

Abstract
Antigen retrieval (AR), in which formalin-fixed paraffin-embedded tissue sections are briefly heated in buffers at high temperature, often greatly improves immunohistochemical staining. An important unresolved question regarding AR is how formalin treatment affects the conformation of protein epitopes and how heating unmasks these epitopes for subsequent antibody binding. The objective of the current study was to use model proteins to determine the effect of formalin treatment on protein conformation and thermal stability in relation to the mechanism of AR. Sodium dodecyl sulfate polyacrylamide gel electrophoresis was used to identify the presence of protein formaldehyde cross-links, and circular dichroism spectropolarimetry was used to determine the effect of formalin treatment and high-temperature incubation on the secondary and tertiary structure of the model proteins. Results revealed that for some proteins, formalin treatment left the native protein conformation unaltered, whereas for others, formalin denatured tertiary structure, yielding a molten globule protein. In either case, heating to temperatures used in AR methods led to irreversible protein unfolding, which supports a linear epitope model of recovered protein immunoreactivity. Consequently, the core mechanism of AR likely centers on the restoration of normal protein chemical composition coupled with improved accessibility to linear epitopes through protein unfolding.
Keywords: antigen retrieval, circular dichroism, formaldehyde, formalin-fixed paraffin-embedded tissue, myoglobin, ribonuclease A, protein unfolding
Formalin fixation and paraffin embedding (FFPE) remains the preeminent technique for processing tissue specimens for pathologic examination, for the study of tissue morphology, and for archival preservation (Fox et al. 1985). The steps of FFPE tissue processing are generally as follows. Following fixation in a 3.7% solution of neutral-buffered formalin (fixation), tissue specimens are dehydrated through a series of solutions with increasing concentrations of alcohol (graded alcohols). Samples are then soaked in a transition solvent such as xylene (cleared) followed by liquid paraffin, which is then cooled for long-term storage as solid blocks that can be cut into thin slices for mounting on slides for microscopy.
The early work of Fraenkel-Conrat (Fraenkel-Conrat et al. 1947; Fraenkel-Conrat and Ollcott 1948; Fraenkel-Conrat and Mecham 1949) and the more recent work of Metz et al. (2004, 2006) have identified four types of chemical modifications following treatment of peptides or proteins with formaldehyde. These modifications are as follows: hydroxymethyl (methylol) adducts, Schiff base adducts, 4-imidazolidinone adducts, and methylene bridges (cross-links). Methylol and Schiff base adducts form rapidly upon reaction with primary amino (lysine) and thiol (cysteine) groups. The 4-imidazolidinone adduct forms at the N-terminal site of proteins, likely by way of a Schiff base intermediate (Fowles et al. 1996). Inter- and intramolecular cross-links are initiated by a fast reaction of formaldehyde with the ε-amino group of lysine or the β-thiol group of cysteine. The resulting methylol groups then form methylene bridges (–CH2–) in a second reaction by attacking available nucleophiles (Kunkel et al. 1981). The amino acids that can serve as nucleophiles for this second reaction are tyrosine, arginine, asparagine, glutamine, histidine, and tryptophan. A second type of cross-link can occur between a secondary amine and a carbonyl compound through the Mannich reaction (Sompuram et al. 2004). Following formalin fixation, ethanol dehydration leads to increased protein aggregation (Fowler et al. 2008).
The great advantage afforded by formalin fixation in preserving tissue morphology is offset by reduced immunohistochemical reactivity (Taylor 1986). The loss of protein immunoreactivity has been attributed to the aforementioned formaldehyde-induced protein modifications (Rait, O’Leary, et al. 2004; Rait, Xu, et al. 2004; Shi et al. 1991; Suurmeijer and Born 1993). In 1991, Shi et al. published their seminal observation that high-temperature incubation of FFPE tissue sections in buffers for short periods (heat-induced antigen retrieval [AR]) led to improved immunohistochemical staining (Shi et al. 1991). However, 20 years later, AR remains largely an empirical method, requiring the optimization of several critical parameters by trial and error (Miller et al. 2000; Shi et al. 1996). Consequently, a greater understanding of the chemistry of formalin fixation and the mechanism of AR would facilitate more rational approaches to improve immunohistochemical and molecular analyses of FFPE tissues.
An important unresolved question regarding AR is the effect of formalin fixation on the conformation of protein epitopes and how heating unmasks these epitopes for subsequent binding to antibodies. In this regard, the various proposed mechanisms of AR fall into four broad categories. The first proposes that formaldehyde treatment “locks in” protein conformation and stabilizes it against thermal denaturation (Bolgiano et al. 2000; Mason and O’Leary 1991; Metz et al. 2003; Rait, O’Leary, et al. 2004; Singh and DasGupta 1989). In this model, AR likely serves to extract diffusible proteins, open gaps in the fixed tissue, extract calcium–protein complexes, break some of the intermolecular protein cross-links, and rehydrate protein epitopes that may have been buried by organic solvents and paraffin (Boon and Kok 1994; Morgan et al. 1994; Morgan et al. 1997; Shi et al. 1997; Suurmeijer and Born 1993). Epitope unmasking is thus achieved by removing steric barriers to antibody penetration into the tissue. The antibodies subsequently bind to epitopes whose native conformation is at least partially preserved by stabilizing cross-links that remain following AR. The second model proposes that formaldehyde treatment reduces immunoreactivity by altering protein conformation (Shi et al. 2001; Volkin and Klibanov 1989). AR serves to reverse a majority of the formaldehyde-induced protein modifications, which allows the protein to reestablish a near-native-like conformation upon cooling. Thus, immunoreactivity is restored by the renaturation of protein epitopes (Rait, O’Leary, et al. 2004; Shi et al. 2001). The third model proposes that AR reverses formaldehyde-induced protein cross-links, thus removing barriers to antibody penetration and allowing access to protein epitopes. However, it is proposed that proteins become denatured (unfolded) at the elevated temperature used during AR and remain so after cooling. The implication of this model, proposed by Bogen and others (Bogen et al. 2009; Sompuram, Vani, and Bogen 2006; Sompuram, Vani, Hafer, et al. 2006), is that antibodies proven successful for immunohistochemistry recognize linear protein epitopes consisting of 5 to 7 amino acid residues. Thus, only the primary sequence of the protein is important, and the secondary and tertiary structures are irrelevant (Bogen et al. 2009). The fourth model proposes that formaldehyde-induced protein modifications reduce immunoreactivity by neutralizing protein electrostatic charges. This model, proposed by Boenisch (2002, 2006), suggests that formaldehyde adducts inhibit antibody binding, even in the absence of steric interference, by their ability to convert charged amino acid side chains, such as positively charged amines, into neutral methylol groups. Such charge neutralization was shown to be predominately a kinetic effect, as antibody staining could be restored by prolonging the antibody incubation time (Boenisch 2002). Thus, an important function of AR may be to restore protein electrostatic charge.
The objective of the current study was to use model proteins to determine the effect of formaldehyde treatment on protein conformation and thermal stability in relation to the mechanism of AR. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was used to identify the presence of protein cross-links (Rait, O’Leary, et al. 2004). Circular dichroism (CD) spectropolarimetry was used to determine the effect of formaldehyde treatment and high-temperature incubation on the conformation of the model proteins (Woody 1995). The near-ultraviolet (UV) CD region refers to wavelengths of approximately 246 to 350 nm, and its spectrum reflects the orientation of aromatic phenylalanine, tyrosine, and tryptophan amino acid side chains, giving a measure of tertiary protein structure. The far-UV region refers to wavelengths of approximately 180 to 240 nm; much of its spectrum comes from the peptide backbone, and it is indicative of secondary protein structure. Our results suggest that AR leads to irreversible protein unfolding, which supports a linear epitope model of recovered protein immunoreactivity. Consequently, the core mechanism of AR likely centers on the restoration of normal protein chemical composition and electrostatic charge coupled with improved accessibility to linear epitopes through protein unfolding.
