used a different Lama2-/- mouse strain (Ringelmann et al. 1999), we observe that TN-C expression typically occurs in focal regions throughout muscles that morphologically appear similar to wild-type. At 1 day of age, TN-C expression extending from myotendinous and osteotendinous junctions into the endomysium is found in both wild-type and Lama2-/- mice, this being somewhat more extensive in the mutant mice (not shown, and Ringelmann et al. 1999). At 7 days of age, wild-type and many of the Lama2-/- muscles express TN-C only in the epimysium around muscle bundles but not in the perimysium or endomysium surrounding individual fibers. The foci of TN-C expression occur in a number of Lama2-/- muscles at 7 days, with higher levels of expression often found in TA, extensor digitorum longus (EDL), and superficial regions of gastrocnemius muscles, and lower prevalence of expression in soleus and other muscles of the lower limb. By 2 weeks of age, the expression of TN-C in Lama2-/- muscle is more widespread, affects more muscle types, and is associated with significant necrotic muscle cell death occurring at this time.
LYVE-1 Expression in Lymphatic Vessels of Normal Muscle Tissue
Pathological conditions often lead to changes in the lymphatic system, including increased lymphatic vessel sprouting in regions of chronic inflammation (Cueni and Detmar 2008; Maby-El Hajjami and Petrova 2008). We sought to determine whether there were changes in muscle lymphatic vessels due to Lama2 deficiency. We first examined muscles with normal Lama2 expression using a double antibody immunostaining protocol and antibodies against the LEC protein LYVE-1, along with antibodies to CD31 (PECAM-1), a pan-endothelial marker expressed on blood vascular endothelial cells and at very low levels on normal adult LECs (Kriehuber et al. 2001). As shown in Figure 2A, LYVE-1 was expressed in the lymphatic capillary vessels interspersed among muscle fibers throughout the muscle tissue. As expected, these vessels expressed very low levels of CD31, and in some cases this was not detectable, particularly in older mice. Blood vessels expressing high levels of CD31 but no LYVE-1 were more prevalent among the muscle fibers, often near lymphatic vessels, consistent with previously observed vascular patterns in skeletal muscle (Kivela et al. 2007). Some larger trunk lymphatic vessels expressed both LYVE-1 and higher levels of CD31 (arrows).
Figure 2.
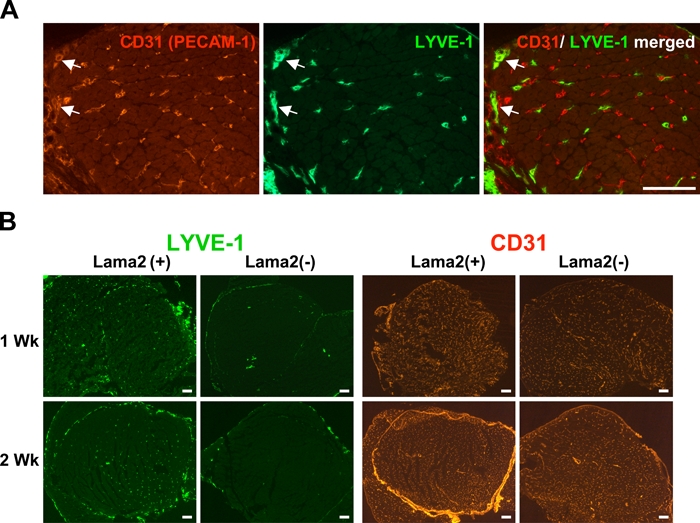
Lymphatic vessel endothelial hyaluronan receptor–1 (LYVE-1) is specifically expressed in lymphatic vessels and is dramatically reduced in vessels of Lama2-deficient muscle. (A) Lymphatic vessels and blood vessels within skeletal muscle tissue can be distinguished by the expression of LYVE-1 and CD31 (PECAM-1). Shown are quadriceps muscles from a 3-day-old Lama2-expressing mouse double immunostained for LYVE-1 and CD31. The hyaluronan receptor LYVE-1 (green) is specifically expressed in lymphatic vessel endothelial cells. CD31, a pan-endothelial marker, is expressed by vascular endothelial cells found in blood capillary vessels (red) but is only expressed at low levels in lymphatic vessel endothelial cells and is often not detectable except in larger trunk lymphatic vessels where expression of both markers can be evident (arrows; scale bar=100 µm). (B) LYVE-1 expression in lymphatic vessels is dramatically reduced in 1- and 2-week-old Lama2(-) muscles compared with those of normal Lama2(+) mice. Quadriceps muscles from Lama2(-) (Lama2-/-) and Lama2(+) (Lama+/+ or +/-) mice were immunostained using antibodies for LYVE-1 (green) or CD31 (red). The pattern of CD31 expression in blood vessels is not affected by Lama2 deficiency (scale bars=100 µm).
LYVE-1 Expression Is Reduced during Early Stages of Lama2-deficient Pathogenesis
We next examined LYVE-1 expression in muscles from Lama2-deficient mice to determine whether early muscle pathology includes changes in lymphatic vessels. Muscles from Lama2-/- mice and siblings expressing Lama2 (Lama2+/- or +/+) at 1 and 2 weeks of age were immunostained for LYVE-1 and CD31 expression. At both ages, LYVE-1 expression was dramatically reduced in lymphatic vessels of Lama2-/- muscles compared with Lama2+/- or +/+ controls (Figure 2B). Muscles from Lama2-expressing mice had numerous LYVE-1(+) small lymphatic capillaries distributed throughout the tissue along with some larger LYVE-1(+) vessels. In contrast, Lama2-/- muscles often had few, if any, LYVE-1(+) small vessels, with LYVE-1 expression limited to a few larger vessel structures. In a manner similar to TN-C expression, the extent of LYVE-1 loss varied among different leg muscles examined at 1 week of age, such that some muscles had normal staining patterns, whereas others (i.e., TA, EDL, superficial gastrocnemius) were typically more severely affected. By 2 weeks of age, most of the leg muscles examined exhibited reduced LYVE-1 staining. In contrast, there was no difference in the expression of CD31 in blood endothelial vessels among different mice (Figure 2B).
Although LYVE-1 expression was lost in Lama2-/- muscles, there was no evidence that there was a complete loss of lymphatic vessels in these tissues. Lama2-/- mice did not exhibit evidence of severe edema, which typically accompanies a loss of lymphatic vessel development and function (Maby-El Hajjami and Petrova 2008). Immunostaining 7-day-old muscles with antibodies for additional LEC markers including the mucin-type transmembrane glycoprotein podoplanin and the transcription factor Prox1 indicated that these continued to be expressed within Lama2-/- muscle tissue in regions where LYVE-1 was downregulated (Dominov, manuscript in preparation). Thus, although LYVE-1 expression was dramatically decreased in early stages of Lama2-/- muscle pathology, at a point before significant muscle degeneration, other lymphatic vessel features and function did not appear to be affected at this early stage by the absence of Lama2.
Loss of LYVE-1 in Lama2-deficient Muscle Is More Widespread Than Is TN-C Expression in Early Pathogenesis
Immunostaining for both LYVE-1 and TN-C revealed that while TN-C appeared in patches within 1-week-old muscles, the loss of LYVE-1 expression was more widespread, generally occurring throughout individual muscles before other obvious signs of pathology (Figures 3 and 4). In some instances, muscles that had no TN-C expression and thus appeared normal nevertheless contained no LYVE-1(+) lymphatic vessels (arrowheads, Figure 3). These results suggested that downregulation of LYVE-1 expression within LECs is a feature of Lama2-/- pathology that precedes the TN-C expression around fibers that increases and affects more muscles as pathogenesis progresses. The absence of LYVE-1 expression thus serves as an early pathological feature of this disease. At 7 days, there were still some Lama2-/- muscles that had LYVE-1 and TN-C expression patterns similar to normal Lama2+/- or +/+ muscles (open arrows, Figure 3), but by 2 weeks of age, TN-C expression and loss of LYVE-1 among Lama2-/- muscles were more uniformly co-localized and widespread (not shown).
Figure 3.
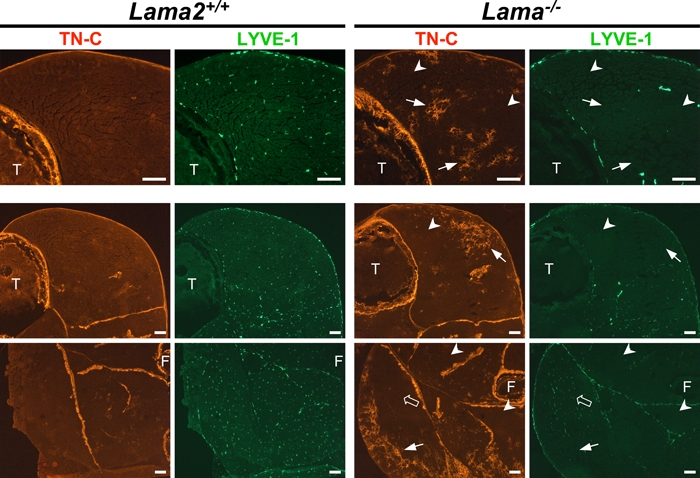
The loss of lymphatic vessel endothelial hyaluronan receptor–1 (LYVE-1) expression in lymphatic endothelial cells is more widespread than is tenascin C (TN-C) expression in Lama2-deficient muscles. Shown are 7-day-old hindlimb muscles from Lama2-/- and Lama+/+ mice double immunostained for TN-C (red) and LYVE-1 (green) expression. The top row of images is at a higher magnification than that of the lower two rows. The high- and low-magnification images are from two different Lama2-/- mice. TN-C is normally expressed in epimysium around large groups of muscles and at sites of tension or stress (e.g., tendons, myotendinous, osteotendinous junctions). In Lama2-/- muscles, TN-C is upregulated in the endomysium around foci of muscle fibers (small arrows). The absence of LYVE-1 in lymphatic vessels of Lama2-/- muscles is more widespread at this age, often observed in muscles where there is no TN-C expression (arrowheads). Some Lama2-/- muscles at this age continue to have normal patterns of LYVE-1 and TN-C expression (open arrows; scale bars=100 µm). F, fibula; T, tibia.
Figure 4.
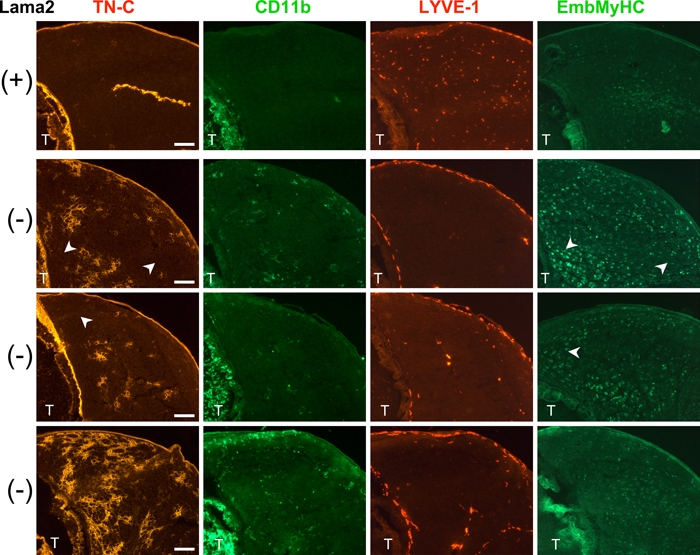
Macrophage infiltration occurs in regions of 7-day-old Lama2-/- muscles where tenascin C (TN-C) is expressed, whereas elevated embryonic myosin heavy chain (EmbMyHC) expression is not restricted to TN-C expressing areas. Shown are the tibialis anterior regions of 7-day-old hindlimbs from a Lama+/+ mouse (+, top row) and three individual Lama2-/- mice (-, three lower rows) immunostained for the proteins indicated. Neighboring tissue sections were immunostained for expression of TN-C (red), double immunostained for the macrophage marker CD11b (green) and lymphatic vessel endothelial hyaluronan receptor–1 (LYVE-1, red), or for EmbMyHC (green). Variability in the expression of these pathology markers is evident among the mutant animals, but in each case, Lama2-/- muscle tissue exhibited localized expression of TN-C that correlated with the presence of CD11b macrophages, and there was an absence of LYVE-1 expression throughout the muscles. EmbMyHC, which is normally downregulated after birth, was low but detectable in some regions of normal muscle but remained more highly expressed in mutant muscles, and this was not restricted to muscle areas expressing TN-C (arrowheads; scale bars=100 µm). T, tibia.