Materials and Methods
Bovine pancreatic RNase A (type XII-A, catalog #R5500, lot #055K7695), myoglobin from equine heart muscle (catalog #M1882, lot #90K7041), and all other proteins used in this study were purchased from Sigma (St Louis, MO). Aqueous formaldehyde (37% w/v) was purchased from Fisher Scientific (Pittsburgh, PA). Formalin solutions were prepared by a 10-fold dilution (10% [v/v] or 3.7% [w/v]) of stock aqueous formaldehyde into water, and the solutions were buffered with potassium phosphate. All other buffers and chemicals were purchased from Sigma unless otherwise noted.
Formaldehyde Treatment and Heat-Induced Antigen Retrieval
Solutions of native RNase A (4 mg/ml) or myoglobin (1.5 mg/ml) in 10 mM potassium phosphate buffer (pH 7.4) were treated with an equal volume of 20% (v/v) formaldehyde in 10 mM potassium phosphate. The RNase A was fixed at ambient temperature overnight, and the myoglobin was fixed at −10C, 4C, 24C, 65C, or 90C for 2 hr and then allowed to equilibrate to room temperature for 2 hr.
Prior to AR, the formaldehyde from aliquots of the fixed RNase A and myoglobin was removed by washing the samples five times with deionized water in a Microcon YM-3 concentrator (Millipore, Billerica, MA). Each of the formalin-treated proteins was added to an equal volume of a recovery buffer containing 4% SDS in 20 mM Tris-HCl (pH 4.0). The formalin-treated protein samples were then heated at 100C for 20 min, followed by 60C for 2 hr, using an established AR protocol described by Shi et al. (2006).
Reduction, Iodoacetamide Capping, and Formalin Treatment
A 3-mg/ml solution of native RNase A in 10 mM potassium phosphate buffer was reduced in 4 mM dithiothreitol (DTT) at 45C for 1 hr. The solution was cooled, and the free cysteine residues were derivatized with iodoacetamide (4 mM final concentration) in the dark for 30 min. The concentration of the protein solution was adjusted to 1.5 mg/ml. An aliquot (1 ml) of the reduced and cysteine-capped RNase A was treated with an equal volume of 20% (v/v) formaldehyde in 10 mM potassium phosphate buffer (pH 7.4) for 2 hr at 24C. Aliquots of native RNase A and reduced and cysteine-capped RNase A that were not treated with formaldehyde were diluted with phosphate buffer to yield 0.75-mg/ml solutions.
Analysis of Protein Composition
After sample processing, 20 µl of each protein preparation was added directly to 5 µl of 4× LDS sample buffer (pH 8.5), containing 50 mM DTT and 2% SDS (Invitrogen, Carlsbad, CA) with no additional heat treatment. SDS-PAGE was performed on precast NuPAGE Bis-Tris 4% to 12% gradient polyacrylamide gels using 2-(N-morpholino) ethanesulfonic acid–SDS running buffer at pH 7.3 (Invitrogen). Benchmark molecular mass standards and the Coomassie blue–based colloidal staining kit were also purchased from Invitrogen. Gel images were documented using a Scanmaker i900 (Microtek, Carson, CA) and annotated in Adobe Photoshop.
Spectroscopic Measurements of Proteins in Solution
All protein samples were dialyzed against three changes of 10 mM potassium phosphate buffer (pH 7.4), using a Pierce Slide-a-Lyzer dialysis cassette (3500 MWCO; Thermo-Fisher Scientific, Rockford, IL). The final concentration of all native and formalin-treated protein solutions was adjusted to 0.75 mg/ml, as determined by absorption at 280 nm, prior to CD analysis. All CD measurements were acquired with a Jasco-715 Spectropolarimeter (Jasco Corporation, Easton, MD). CD spectral and thermal scans were acquired from the native and formalin-treated RNase A and myoglobin preparations. CD spectral scans were also acquired for the reduced RNase A preparations. CD scans in the near-UV (246–350 nm) region were recorded using a 10-mm path length water-jacketed cell, and those in the far-UV region (180–240 nm) were recorded using a 0.02-mm path length water-jacketed cell. Each CD spectral scan was an average of 10 measurements taken under identical conditions. The following settings were used: the wavelength scan speed was 50 nm/min, the bandwidth was 1 nm, the step resolution was 0.2 nm, and the response time was 8 sec. Isothermal sample temperature was maintained using a Neslab RTE-111 programmable water bath (Thermo-Fisher Scientific) under control of the spectropolarimeter. CD thermal scans were run from 30 to 95C using a scan rate of 20C/h, a bandwidth of 2 nm, and a response time of 2 sec. The effective thermal resolution was ~0.15C. The near-UV thermal scans were acquired at 275 nm for RNase A and 274 nm for myoglobin. The far-UV thermal scans were acquired at 222 nm for RNase A and 192 nm for myoglobin.
A reference spectrum for buffer was subtracted from the far-UV CD spectral scans, and the far- and near-UV spectral and thermal scans were converted to molar ellipticity prior to graphing the CD data using Origin 7 (OriginLab, Northampton, MA). The far-UV spectral scans were also converted to units of mean residue ellipticity, and the protein secondary structure of each native, fixed, and reduced protein was estimated from its far-UV spectral data using the CONTIN program of the CDPro software package (Sreerama and Woody 2000). The CLUSTER program of CDPro was used to predict the structure class of the proteins from the far-UV CD data (Sreerama et al. 2001).
Spectroscopic Measurements of Solid Tissue Surrogate Protein Films
To better mimic archival tissue, 10 µl of a 150-mg/ml solution of RNase A in PBS was spotted onto the center of several quartz slides. The quartz microscope slides (1 × 1 × 0.04 in. thick) were purchased from AdValue Technology (Tucson, AZ) and cleaned with ethanol and air-dried prior to use. An equal volume of 20% neutral-buffered formalin was then added to the RNase A solutions, and the mixtures were spread evenly over the surface of the slides with the end of a flexible plastic spatula. A thin, semi-opaque “tissue surrogate” film formed within 2 min (Fowler et al. 2007). The slides were allowed to dry overnight at room temperature. Unfixed RNase A (20 µl of a 75-mg/ml solution in PBS) was also streaked onto a quartz slide and allowed to dry overnight. Two sets of slides containing formalin-treated tissue surrogate films were rinsed briefly with distilled water and then incubated through a series of graded alcohols, 85% ethanol, and two changes of 100% ethanol (15 min each), followed by two changes of xylene (15 min each). The xylene was then removed by incubating the slides through two changes of 100% ethanol (15 min each) followed by brief rinsing in PBS. One of theses slides was subsequently processed at 121C for 15 min in a model 2100 steam antigen retrieval unit (PickCell Laboratories, Leiden, the Netherlands). Finally, each of the dried protein films was covered with a second clean quartz slide, and the pair of slides was held together using the sample holder of the CD spectropolarimeter.