Macrophage Infiltration Is Associated with Foci of TN-C Expression in Lama2-/- Muscle
TN-C expression is elevated in pathological conditions and is used as an indicator of inflammation within tissues. However, elevated levels of TN-C are present in 1-day-old Lama2-/- muscles compared with normal Lama2-expressing siblings (not shown and Ringelmann et al. 1999), and Ringelmann et al. suggested that this and elevated TN-C in foci around 7-day-old 129ReJ dy/dy muscle fibers might be a consequence of delayed muscle maturation during which TN-C normally becomes restricted to the epimysium and is not in endomysium around fibers. This was supported by work showing prolonged expression of neonatal or embryonic myosin heavy chain in 129ReJ dy/dy muscles at 10 days after birth compared with normal siblings (Reggiani et al. 1992).
To determine whether TN-C expression in 7-day-old Lama2-/- muscles was due to inflammation or delayed muscle maturation, we immunostained muscle tissues with antibodies to the macrophage marker CD11b to evaluate macrophage infiltration, along with TN-C and LYVE-1. In addition, we used antibodies for EmbMyHC as an indicator of muscle maturation, as this protein is highly expressed at birth but is replaced by mature MHC isoforms within 1 to 2 weeks after birth. As shown in Figure 4, there was no TN-C surrounding 7-day-old wild-type muscle fibers and no evidence of macrophage infiltration. LYVE-1(+) lymphatic endothelial vessels were present throughout the muscle, and there is a low level of residual EmbMyHC expression within some muscle fibers, similar to that previously described for 10-day-old 129ReJ dy/dy muscles (Reggiani et al. 1992). In 7-day-old Lama2-/- mice, however, CD11b(+) macrophages were present and tended to be localized in or near regions of TN-C expression (Figure 4). LYVE-1 was absent throughout most of these muscles. EmbMyHC expression was variable among animals (as was TN-C expression) but was generally higher in Lama2-/- mice than in wild-type. This EmbMyHC expression was not localized only to areas of TN-C expression but was throughout the muscle, even in areas with no detectable TN-C (Figure 4, arrowheads). In occasional areas with extensive TN-C expression and morphological evidence of fiber disruption, small intensely stained EmbMyHC(+) cells were observed, suggesting the presence of nascent regenerating fibers, which become prevalent in mutant mice by 2 weeks of age (not shown). The co-localization of TN-C and macrophages indicated that TN-C was expressed as part of an inflammatory process in these muscles and not due only to maturation defects.
Inflammatory Cytokine Expression Is Elevated during Early Stages of Lama2-deficient Muscle Pathogenesis
We used quantitative RT-PCR to determine whether inflammatory cytokine expression was elevated during the early stages of Lama2-/- muscle pathology. As shown in Figure 5, we found that both TNFα and IL-1β mRNA levels, along with TN-C mRNA levels, were significantly elevated in 1-week-old Lama2-/- muscles compared with unaffected age-matched siblings. These data reveal that inflammatory signaling is a very early feature of Lama2-/- disease progression that could contribute to muscle degeneration.
Figure 5.
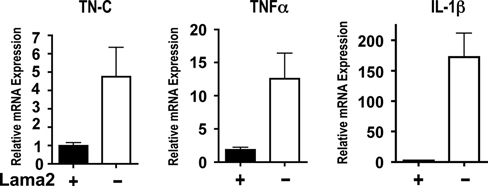
The expression of tenascin C (TN-C), tumor necrosis factor α and IL-1β mRNAs are upregulated in Lama2-/- tibialis anterior muscles at 7 days of age. Quantitative PCR methods were used to measure the relative levels of mRNA in 7-day-old muscle of the indicated genotypes. The relative expression levels of each gene were normalized to glyceraldehyde-3-phosphate dehydrogenase expression and relative expression calculated in relationship to one of the wild-type samples. In each case, there was a significant (p<0.04, n=3 Lama2(-), n=4 Lama2(+)) increase in transcript levels in Lama2-deficient muscles compared with Lama2-expressing siblings. Data shown as mean ± SE.
LYVE-1 Is Downregulated in Other Diseased and Damaged Muscles
We examined additional muscle samples to determine whether the loss of LYVE-1 expression was a unique feature of Lama2-/- muscles or was common in other muscle pathologies. We immunostained limb muscles from dystrophin-deficient mdx mice, which undergo severe apoptotic and necrotic muscle degeneration between 3 and 4 weeks of age due to the absence of dystrophin and its associated proteins at the sarcolemma. LYVE-1 expression appeared fairly normal in dystrophin-deficient limb muscle tissues at 3 weeks of age, a point when muscle degeneration is just beginning to occur (Figures 6A and B). However, at 4 weeks of age, a point where significant muscle degeneration, accompanied by necrosis and infiltration of inflammatory cells had occurred, the expression of LYVE-1 was significantly reduced in lymphatic vessel capillaries (Figures 6C and D). Similarly, in an acute muscle damage model that also elicits inflammation, LYVE-1 expression was much reduced in lymphatic capillaries of muscles after degeneration induced by cardiotoxin injection (Figure 6E), coincident with TN-C expression (Figure 6F). Thus, loss of LYVE-1 in the lymphatic vasculature was not restricted to Lama2-deficient muscles but also occurred in other muscle pathologies characterized by muscle degeneration and tissue inflammation.
Figure 6.
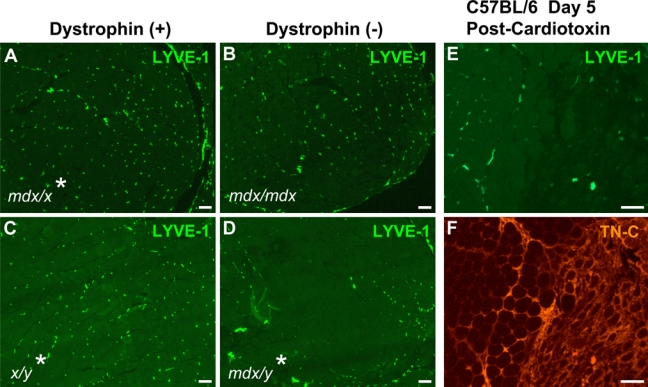
Lymphatic vessel endothelial hyaluronan receptor–1 (LYVE-1) expression is reduced in lymphatic vessels of dystrophin-deficient mdx muscles and normal muscles regenerating after cardiotoxin injury. Quadriceps muscles from 3-week-old (A, B) or 4-week-old (C, D) mice were immunostained for LYVE-1 expression. At 3 weeks, LYVE-1 expression in lymphatic vessels of affected mdx muscle (B) appeared similar to that of unaffected sibling muscle (A). At 4 weeks, mdx muscle undergoing degeneration and regeneration (D) had reduced LYVE-1 expression compared with unaffected sibling muscle (C). Regenerating tibialis anterior muscle of normal C57BL/6 mice 5 days after cardiotoxin injury exhibited reduced LYVE-1 (E) and elevated tenascin C levels (F) in regions of tissue damage and regeneration (double immunostained sample). *MyoD-hBcl-2 tg(+). (Scale bars=100 µm.)
Discussion
Defining the early events in disease progression could identify molecular mechanisms contributing to pathology and lead to new therapeutic strategies. In this study, we have shown that inflammatory cytokines are expressed very early in the pathogenesis of Lama2-deficient mouse muscle, before overt muscle degeneration. TN-C, which is induced during inflammation and tissue remodeling, is expressed within a week after birth along with macrophage infiltration in these regions. LYVE-1, an HA receptor specifically expressed on lymphatic vessel endothelial cells and inhibited by TNFα, is dramatically reduced in vessels within Lama2-/- muscles at this age. The early loss of LYVE-1 expression in Lama2-/- muscles is more widespread than is TN-C expression, affecting a greater area within individual muscles. This suggests that LYVE-1 loss precedes the increase in TN-C expression, which affects an increasing proportion of muscles as pathology progresses during 1 to 2 weeks after birth. LYVE-1 expression is also decreased in other muscle pathologies, including degenerating mdx muscle and muscle damaged by cardiotoxin treatment. LYVE-1 expression therefore provides a sensitive biomarker to monitor active inflammatory or other cues that are present within tissues before morphological signs of cellular damage. Our data suggest that molecular pathways associated with inflammation could contribute to the initial degeneration of Lama2-/- muscle.
The Lama2dy-W mouse model for MDC1A along with other Lama2-deficient strains have been widely used to study features of Lama2-deficient muscle pathology, including muscle apoptosis, necrosis, regeneration, and various molecular and pharmacological interventions that can affect these. Most studies have evaluated later stages of pathology in mice 3 to 4 weeks of age and beyond (Kuang et al. 1998; Kuang et al. 1999; Moll et al. 2001; Girgenrath et al. 2004; Bentzinger et al. 2005; Dominov et al. 2005; Li et al. 2005; Qiao et al. 2005; Gawlik et al. 2006; van Lunteren et al. 2006; Meinen et al. 2007; Erb et al. 2009; Girgenrath et al. 2009). By 4 weeks of age in this model, significant muscle degeneration has occurred, apoptosis of regenerating fibers has led to poor regeneration, and fibrosis has become a prominent feature. Most studies have thus focused on these later aspects of disease progression and have not investigated the molecular events involved in the initial loss of muscle in this disease. The molecular mechanisms that contribute to initial Lama2-/- muscle degeneration are not known and could be distinct from mechanisms involved in later disease stages where significant tissue remodeling has occurred and cues from the extracellular matrix could be different. A better understanding of early pathogenesis would be helpful in developing new therapeutic approaches to treat MDC1A (or other diseases) in the earliest stages of disease progression.