CD spectra of the tissue surrogate films were obtained at room temperature with a Jasco-715 spectropolarimeter. The CD spectral scans were acquired from 400 to 180 nm using a scan speed of 50 nm/min, a bandwidth of 5 nm, a step resolution of 0.2 nm, and a response time of 4 sec. Molar ellipticities could not be calculated for the dried protein films because of the uncertainty of the protein concentrations and effective path lengths for the films. Consequently, the data were plotted as raw ellipticity, meaning that the shapes of the CD spectra of the dried films were comparable, but not their intensities.
Results
The Effect of Formalin on Protein Conformation and Cross-Linking
Treatment of RNase A with 10% (v/v) formaldehyde in 10 mM potassium phosphate buffer (pH 7.4; formalin) for 24 hr resulted in the CD spectra shown in Figure 1A (far UV) and Figure 1B (near UV). The CD spectra of the native and formalin-treated proteins were almost identical. The formalin-treated RNase A exhibited a slight decrease in positive band intensity at 190 nm and a minor decrease in negative band intensity between 205 and 220 nm in the far-UV region relative to the native protein. This likely reflects a subtle perturbation of secondary structure due to formalin treatment (Rait, O’Leary, et al. 2004), which resulted in a 7% increase in the α-helix content of the protein (Table 1). The formalin-treated RNase A exhibited a 10% decrease in band intensity at 278 nm relative to the native protein in the near-UV region. The near-UV band in RNase A is attributed to the six tyrosine residues, three of which are located on the surface of the protein (Woody and Dunker 1996). This subtle decrease reflects either a chemical modification of the microenvironment of one or more of the tyrosine residues, due to fixation, or a small change in tertiary structure (Rait, O’Leary, et al. 2004; Woody and Dunker 1996). However, the structural class (α + β) remains identical to that of native RNase A (Table 1).
Figure 1.
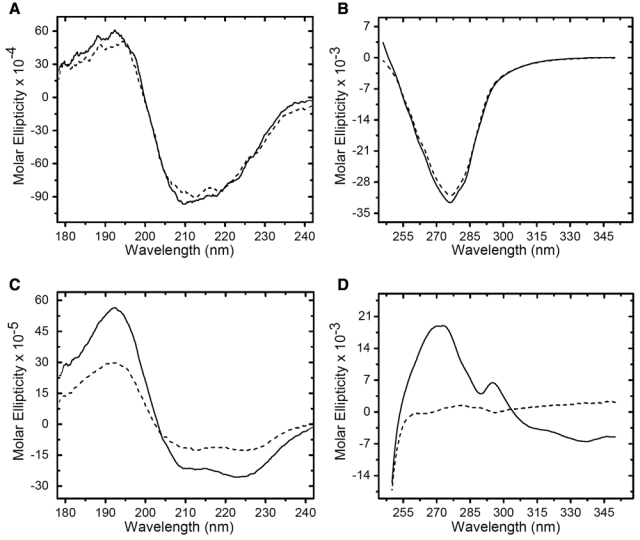
Circular dichroism (CD) spectral scans of RNase A and myoglobin with and without treatment with formalin. (A) Far-ultraviolet (UV) CD spectra of native RNase A (solid line) and formalin-treated RNase A (dashed line). (B) Near-UV CD spectra of native RNase A (solid line) and formalin-treated RNase A (dashed line). (C) Far-UV CD spectra of native myoglobin (solid line) and formalin-treated myoglobin (dashed line). (D) Near-UV CD spectra of native myoglobin (solid line) and formalin-treated myoglobin (dashed line). The units of molar ellipticity are deg · cm2 · decimole−1.
Table 1.
Results of conformational analysis of far-UV CD spectra of myoglobin and RNase A by CD PROa.
| PROTEIN | Condition | CONTIN resultb(α-helix/β-strand) | CLUSTER structure class prediction |
|---|---|---|---|
| Myoglobin | Native | 5.45/0.237 | all-α |
| Myoglobin | Fixed at -10C | 4.71/2.22 | α/β |
| Myoglobin | Fixed at 24C | 3.78/2.73 | α/β |
| Myoglobin | Fixed at 65C | 3.88/3.34 | α/β |
| Myoglobin | Fixed at 90C | 4.10/2.7 | α/β |
| RNase A | Native | 2.69/4.87 | α + β |
| RNase A | Fixed at 24C | 2.89/4.73 | α + β |
| RNase A | Reduced | 2.58/4.74 | α + β |
| RNase A | Reduced and Fixed | 2.09/5.78 | all-β |
Reference: Sreerama et al. (2000, 2001).
Number of α-helices/β-strands per 100 residues.
The RNase A samples used for CD analysis were analyzed by SDS-PAGE, as shown in Figure 2A. The native protein (Fig. 2A, lane 1) revealed a single band corresponding to a molecular weight of ~14.4 kDa (Rait, O’Leary, et al. 2004). The formalin-treated RNase A (Fig. 2A, lane 2) revealed the formation of a series of high molecular weight oligomers consisting of two to eight cross-linked proteins, which reflect the formation of intermolecular methylene bridges (Rait, O’Leary, et al. 2004; Rait, Xu, et al. 2004). The formation of intramolecular protein cross-links was manifested as an increase in the mobility of the formalin-treated RNase A monomer relative to the native protein. In the absence of disulfide bridges, because of the reducing conditions of the SDS-PAGE, intramolecular cross-linking caused the formalin-treated protein to remain more compact than the native protein; thus, it ran slightly lower on the gel (Rait, O’Leary, et al. 2004). AR recovery of the fixed RNase A sample (Fig. 2A, lane 3) resulted in a significant reversal of intermolecular protein cross-links as only dimer, trimer, and a trace amount of tetramer remained. The reversal of intramolecular protein cross-links is manifested in the monomer and oligomer gel bands as a decrease in the mobility of the recovered RNase A relative to the formalin-treated protein.
Figure 2.
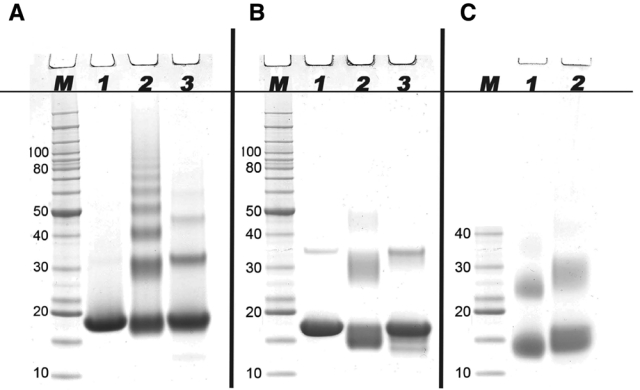
Results of SDS-PAGE analysis of formalin-treated RNase A and myoglobin before and after antigen retrieval (AR). For AR, the formalin-treated proteins (2 mg/ml) were heated at 100C for 20 min followed by heating at 60C for 2 hr in 20 mM Tris-HCl (pH 4.0), 2% (w/v) SDS. (A) Lane M: molecular weight marker; lane 1: native RNase A; lane 2: RNase A treated with formalin for 24 hr at 24C; lane 3: formalin-treated RNase A from lane 2 after AR. (B) Lane M: molecular weight marker; lane 1: native myoglobin; lane 2: myoglobin treated with formalin for 2 hr at 24C; lane 3: the formalin-treated myoglobin from lane 2 after AR. (C) Lane 1: myoglobin treated with formalin for 2 hr at 90C; lane 2: the formalin-treated myoglobin from lane 1 after AR.