A few studies have analyzed the initial features of Lama2-/- muscle pathology. Initial muscle degeneration in Lama2-null was observed between 9 and 11 days after birth, with apoptosis evident in muscle tissues by 11 to 14 days of age (Miyagoe et al. 1997; Mukasa et al. 1999). In neonatal mice, Ringelmann et al. (1999) noted elevated expression of laminin α5, fibronectin, TN-C, VCAM-1, and ICAM-1 in Lama2-deficient muscles 1 day after birth compared with normal siblings, which they suggested could be due to delayed muscle development because these proteins are normally expressed during embryogenesis. By 7 days of age, they found only TN-C and, to a lesser extent, fibronectin were elevated in foci within the Lama2-deficient muscles, which was attributed to a possible continuation of fiber formation or maturation delay because there was no evidence of fiber degeneration at this age, and others have shown a delay in the normal developmental loss of neonatal or embryonic MHC at 10 days in 129ReJ dy/dy muscle (Reggiani et al. 1992). Our results are consistent with these studies in that we found foci of TN-C expression around Lama2-/- muscle fibers along with a higher prevalence of fibers that express EmbMyHC relative to Lama2+/+ or +/- muscles of the same age. In addition, however, we have found that macrophage infiltration, an indicator of an active inflammatory response, occurs in regions of 7-day-old Lama2-/- muscles where TN-C is expressed, with few if any macrophages in areas where TN-C is undetectable. TN-C is strongly induced by inflammatory cytokines and factors associated with inflammation or pathological stress and serves as a pathological indicator when upregulated around muscle fibers (Muller-Felber et al. 1998; Jarvinen et al. 2000; Chiquet-Ehrismann and Chiquet 2003; Tucker and Chiquet-Ehrismann 2009). In contrast, elevated EmbMyHC expression was not restricted to TN-C expressing regions but rather was observed to varying degrees in a more widespread pattern throughout the muscles at this age. These data, along with our observations of elevated TNFα and IL-1β expression, support the conclusion that the foci of TN-C expression in Lama2-/- muscle within a week after birth reflects the activation of inflammatory pathways in these muscles, occurring before a stage of significant muscle apoptosis or degeneration (Miyagoe et al. 1997), and is not due to a developmental delay alone.
In addition to the aberrant expression of TN-C, we have shown that LYVE-1 expression by LECs is dramatically reduced within Lama2-/- muscles very early in disease progression. Lymphatic vessel capillaries are normally present throughout muscle tissue, as in other tissues, where they play a critical role in maintenance of fluid balance and clearance of cells, degraded proteins, and lipids from normal as well as diseased or damaged tissues (Cueni and Detmar 2008; Maby-El Hajjami and Petrova 2008). Although LYVE-1 was absent from LECs, there was no evidence that lymphatic function was affected in young Lama2-/- muscles because we did not observe significant edema, which is typical of abnormal lymphatic vasculature, and additional lymphatic vessel markers such as podoplanin and Prox1 continued to be expressed.
The HA receptor LYVE-1 serves as a very specific marker for lymphatic vessels because it is only expressed by the LECs and by a unique population of hepatic sinusoidal vessels endothelial cells restricted to the liver (Mouta Carreira et al. 2001; Jackson 2004). The glycosaminoglycan HA plays an important role in normal tissues as well as in modulating pathogenesis (Jackson 2009). HA is ubiquitously expressed in the extracellular matrix of tissues where it interacts with numerous proteins to form a hygroscopic matrix important for cell adhesion and migration. High molecular weight forms of HA can have a tissue protective function and can be anti-inflammatory, whereas lower molecular weight cleaved forms can induce the expression of proinflammatory cytokines and chemokines by blood and endothelial cells. The turnover rate of HA is high in some tissues, and its uptake and removal through the lymphatic vasculature are important for healthy tissue homeostasis.
LYVE-1 is normally heavily sialylated, blocking its ability to bind to HA, but neuraminidases can unmask LYVE-1 sites allowing HA binding to occur (Nightingale et al. 2009). In addition, and of particular importance to our studies, LYVE-1 becomes rapidly internalized and degraded when LECs are exposed to TNFα (Johnson et al. 2007). In contrast, another LEC marker, podoplanin, is not affected by TNFα in these cells (Johnson et al. 2007). We have shown that the proinflammatory cytokines TNFα and IL-1β are both upregulated within 7 days of birth in Lama2-/- muscles. Therefore, the loss of LYVE-1 in these muscles is likely due to the expression of these proinflammatory cytokines soon after birth. Our results are consistent with a recent observation that LYVE-1 is lost from hepatic sinusoidal vessel endothelial cells during inflammation (Arimoto et al. 2010). This tight regulation suggests that LYVE-1 provides a specific function, yet to be determined, that is modulated by inflammation. Although LYVE-1 is present on LECs and its function is tightly regulated by TNFα and sialylation, studies of LYVE-1 and CD44 (a homologous HA receptor) knockout mice indicate that LYVE-1 is not essential for LEC function (Gale et al. 2007; Luong et al. 2009); thus, a complete understanding of its role has yet to be determined. The restricted expression of LYVE-1 provides a valuable tool, however, to study LECs both in vivo and in vitro.
The loss of LYVE-1 from LECs was also observed in 4-week-old mdx muscles and wild-type muscles undergoing regeneration after cardiotoxin injury. Therefore, the loss of LYVE-1 was not limited to Lama2-deficient muscles and did not require the absence of Lama2 in the extracellular environment around lymphatic vessels. LYVE-1 loss was most likely due to proinflammatory cytokines or other aspects of the proinflammatory environment that have been extensively studied and are known to be present in mdx and damaged muscles (Lundberg et al. 1995; Evans et al. 2009; Tidball and Villalta 2010). Additional studies of mdx and Lama2-/- muscles at different ages are necessary to determine if disease-specific mechanisms that underlie pathogenesis lead to differences in the temporal progression of LYVE-1 changes in LECs.