Treatment of myoglobin with formalin for 2 hr resulted in the CD spectra shown in Figure 1C (far UV) and Figure 1D (near UV). In contrast to RNase A, substantial changes were seen in both CD spectra of the formalin-treated myoglobin. The formalin-treated protein exhibited a 53% decrease in intensity for the positive band at 192 nm in the far UV but with no shift in the peak maximum. There was a 45% decrease in the intensity of the negative band between 204 and 234 nm, with the greatest change centered near 222 nm, which serves as an α-helix marker (Wang et al. 2000). Taken together, these changes suggest that the secondary structure of the formalin-treated myoglobin remained highly ordered but that the β-sheet content was increased at the expense of α-helix relative to the native protein. This was confirmed by the conformational analysis, which showed a ~10-fold increase in the β-sheet content of the formalin-treated myoglobin at 24C (Table 1). The near-UV spectrum of native myoglobin exhibited a broad asymmetric positive band between 255 and 340 nm. This band consists of contributions from phenylalanine and tyrosine (255–289 nm) and a partially resolved peak at 293 nm arising from tryptophan (Irace et al. 1986). In contrast, the near-UV spectrum of the formalin-treated myoglobin was essentially absent, exhibiting only a very small broad positive peak centered at 280 nm. These results were in agreement with previous studies (Dolgikh et al. 1981; Rott and Poverennyi 1982). The absorptive bands in the near-UV region originate from chromophoric amino acid side chains that experience an asymmetric electric field, which only occurs if the protein tertiary structure is highly ordered. The collapse of the near-UV absorptive band results from field averaging that originates from a fluctuating tertiary structure associated with a non-ridged packing of amino acid side chains. The loss of vibrational structure in the near UV coupled with more modest changes in the far UV is the CD spectral signature characteristic of the molten globule protein state (Cort and Andersen 1997; Dolgikh et al. 1981). Furthermore, the collapse of the tertiary structure coupled with the increase in the β-sheet content of the secondary structure was sufficient to change the predicted structural class from the all-α of native myoglobin to an α/β conformation for the formalin-treated protein at all temperatures that were analyzed (Table 1).
The myoglobin samples used for CD analysis were analyzed by SDS-PAGE as shown in Figure 2B,C. The native protein (Fig. 2B, lane 1) revealed a prominent band with a molecular weight of ~17 kDa and a faint band corresponding to a trace amount of myoglobin dimer, which is typically present in myoglobin preparations (van der Oord et al. 1969; Zaia et al. 1992). The different effects of formalin treatment on RNase A and myoglobin seen by CD spectropolarimetry were reflected in the electrophoretic behavior of the proteins. Formalin-treated myoglobin (Fig. 2B, lane 2) revealed the formation of only intermolecular cross-linked dimers and a very faint trimer band. These bands, as well as the intramolecular cross-linked monomer band, were considerably more diffuse that those seen for formalin-treated RNase A, with a number of cross-linked species running well below the native protein bands. The AR treatment (Fig. 2B, lane 3) resulted in bands that were almost identical to those of the native protein. We speculated that these diffuse gel bands reflected an increase in the heterogeneity of cross-linked proteins arising from the increased conformational space sampled by the molten globule state of the formalin-treated myoglobin. To test this idea, we fixed myoglobin at several temperatures between −10 and 90C, theorizing that a progressive increase in conformational flexibility with temperature would lead to both a greater heterogeneity and a more compact average size for the myoglobin oligomeric species. This was indeed the case, as shown in Figure 2C (lane 1), for myoglobin treated with formalin at 90C. Both the fixed myoglobin monomer and dimer displayed gels bands that were shaper and ran below the positions of corresponding bands for myoglobin fixed at 24C (Fig. 2B, lane 2). It is also interesting to note that this sample resulted in a reduced recovery of native protein following AR treatment (Fig. 2C, lane 2) than did the sample treated with formalin at 24C (Fig. 2B, lane 3).
Effect of Disulfide Bridges on the Conformation of Formalin-Treated RNase A
One seemingly obvious explanation for the difference in behavior of formalin-treated RNase A and myoglobin is that disruption of RNase A tertiary structure in the presence of formalin was prevented by disulfide bonds. Specifically, RNase A has four disulfide bonds, whereas myoglobin has none. To examine the role of the disulfide bonds on conformational stability in the presence of formaldehyde, the disulfide bridges in RNase A were reduced with DTT and end-capped with iodoacetamide. The result of eliminating the disulfide bonds on the conformation of RNase A in both the absence and presence of formalin is shown in Figure 3A (far UV) and Figure 3B (near UV). The CD spectra of the native protein (solid line) and the reduced and capped RNase A (dashed line) showed modest changes. The conformational analysis suggested that eliminating the disulfide bonds increased protein chain disorder at the equal expense of α-helix (4% reduction) and β-sheet (3% reduction), but the structural class remained α + β (Table 1). Formalin treatment of the reduced and capped RNase A (dash-dot line) increased the β-sheet content of the protein by 16% at the expense of α-helix (22% decrease), as evidenced by the decrease in intensity of the positive band at 190 nm and a decrease in intensity of the negative band between 202 and 225 nm (Woody and Dunker 1996). This transformation was also reflected in the conformational analysis, which predicted an all-β structural class for the reduced and formalin-treated protein (Table 1). In the near-UV region, formalin treatment of the reduced and capped RNase A resulted in a 33% decrease in band intensity. The key result of this study is that the absence of disulfide bridges alone does not explain the different effects of formalin on the tertiary structure of RNase A and myoglobin. This does not preclude the possibility that disulfide bridges could be important in the stabilization of other proteins against the denaturing effects of formalin.
Figure 3.
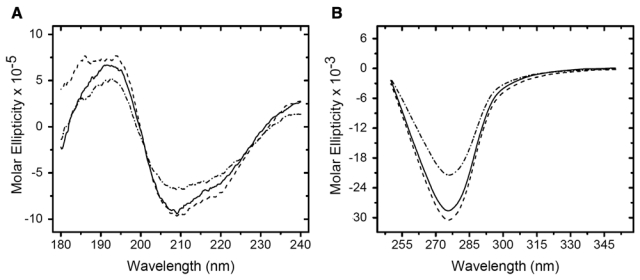
Circular dichroism (CD) spectra of RNase A: effect of reduction and fixation on protein structure. Native RNase A in 10 mM potassium phosphate buffer was reduced in 4 mM dithiothreitol (DTT) at 45C for 1 hr. The solution was cooled, and the free cysteine residues were derivatized with 4 mM iodoacetamide in the dark for 30 min. Aliquots of the RNase A solution were fixed with an equal volume of 20% (v/v) formaldehyde in 10 mM potassium phosphate buffer (pH 7.4) for 2 hr at 24C. (A) Far-ultraviolet (UV) and (B) near-UV CD spectra of native RNase A (solid line), RNase A reduced and capped without formalin treatment (dashed line), and RNase A reduced, capped, and subsequently treated with formalin (dash-dot line). The units of molar ellipticity are as follows: deg · cm2 · decimole−1.
The Effect of AR on Protein Conformation
The effect of heating on the conformational properties of native and formalin-treated RNase A was assessed in the CD thermal scans shown in Figure 4A (far UV) and Figure 4B (near UV). In Figure 4A, the far-UV wavelength of 222 nm (the α-helix marker) was monitored as the native (solid line) and formalin-treated (dashed line) samples were heated from 30 to 95C at a rate of 20C/h. The native protein exhibited a cooperative melting transition centered at ~63C. This corresponds to the phase transition temperature of RNase A at neutral pH as determined by scanning calorimetry (Rait, O’Leary, et al. 2004). In contrast, the formalin-treated RNase A exhibited a progressive, nearly linear decrease in the negative intensity of the 222-nm band with increasing temperature. The derivative of this plot (not shown) revealed a very broad non-cooperative transition centered near 59C. This behavior was repeated in the thermal scans shown in Figure 4B, where the near-UV wavelength of 275 nm was monitored. The derivative of this plot (not shown) revealed a very broad phase transition centered near 59C. With regard to the mechanism of AR, the key observation is that the degree of secondary and tertiary conformational disorder, as measured by CD spectropolarimetry, is nearly identical for the native and formalin-treated RNase A at 95C, which is near the lower temperature limit of most AR protocols (Shi et al. 2001; Shi et al. 2006).
Figure 4.
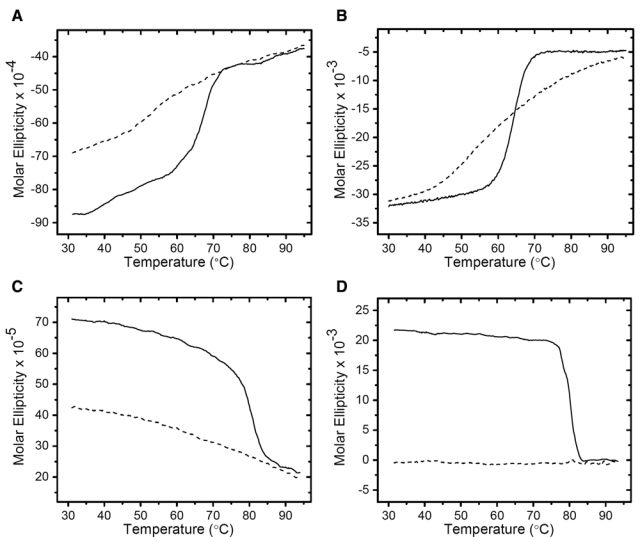
Circular dichroism (CD) thermal scans of RNase A and myoglobin with and without treatment with formalin. Ascending-temperature scans of native RNase A (solid line) and formalin-treated RNase A (dashed line) were run from 30 to 95C at a scan rate of 20C/hr. (A) Far-ultraviolet (UV) thermal scan monitored at 192 nm and (B) near-UV thermal scan monitored at 274 nm. Ascending-temperature scans of native myoglobin (solid line) and formalin-treated myoglobin (dashed line) were run from 30 to 95C at a scan rate of 20C/hr. (C) Far-UV thermal scan monitored at 222 nm and (D) near-UV thermal scan monitored at 275 nm. The units of molar ellipticity are as follows: deg · cm2 · decimole−1.
The thermal analysis was repeated for native and formalin-treated myoglobin, as shown in Figure 4C (far UV) and Figure 4D (near UV). The native protein (solid line) exhibited a cooperative melting transition centered at ~75C when monitored at 192 nm in the far UV. This corresponds to the phase transition temperature of myoglobin at neutral pH, as determined by infrared spectroscopy (Mason and O’Leary 1991) and thermal scanning CD spectropolarimetry (Mehl et al. 2009). The formalin-treated myoglobin (dashed line) was significantly more disordered at 24C than the native protein, as demonstrated in the far-UV spectral scan at 192 nm (Figure 1C), and exhibited a further nearly linear increase in disorder with increasing temperature. In the near-UV region, the native myoglobin revealed a highly cooperative melting transition centered at 78C when monitored at 274 nm. In contrast, the fixed myoglobin, already highly disordered at 24C (Fig. 1D), exhibited essentially no change in disorder over the scanned temperature range of 30 to 95C. Again, as with RNase A, the degree of secondary and tertiary conformational disorder for myoglobin at 95C was nearly identical for the native and formalin-treated proteins.
Changes in Conformation of Recovered Proteins upon Cooling to Room Temperature
Upon completion of the CD thermal scans at 95C, the proteins were held at this temperature for ~10 min while spectral scans were acquired. The proteins were then cooled to 24C at 20C/h. After a 30-min equilibration, CD spectral scans in the far-UV and near-UV regions were acquired to determine the conformation of the recovered proteins. The far-UV and near-UV CD spectral scans for RNase A are shown in Figure 5A,B. For both CD regions, the spectral scans of the formalin-treated RNase A prior to the initiation of the thermal scan are shown as solid lines, and the spectral scans of the protein after heating to 95C are shown as dashed lines. The spectral scans of RNase A heated to 95C and then cooled to 24C are shown as dash-dot lines. The heated protein was observed to recover only a modest amount of its native conformation upon cooling. Specifically, there was only a ~12% recovery of the tertiary protein conformation based on the negative band intensity at 275 nm in the near-UV spectrum. The shape and intensity of the bands in the far-UV spectrum suggest that both the heated and cooled proteins have a higher percentage of β-sheet conformation and reduced percentage of α-helix structure relative to the native protein. This behavior was independent of the rate at which the heated RNase A was cooled to room temperature from 95C. Also, the CD spectra did not change if the equilibration time at 24C was increased. Myoglobin exhibited a similar pattern of behavior; it regained little of the native myoglobin conformation upon cooling to 24C, as shown in Figure 5C (far UV) and Figure 5D (near UV). We performed the above CD spectral analysis with additional proteins and obtained similar results (Table 2). The key observations of these studies are, first, that formalin-treated proteins achieved the same level of conformational disorder as the corresponding native proteins at 95C. Second, there was practically no recovery of native tertiary structure and only modest recovery of native secondary structure upon cooling the proteins to room temperature.
Figure 5.
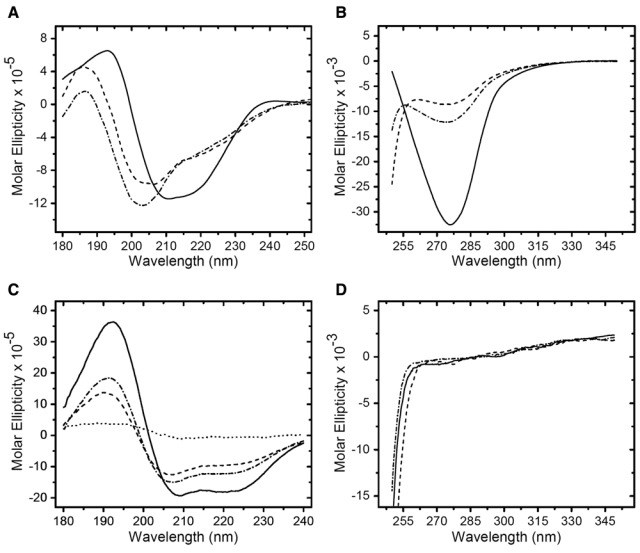
Circular dichroism (CD) spectral scans of formalin-treated RNase A and myoglobin acquired during the thermal scans of Figure 4. Far-ultraviolet (UV) (A) and near-UV (B) spectral scans of formalin-treated RNase A at 24C prior to the start of the thermal scan (solid line), after a 10-min equilibration at 95C following completion of the thermal scan (dashed line), and after cooling to 24C at a rate of 20C/hr and equilibrating for 30 min (dash-dot line). Far-UV (C) and near-UV (D) spectral scans of formalin-treated myoglobin at 24C prior to the start of the thermal scan (solid line), after a 10-min equilibration at 95C following completion of the thermal scan (dashed line), and after cooling to 24C at a rate of 20C/hr and equilibrating for 30 min (dash-dot line). The spectral scan shown by the dotted line in (C) is myoglobin denatured in 5% (w/v) SDS at room temperature. The units of molar ellipticity are as follows: deg · cm2 · decimole−1.
Table 2.