Our observation that inflammation is among the earliest features of Lama2-/- mouse pathology is consistent with studies of early human MDC1A pathology. In MDC1A, acute inflammation reminiscent of an inflammatory myopathy has been observed in patients between 2 and 6 months of age, with lymphocyte infiltration and evidence of complement activation on muscle fibers that could contribute to fiber necrosis (Pegoraro et al. 1996; Mrak 1998; Hayashi et al. 2001). This inflammation is transient, however, as later muscle biopsies of the same patients or other patients (e.g., 9+ months) exhibited less inflammation and more features of degenerated and regenerated muscle, including centrally nucleated fibers, fiber size variation, fibrosis, and fat accumulation (Pegoraro et al. 1996; Mrak 1998; Hayashi et al. 2001). Thus, in both MDC1A and mouse models of this disease, early pathology differs from later pathology, and the mechanisms that lead to muscle degeneration could vary at different disease stages.
It is not clear how the absence of LAMA2 in the basement membrane leads to the induction of inflammatory signals in muscle tissue or if these signals have a direct impact on early muscle degeneration. Nor is it clear why some muscles, such as the TA, EDL, and superficial gastrocnemius, tend to exhibit this inflammatory pathology earlier than do others (e.g., soleus) in this model, although eventually all of the limb muscles examined exhibit some pathology. Differences in muscle activity as neonatal mice become active could contribute to these differences, or there could be differences in fiber-type physiology that affect these aspects of pathogenesis, but these have yet to be explored. For example, we find that early changes in TN-C and LYVE-1 expression are less prevalent in the soleus muscle, which unlike most other muscles of the limb is composed of about 50% slow myosin fibers in adult mice. In 129ReJ dy/dy mice, the loss of neonatal/embryonic MHC after birth is delayed in all muscles except the soleus, and there is a preferential loss of fast type II-B and survival of slow type I fibers within 2 weeks after birth (Reggiani et al. 1992). Whether human MDC1A patients exhibit a similar pattern of muscle variability in the expression of these early pathogenic markers also needs to be studied to gain more insight on human pathogenesis.
Early, presymptomatic activation of inflammatory pathways also occurs in other muscular dystrophies including Duchenne muscular dystrophy, mdx mice, and dysferlin-deficient LGMD2B, where early inflammatory pathways might be promoted by the activation of Toll-like receptors, NF-κβ, NFAT, and AP-1 signaling pathways (Nagaraju et al. 2008; Evans et al. 2009). It will be important to determine which, if any, of these signaling pathways is activated in the earliest stages of Lama2-/- muscular dystrophy and how these pathways are induced by the disruption of interactions between laminin 211 and its key receptors (α-dystroglycan, α7β1 integrin) on the surface of Lama2-/- muscle fibers. In addition, it will be important to determine how inflammatory signals might contribute to the apoptotic mechanisms that play an important role in disease progression.
To develop therapeutics to treat muscular dystrophies, the molecular mechanisms that contribute to muscle pathogenesis must be identified, recognizing that these could vary with the stage of disease progression. A number of studies have demonstrated that apoptotic mechanisms are involved in the death of Lama2-deficient muscle fibers (Miyagoe et al. 1997; Kuang et al. 1999; Mukasa et al. 1999). Genetic experiments designed to inhibit apoptosis in Lama2-deficient mice have led to improved long-term muscle function and survival (Girgenrath et al. 2004; Dominov et al. 2005). Pharmacological studies have also shown that long-term Lama2-/- disease symptoms can be reduced by drugs that can inhibit apoptosis, such as omigapil (Erb et al. 2009) and doxycycline (Girgenrath et al. 2009), which can also inhibit inflammation at later stages in Lama2-/- muscle (Sapadin and Fleischmajer 2006; Girgenrath et al. 2009). Minimizing inflammation damage by breeding Lama2-deficient mice with a complement C3-deficient strain or treating mice with prednisolone also reduced later-stage disease symptoms (Connolly et al. 2002). Additional approaches to block early inflammatory mechanisms, possibly in conjunction with apoptosis inhibition, could enhance muscle survival further.
A better understanding of the early events in Lama2-deficent pathogenesis will be useful in the development of new strategies for early intervention in this and possibly other muscular dystrophies. This is particularly important for congenital diseases such as MDC1A, where severe symptoms occur early, and patients would benefit greatly from early intervention.
Acknowledgments
We thank Dr Eva Engvall (Sanford-Burnham Institute) for the Lama2dy-W mouse strain used in these studies; Drs Jennifer Chen, Charles Emerson, and Moonkyoung Um (BBRI) for helpful comments on the manuscript; and Dr Chen for immunohistochemistry advice.
Footnotes
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by grants to JAD from the National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01 AR051368), and the John W. Alden Trust.