Effect of Formalin Treatment on the Conformation of Selected Proteins
| Protein | Mass (kDa) | Classa | β/α/otherb | S-Sc | 2°Structured | 3°Structuree | Formalinf | Folded 95/24Cg |
|---|---|---|---|---|---|---|---|---|
| RNase A | 13.7 | α + β | 33/20/47 | 4 | 1 | 1 | 1 | +/+ |
| Lysozyme | 14.3 | α + β | 10/41/49 | 4 | 2 | 2 | 2 | +/+ |
| α-Lactalbumin | 14.2 | α + β | 11/43/46 | 4 | 2 | 3 | 3 | +/+ |
| Myoglobin | 17 | all-α | 00/76/24 | 0 | 3 | 3 | 3 | +/+ |
| Hemoglobin | 64.5 | all-α | 00/76/24 | 0 | 1 | 2 | 2 | +/+ |
| Bovine serum albumin | 66.7 | all-α | 00/70/30 | 17 | 1 | 1 | 1 | +/+ |
| Concanavalin A | 104 | all-β | 46/03/51 | 0 | 1 | 2 | 2 | ND |
| Carbonic anhydrase | 29 | all-β | 30/15/55 | 0 | 1 | 1 | 1 | +/+ |
| Pepsin | 25 | all-β | 45/11/44 | 3 | 1 | 1 | 1 | +/+ |
Protein structural classification from the Protein Data Bank (PDB); bPercentage of β-sheet/ α-helix/other secondary structural elements, where “other” refers to β-turns, γ-turns, β-hairpin loops, and disordered segments; cNumber of disulfide bonds in protein; dRefers to a number score characterizing the increase in β-sheet content of the formalin-treated protein relative to the native protein as determined from their far-UV CD spectra (3 = >66% increase, 2 = 66-20% increase, 1 = 20 to 0% increase); eRefers to a number score characterizing the loss of integrated intensity (loss of tertiary structural order) of the near-UV CD spectrum of the formalin-treated protein relative to the native protein (3 = >66% decrease, 2 = 66-20% decrease, 1 = 20-0% decrease); fRefers to a number score characterizing the overall effect of formalin treatment on protein unfolding at 24C (3 = unfolded molten globule-like conformation, 2 = intermediate unfolded state between a molten globule and native protein, 1 = native-like folded conformation); gConformation of the formalin-treated protein at 95C and after cooling to 24C (+ = same degree of unfolding as the native protein, ND = not determined).
Discussion
Formalin treatment at room temperature was observed to have a different effect on the conformations of RNase A and myoglobin. This difference arises from both the nature of formalin as a solvent and from the conformational properties of the proteins. When formaldehyde is dissolved in buffer to prepare formalin, it reacts instantaneously with water and exists predominately in the form of methylene glycol (Fox et al. 1985; Le Botlan et al. 1983). Methylene glycol, like other low molecular weight alcohols, can alter protein structure (Brandts et al. 1989; Kundu et al. 2002; Naseem and Khan 2003) through at least two mechanisms. The first mechanism involves promotion of protein unfolding through weakening of the hydrophobic stabilization within the protein core (Brandts et al. 1989; Kundu et al. 2002). The second mechanism involves the energetically unfavorable interaction of alcohols with the peptide backbone (Pace et al. 2004). The response of most proteins to this situation is to form β-sheets to sequester the peptides bonds away from the solvent while exposing non-polar side chains to the alcohol (Cao et al. 2004). Such a secondary structural change is typically accompanied by a significant disruption (collapse) of tertiary structure to form the molten globule protein state (Kamatari et al. 1998). This is the pattern of behavior seen with myoglobin as formalin treatment resulted in a 10-fold increase in the β-sheet content of the secondary structure, at the expense of α-helix, and a near-total loss of tertiary structure. Myoglobin can be induced to form the molten globule state at very low alcohol concentrations, for example, in 4% (v/v) aqueous hexafluoroisopropanol (Cort and Andersen 1997). Formalin-treated RNase A can also adopt the molten globule state but only in the presence of ≥80% aqueous ethanol (Fowler et al. 2008). In formalin, the alcohol content is too low to significantly perturb the secondary structure of RNase A because this protein already has high β-sheet content (Rait, O’Leary, et al. 2004). Consequently, the native conformation of RNase A is only modestly perturbed in the presence of formalin. The significance of β-sheet structure in protecting RNase A from denaturation by formalin is further emphasized by the disulfide reduction experiment. Treatment of the reduced and thiol-capped RNase A with formalin resulted in a 16% increase in β-sheet structure, but the tertiary conformation remained essentially intact. Thus, it is likely that the high β-sheet content of RNase A is more important than the disulfide bonds in protecting against formalin-induced unfolding. As discussed below, disulfide bonds may play a more prominent role in stabilizing low molecular weight proteins and those with low β-sheet content.
We performed similar analyses on a variety of proteins differing in structural class and molecular weight (Table 2). These studies reveal that the effect of formalin on protein conformation is more complex than suggested by the above examples. For instance, hemoglobin and bovine serum albumin are both in the all-α structural class with myoglobin, but they do not form the molten globule state in the presence of formalin. This difference is likely due to the increased stability gained by the size of the hydrophobic protein core and the presence of 17 disulfide bonds in the case of bovine serum albumin. For hemoglobin, the increased stability likely arises from the large contact surface area that exists between the four protein subunits (quaternary structure). Another interesting finding was that α-lactalbumin formed the molten globule state despite its similarity to RNase A in molecular weight, number of disulfide bonds, and structural class. A key difference is that β-sheets constitute 33% of the secondary structure of RNase A but only 11% for α-lactalbumin, which may be insufficient to stabilize the protein against formalin denaturation. Another example is concanavalin A, which belongs to the all-β structural class and has a high molecular weight yet is partially unfolded by formalin. Concanavalin A has no stabilizing disulfide bridges, and the center of the protein forms a solvent-accessible pore, which reduces the size of the hydrophobic core. The loss of these stabilizing elements is likely responsible for the partial loss of tertiary structure in the presence of formalin. From the point of view of the mechanism of AR, the important conclusion from these studies is that formalin treatment does not always lock in protein structure. It can alter or completely disrupt the tertiary structure of some proteins, thus destroying conformational protein epitopes.
The SDS-PAGE analyses provided additional information about the formation of formaldehyde cross-links in these proteins. In the case of RNase A, the highly ordered protein conformation likely limits the formation of intramolecular cross-links to the fortuitous spatial orientation of cross-link acceptor–donor side chain pairs. For example, human insulin treated with formaldehyde formed only seven intramolecular cross-links, and eight formaldehyde-reactive amino acid residues were not modified (Metz et al. 2006). The study concluded that protein conformation affected the accessibility and reactivity of the amino acid residues. Analogously, the ordered protein conformation limits the number of ways that RNase A can arrange to form intermolecular cross-links. The result of these constraints is a lack of heterogeneity for both intra- and intermolecular cross-linked species, which results in tight protein bands on the SDS-PAGE gel. In contrast, formalin unfolds myoglobin, giving rise to a fluctuating tertiary structure and a secondary structure consisting of α-helix and β-sheet secondary elements connected by random coil strands (Cort and Andersen 1997). This disorder causes the protein to sample a broad conformational space due to thermal motion, which allows amino acid residues not normally in close proximity to come together to form cross-links. The likely result is the formation of more intramolecular cross-links, a more compressed protein conformation, and a more heterogeneous distribution of cross-linked conformers than was seen with RNase A. This heterogeneity would extend to the intermolecular cross-linked myoglobin oligomers as well. Hence, formalin-treated myoglobin yields diffuse bands that run well below the corresponding native protein on the SDS-PAGE gel, and this behavior becomes more pronounced with increasing temperature. The SDS-PAGE gel also revealed that significantly less monomeric protein was recovered by AR after formalin treatment at 90C than at 24C. The origin of this phenomenon is not clear but may result from an increase in the number of protein modifications that form in the unfolded myoglobin at 90C relative to the molten globule protein at 24C.
The CD thermal scans carried out with formalin-treated RNase A revealed that the conformational disorder of the protein increased in a roughly linear fashion with increasing temperature. This behavior was seen in thermal scans monitored at wavelengths in both the far-UV (secondary structure) and near-UV (tertiary structure) spectral regions. In contrast, native RNase A displayed the expected cooperative melting transition centered at 65C (Brandts et al. 1989; Rait, O’Leary, et al. 2004) in both the far-UV and near-UV thermal scans. Thus, it appears that formaldehyde modifications “spread out” the RNase A melting transition over a broad temperature range. There are several mechanisms that could explain this loss of cooperativity. One explanation is that intramolecular cross-links divide the protein into multiple independent structural domains, each of which unfolds at a unique temperature. This, in combination with the heterogeneity in the number and location of cross-links within individual protein molecules, could result in a continuous non-cooperative melting transition. A second explanation is that the broad melting transition results from the progressive hydrolysis of formaldehyde cross-links with increasing temperature. SDS-PAGE studies of formalin-treated RNase A following calorimetry scans from 20 to 100C revealed the recovery of mostly native protein (Rait, O’Leary, et al. 2004), meaning that the majority of the cross-links hydrolyzed during the thermal scan. In the case of myoglobin, the formalin-treated protein exhibited a highly disordered tertiary structure at room temperature, and no further loss of conformational order was observed upon heating to 95C. Similar behavior was observed in the far-UV CD thermal scan of formalin-treated myoglobin, where a modest linear increase in secondary structure disorder with increasing temperature was observed. These thermal studies provide evidence that any tendency of the formaldehyde to lock in protein secondary and tertiary structure is largely overcome at the high temperatures typically used in AR procedures.
A key observation for both formalin-treated RNase A and myoglobin is that their secondary and tertiary conformational disorder was nearly identical to that of the corresponding native proteins at 95C. Upon cooling to room temperature, there was a modest recovery of protein secondary structure but very little recovery of the protein’s native tertiary structure. In short, both proteins more closely resembled the molten globule conformational state than the native protein conformation. This behavior was independent of the rate at which the proteins were cooled from 95C or how long the proteins were allowed to equilibrate at room temperature prior to the CD spectral scans. In fact, most proteins denature irreversibly when heated above their melting transition temperatures (Freire et al. 1990). Irreversible denaturation of heated proteins typically results from thermally induced covalent protein modifications (Tomizawa et al. 1994; Zale and Klibanov 1986) or from hydrophobic aggregation of the unfolded protein (Freire et al. 1990; Jackson and Brandts 1970). Reversible protein denaturation typically occurs under carefully controlled conditions that cannot be achieved using AR methods or FFPE tissues (Jackson and Brandts 1970). Consequently, all of the formalin-treated proteins we examined (Table 2) exhibited similar levels of conformational disorder at 95C and resembled molten globule proteins following cooling to room temperature.
The study reported here lacks support for those models of AR proposing (1) that cross-links protect proteins from thermal denaturation at a temperature typically used in AR procedures or (2) that heat treatment reverses formaldehyde adducts, allowing the protein and its epitopes to refold so as to regenerate their native conformation upon cooling to room temperature. This study does support a linear epitope model described by Sompuram et al. (Sompuram et al. 2004; Sompuram, Vani, and Bogen 2006; Sompuram, Vani, Hafer, et al. 2006) and Bogen et al. (2009), proposing that the most useful antibodies for immunohistochemistry recognize linear epitopes that become exposed in proteins unfolded by AR methods. Although the study reported here does not specifically address the fourth model, it is likely that AR contributes to the recovery of protein immunoreactivity by restoring the native-like distribution of electrostatic charge within and surrounding the protein epitope (Boenisch 2002, 2006; Rait, O’Leary, et al. 2004).
Considerable secondary structure was maintained in the proteins that we recovered by AR. This may appear to contradict the linear epitope model, which anticipates that only primary protein structure is important for immunohistochemistry following AR (Bogen et al. 2009). To clarify this point, Figure 5C shows the far-UV spectrum of myoglobin cooled to 24C after AR (dash-dot line) and myoglobin denatured in 5% (w/v) SDS at room temperature (dotted line). In SDS, myoglobin is almost completely unfolded and, as a result, yields a very weak far-UV spectrum. In contrast, the far-UV spectrum of recovered myoglobin retains a considerable secondary structure in the molten globule state and can be characterized as α-helix and β-sheet secondary structural elements interconnected by disordered chain segments (Cort and Andersen 1997). Thus, linear epitopes could consist of consecutive amino acids along a disordered chain segment, loop, or turn (primary structure) but could also consist of non-consecutive amino acids positioned along the axis of an α-helix or across the surface of a β-sheet (secondary structure). One consequence of this observation is that the secondary structure of a protein (if known) should be taken into account when using peptide segments derived from that protein to induce the formation of antibodies against linear chain segments (Sompuram et al. 2002).
This model can explain many of the observations reported in the literature regarding immunoreactivity and AR. For example, AR has been reported to improve immunoreactivity in some fresh tissues not treated with formalin (Kakimoto et al. 2008). This can be explained by improved exposure of buried epitopes that results from the unfolding of proteins during AR. In some cases, the amino acids that constitute an epitope may not contain any formaldehyde-reactive side chains or be sterically blocked by nearby cross-linked molecules. In such cases, the use of protein denaturants, such as guanidine hydrochloride and urea (Fowler et al. 2007), or detergents, such as SDS or Triton X-100 (Fowler et al. 2007; Robinson and Vandre 2001), to open gaps in the tissue and unfold the proteins may be sufficient to recover immunoreactivity. Alternately, if such an epitope were to be located on the protein surface and easily accessible, unfolding might be unnecessary. In this event, the opening of channels into the tissue through enzymatic (Jasani and Rhodes 2001; Rojo et al. 2006) or chemical (Lobo et al. 2002; Roth 1986) etching, or simply increasing the time of incubation of antibodies with the FFPE tissue sections in buffer (Boenisch 2002; Puchtler and Meloan 1985), may be all that is required to restore immunoreactivity.
Finally, it is appropriate to question the relevance of the in vitro studies reported here to the behavior of proteins in FFPE tissues. In tissues, macromolecules can account for up to 40% of the total cellular volume (Homouz et al. 2009), which corresponds to a concentration of ~200 g/l (Swaminathan et al. 1997). Such an environment is referred to as a “crowded” solution (Minton 1997). At physiological temperature, the effect of crowded solutions on protein structure is dependent on a number of factors. In general, high concentrations of large volume-excluding co-solutes stabilize the native protein conformation, whereas high concentrations of smaller co-solutes stabilize the unfolded state (Minton 2000; Stagg et al. 2007). However, studies of the pathophysiology of burn injuries (Despa, Orgill, and Lee 2005a, 2005b; Despa, Orgill, Neuwalder, et al. 2005) suggest that virtually all tissue proteins in crowded solutions are denatured and exist as irreversibly unfolded aggregates at temperatures above 60C.