References
- Arimoto J, Ikura Y, Suekane T, Nakagawa M, Kitabayashi C, Iwasa Y, Sugioka K, Naruko T, Arakawa T, Ueda M. 2010. Expression of LYVE-1 in sinusoidal endothelium is reduced in chronically inflamed human livers. J Gastroenterol. 45(3):317-325 [DOI] [PubMed] [Google Scholar]
- Bentzinger CF, Barzaghi P, Lin S, Ruegg MA. 2005. Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-alpha2-deficient mice. FASEB J. 19:934-942 [DOI] [PubMed] [Google Scholar]
- Bushby K, Lochmuller H, Lynn S, Straub V. 2009. Interventions for muscular dystrophy: molecular medicines entering the clinic. Lancet. 374:1849-1856 [DOI] [PubMed] [Google Scholar]
- Chiquet M, Fambrough DM. 1984. Chick myotendinous antigen, I: a monoclonal antibody as a marker for tendon and muscle morphogenesis. J Cell Biol. 98:1926-1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiquet-Ehrismann R, Chiquet M. 2003. Tenascins: regulation and putative functions during pathological stress. J Pathol. 200:488-499 [DOI] [PubMed] [Google Scholar]
- Connolly AM, Keeling RM, Streif EM, Pestronk A, Mehta S. 2002. Complement 3 deficiency and oral prednisolone improve strength and prolong survival of laminin alpha2-deficient mice. J Neuroimmunol. 127:80-87 [DOI] [PubMed] [Google Scholar]
- Cueni LN, Detmar M. 2008. The lymphatic system in health and disease. Lymphat Res Biol. 6:109-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominov JA, Kravetz AJ, Ardelt M, Kostek CA, Beermann ML, Miller JB. 2005. Muscle-specific BCL2 expression ameliorates muscle disease in laminin {alpha}2-deficient, but not in dystrophin-deficient, mice. Hum Mol Genet. 14:1029-1040 [DOI] [PubMed] [Google Scholar]
- Durbeej M. 2010. Laminins. Cell Tissue Res. 339:259-268 [DOI] [PubMed] [Google Scholar]
- El-Karef A, Yoshida T, Gabazza EC, Nishioka T, Inada H, Sakakura T, Imanaka-Yoshida K. 2007. Deficiency of tenascin-C attenuates liver fibrosis in immune-mediated chronic hepatitis in mice. J Pathol. 211:86-94 [DOI] [PubMed] [Google Scholar]
- Erb M, Meinen S, Barzaghi P, Sumanovski LT, Courdier-Fruh I, Ruegg MA, Meier T. 2009. Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-alpha2 deficiency. J Pharmacol Exp Ther. 331:787-795 [DOI] [PubMed] [Google Scholar]
- Evans NP, Misyak SA, Robertson JL, Bassaganya-Riera J, Grange RW. 2009. Dysregulated intracellular signaling and inflammatory gene expression during initial disease onset in Duchenne muscular dystrophy. Am J Phys Med Rehabil. 88:502-522 [DOI] [PubMed] [Google Scholar]
- Flister MJ, Wilber A, Hall KL, Iwata C, Miyazono K, Nisato RE, Pepper MS, Zawieja DC, Ran S. 2010. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1. Blood. 115:418-429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale NW, Prevo R, Espinosa J, Ferguson DJ, Dominguez MG, Yancopoulos GD, Thurston G, Jackson DG. 2007. Normal lymphatic development and function in mice deficient for the lymphatic hyaluronan receptor LYVE-1. Mol Cell Biol. 27:595-604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawlik KI, Mayer U, Blomberg K, Sonnenberg A, Ekblom P, Durbeej M. 2006. Laminin alpha1 chain mediated reduction of laminin alpha2 chain deficient muscular dystrophy involves integrin alpha7beta1 and dystroglycan. FEBS Lett. 580:1759-1765 [DOI] [PubMed] [Google Scholar]
- Girgenrath M, Beermann ML, Vishnudas VK, Homma S, Miller JB. 2009. Pathology is alleviated by doxycycline in a laminin-alpha2-null model of congenital muscular dystrophy. Ann Neurol. 65:47-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girgenrath M, Dominov JA, Kostek CA, Boone Miller J. 2004. Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J Clin Invest. 114:1635-1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi YK, Tezak Z, Momoi T, Nonaka I, Garcia CA, Hoffman EP, Arahata K. 2001. Massive muscle cell degeneration in the early stage of merosin-deficient congenital muscular dystrophy. Neuromuscul Disord. 11:350-359 [DOI] [PubMed] [Google Scholar]
- Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tome FM, Schwartz K, Fardeau M, Tryggvason K, et al. 1995. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. 11:216-218 [DOI] [PubMed] [Google Scholar]
- Jackson DG. 2004. Biology of the lymphatic marker LYVE-1 and applications in research into lymphatic trafficking and lymphangiogenesis. APMIS. 112:526-538 [DOI] [PubMed] [Google Scholar]
- Jackson DG. 2009. Immunological functions of hyaluronan and its receptors in the lymphatics. Immunol Rev. 230:216-231 [DOI] [PubMed] [Google Scholar]
- Jarvinen TA, Kannus P, Jarvinen TL, Jozsa L, Kalimo H, Jarvinen M. 2000. Tenascin-C in the pathobiology and healing process of musculoskeletal tissue injury. Scand J Med Sci Sports. 10:376-382 [DOI] [PubMed] [Google Scholar]
- Johnson LA, Prevo R, Clasper S, Jackson DG. 2007. Inflammation-induced uptake and degradation of the lymphatic endothelial hyaluronan receptor LYVE-1. J Biol Chem. 282:33671-33680 [DOI] [PubMed] [Google Scholar]
- Kivela R, Havas E, Vihko V. 2007. Localisation of lymphatic vessels and vascular endothelial growth factors-C and -D in human and mouse skeletal muscle with immunohistochemistry. Histochem Cell Biol. 127:31-40 [DOI] [PubMed] [Google Scholar]
- Kriehuber E, Breiteneder-Geleff S, Groeger M, Soleiman A, Schoppmann SF, Stingl G, Kerjaschki D, Maurer D. 2001. Isolation and characterization of dermal lymphatic and blood endothelial cells reveal stable and functionally specialized cell lineages. J Exp Med. 194:797-808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang W, Xu H, Vachon PH, Liu L, Loechel F, Wewer UM, Engvall E. 1998. Merosin-deficient congenital muscular dystrophy: partial genetic correction in two mouse models. J Clin Invest. 102:844-852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang W, Xu H, Vilquin JT, Engvall E. 1999. Activation of the lama2 gene in muscle regeneration: abortive regeneration in laminin alpha2-deficiency. Lab Invest. 79:1601-1613 [PubMed] [Google Scholar]
- Li ZF, Shelton GD, Engvall E. 2005. Elimination of myostatin does not combat muscular dystrophy in dy mice but increases postnatal lethality. Am J Pathol. 166:491-497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg I, Brengman JM, Engel AG. 1995. Analysis of cytokine expression in muscle in inflammatory myopathies, Duchenne dystrophy, and non-weak controls. J Neuroimmunol. 63:9-16 [DOI] [PubMed] [Google Scholar]
- Luong MX, Tam J, Lin Q, Hagendoorn J, Moore KJ, Padera TP, Seed B, Fukumura D, Kucherlapati R, Jain RK. 2009. Lack of lymphatic vessel phenotype in LYVE-1/CD44 double knockout mice. J Cell Physiol. 219:430-437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maby-El Hajjami H, Petrova TV. 2008. Developmental and pathological lymphangiogenesis: from models to human disease. Histochem Cell Biol. 130:1063-1078 [DOI] [PubMed] [Google Scholar]
- Meinen S, Barzaghi P, Lin S, Lochmuller H, Ruegg MA. 2007. Linker molecules between laminins and dystroglycan ameliorate laminin-alpha2-deficient muscular dystrophy at all disease stages. J Cell Biol. 176:979-993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, Takeda S. 1997. Laminin alpha2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 415:33-39 [DOI] [PubMed] [Google Scholar]
- Moll J, Barzaghi P, Lin S, Bezakova G, Lochmuller H, Engvall E, Muller U, Ruegg MA. 2001. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature. 413:302-307 [DOI] [PubMed] [Google Scholar]
- Mouta Carreira C, Nasser SM, di Tomaso E, Padera TP, Boucher Y, Tomarev SI, Jain RK. 2001. LYVE-1 is not restricted to the lymph vessels: expression in normal liver blood sinusoids and down-regulation in human liver cancer and cirrhosis. Cancer Res. 61:8079-8084 [PubMed] [Google Scholar]
- Mrak RE. 1998. The pathologic spectrum of merosin deficiency. J Child Neurol. 13:513-515 [DOI] [PubMed] [Google Scholar]
- Mukasa T, Momoi T, Momoi MY. 1999. Activation of caspase-3 apoptotic pathways in skeletal muscle fibers in laminin alpha2-deficient mice. Biochem Biophys Res Commun. 260:139-142 [DOI] [PubMed] [Google Scholar]
- Muller-Felber W, Toepfer M, Muller T, Muller-Hocker J, Fischer P, Lochmuller H, Pongratz D. 1998. Tenascin is a useful marker in the diagnosis of inflammatory myopathies. Eur J Med Res. 3:281-287 [PubMed] [Google Scholar]
- Nagaraju K, Rawat R, Veszelovszky E, Thapliyal R, Kesari A, Sparks S, Raben N, Plotz P, Hoffman EP. 2008. Dysferlin deficiency enhances monocyte phagocytosis: a model for the inflammatory onset of limb-girdle muscular dystrophy 2B. Am J Pathol. 172:774-785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale TD, Frayne ME, Clasper S, Banerji S, Jackson DG. 2009. A mechanism of sialylation functionally silences the hyaluronan receptor LYVE-1 in lymphatic endothelium. J Biol Chem. 284:3935-3945 [DOI] [PubMed] [Google Scholar]
- Overbergh L, Valckx D, Waer M, Mathieu C. 1999. Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine. 11:305-312 [DOI] [PubMed] [Google Scholar]
- Pegoraro E, Mancias P, Swerdlow SH, Raikow RB, Garcia C, Marks H, Crawford T, Carver V, Di Cianno B, Hoffman EP. 1996. Congenital muscular dystrophy with primary laminin alpha2 (merosin) deficiency presenting as inflammatory myopathy. Ann Neurol. 40:782-791 [DOI] [PubMed] [Google Scholar]
- Qiao C, Li J, Zhu T, Draviam R, Watkins S, Ye X, Chen C, Li J, Xiao X. 2005. Amelioration of laminin-alpha2-deficient congenital muscular dystrophy by somatic gene transfer of miniagrin. Proc Natl Acad Sci U S A. 102:11999-12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiani C, Brocks L, Wirtz P, Loermans H, te Kronnie G. 1992. Myosin isoforms in hindlimb muscles of normal and dystrophic (ReJ129 dy/dy) mice. Muscle Nerve. 15:199-208 [DOI] [PubMed] [Google Scholar]
- Ringelmann B, Roder C, Hallmann R, Maley M, Davies M, Grounds M, Sorokin L. 1999. Expression of laminin alpha1, alpha2, alpha4, and alpha5 chains, fibronectin, and tenascin-C in skeletal muscle of dystrophic 129ReJ dy/dy mice. Exp Cell Res. 246:165-182 [DOI] [PubMed] [Google Scholar]
- Ristimaki A, Narko K, Enholm B, Joukov V, Alitalo K. 1998. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem. 273:8413-8418 [DOI] [PubMed] [Google Scholar]
- Sapadin AN, Fleischmajer R. 2006. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 54:258-265 [DOI] [PubMed] [Google Scholar]
- Tammela T, Alitalo K. 2010. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 140:460-476 [DOI] [PubMed] [Google Scholar]
- Tidball JG, Villalta SA. 2010. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol. 298:R1173-1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker RP, Chiquet-Ehrismann R. 2009. The regulation of tenascin expression by tissue microenvironments. Biochim Biophys Acta. 1793:888-892 [DOI] [PubMed] [Google Scholar]
- Vachon PH, Loechel F, Xu H, Wewer UM, Engvall E. 1996. Merosin and laminin in myogenesis; specific requirement for merosin in myotube stability and survival. J Cell Biol. 134:1483-1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lunteren E, Moyer M, Leahy P. 2006. Gene expression profiling of diaphragm muscle in alpha2-laminin (merosin)-deficient dy/dy dystrophic mice. Physiol Genomics. 25:85-95 [DOI] [PubMed] [Google Scholar]
- Xu H, Wu XR, Wewer UM, Engvall E. 1994. Murine muscular dystrophy caused by a mutation in the laminin alpha 2 (Lama2) gene. Nat Genet. 8:297-302 [DOI] [PubMed] [Google Scholar]