We examined this question further by using CD spectropolarimetry to study the effect of histological processing and heat treatment on the conformation of RNase A tissue surrogates. Tissue surrogates are highly concentrated proteins solutions (≥75 mg/mL) that, upon treatment with 10% formaldehyde, form gels sufficiently rigid that they can be processed, sectioned, and subjected to AR using normal histological methods (Fowler et al. 2007). Because CD requires a nearly transparent solution for light transmission, the tissue surrogates were prepared as thin films on quartz slides. This precludes the accurate determination of the protein concentration and effective path length of the protein film in the beam path. Thus, only the shapes of the CD spectra were compared. The far-UV CD spectra of the dried protein films are shown in Figure 6A. The dried film of native RNase A revealed a significant secondary structure but with an increase in α-helix content relative to RNase A in solution. This is evident by the loss of negative band intensity at 210 nm (see Fig. 1A). When the dried film of RNase A was treated with formalin, there was an increase in the β-sheet content, which can be seen by the increase in negative band intensity at 210 nm relative to that at 222 nm. A similar pattern of behavior was seen for the dried film of formalin-treated RNase A incubated in 100% ethanol. The dried film of RNase A that had been treated with 10% formalin and then processed using conventional histology followed by AR revealed a significant loss of protein secondary structure. The loss in secondary structure was greater than that seen for fixed RNase A heated in solution. In fact, the loss of secondary structure was similar to that of myoglobin denatured in 5% (w/v) SDS (see Fig. 5C). What little secondary structure remained appeared to be essentially all β. As seen in Figure 6B, all of the RNase A dried films revealed a significant loss of tertiary structure regardless of the treatment protocol. The native, formalin-treated, and 100% ethanol-incubated films demonstrated a reduction in tertiary structure similar to that of formalin-treated RNase A solutions following AR (see Fig. 5B). Again, the loss of tertiary structure of the dried film following AR is similar to that of myoglobin denatured in 5% (w/v) SDS (not shown).
Figure 6.
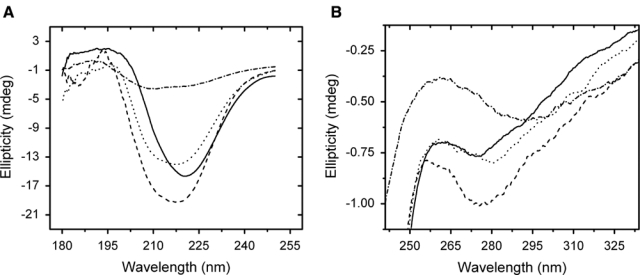
Circular dichroism (CD) spectral scans of RNase A tissue surrogates prepared as dried thin films on quartz slides. Far-ultraviolet (UV) CD spectra (A) and near-UV CD spectra (B) of native RNase A (solid line), formalin-treated RNase A (dashed line), formalin-treated RNase A processed through 100% ethanol (dotted line), and formalin-treated RNase A processed by histology and subjected to antigen retrieval at 121C for 15 min (dash-dot line).
In summary, although the RNase A tissue surrogates do not represent true FFPE tissues, they do reveal the behavior of a dehydrated protein film that was treated with formalin, processed using standard histology, and subjected to AR. The results indicate that following AR, these protein films reveal a loss of secondary and tertiary structure greater than that of the corresponding proteins in solution. Accordingly, the conclusions reached by our in vitro study are likely applicable to FFPE tissues.
Acknowledgments
We thank the American Registry of Pathology for its support and Iosif Gurevich for translating Russian to English.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: The National Cancer Institute supported this work with grants R21 CA134359-01 and R33 CA107844-01. The content of this publication does not necessarily reflect the views or policies of the Department of Defense or the Veterans Health Administration, nor does mention of trade names, commercial products, or organizations imply endorsement by the United States Government.
References
- Boenisch T. 2002. Heat-induced antigen retrieval restores electrostatic forces: prolonging the antibody incubation as an alternative. Appl Immunohistochem Mol Morphol. 10:363–367 [DOI] [PubMed] [Google Scholar]
- Boenisch T. 2006. Heat-induced antigen retrieval: what are we retrieving? J Histochem Cytochem. 54:961–964 [DOI] [PubMed] [Google Scholar]
- Bogen SA, Vani K, Sompuram SR. 2009. Molecular mechanisms of antigen retrieval: antigen retrieval reverses steric interference caused by formalin-induced cross-links. Biotech Histochem. 84:207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolgiano B, Fowler S, Turner K, Sesardic D, Xing DK, Crane DT, Corbel MJ. 2000. Monitoring of diphtheria, pertussis and tetanus toxoids by circular dichroism, fluorescence spectroscopy and size-exclusion chromatography. Dev Biol (Basel). 103:51–59 [PubMed] [Google Scholar]
- Boon ME, Kok LP. 1994. Microwaves for immunohistochemistry. Micron. 25:151–170 [DOI] [PubMed] [Google Scholar]
- Brandts JF, Hu CQ, Lin LN, Mos MT. 1989. A simple model for proteins with interacting domains: applications to scanning calorimetry data. Biochemistry. 28:8588–8596 [DOI] [PubMed] [Google Scholar]
- Cao A, Hu D, Lai L. 2004. Formation of amyloid fibrils from fully reduced hen egg white lysozyme. Protein Sci. 13:319–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cort JR, Andersen NH. 1997. Formation of a molten-globule-like state of myoglobin in aqueous hexafluoroisopropanol. Biochem Biophys Res Commun. 233:687–691 [DOI] [PubMed] [Google Scholar]
- Despa F, Orgill D, Lee R. 2005a. Effects of crowding on the thermal stability of heterogeneous protein solutions. Ann Biomed Eng. 33:1125–1131 [DOI] [PubMed] [Google Scholar]
- Despa F, Orgill D, Lee R. 2005b. Molecular crowding effects on protein stability. Ann N Y Acad Sci. 1066:54–66 [DOI] [PubMed] [Google Scholar]
- Despa F, Orgill D, Neuwalder J, Lee R. 2005. The relative thermal stability of tissue macromolecules and cellular structure in burn injury. Burns. 31:568–577 [DOI] [PubMed] [Google Scholar]
- Dolgikh DA, Gilmanshin RI, Brazhnikov EV, Bychkova VE, Semisotnov GV, Venyaminov SY, Ptitsyn OB. 1981. Alpha-lactalbumin: compact state with fluctuating tertiary structure? FEBS Lett. 136:311–315 [DOI] [PubMed] [Google Scholar]
- Fowler CB, Cunningham RE, O’Leary TJ, Mason JT. 2007. “Tissue surrogates” as a model for archival formalin-fixed paraffin-embedded tissues. Lab Invest. 87:836–846 [DOI] [PubMed] [Google Scholar]
- Fowler CB, O’Leary TJ, Mason JT. 2008. Modeling formalin fixation and histological processing with bovine ribonuclease A: effects of ethanol dehydration on reversal of formaldehyde-induced cross-links. Lab Invest. 88:785–791 [DOI] [PubMed] [Google Scholar]
- Fowles LF, Beck E, Worrall S, Shanley BC, de Jersey J. 1996. The formation and stability of imidazolidinone adducts from acetaldehyde and model peptides: a kinetic study with implications for protein modification in alcohol abuse. Biochem Pharmacol. 51:1259–1267 [DOI] [PubMed] [Google Scholar]
- Fox CH, Johnson FB, Whiting J, Roller PP. 1985. Formaldehyde fixation. J Histochem Cytochem. 33:845–853 [DOI] [PubMed] [Google Scholar]
- Fraenkel-Conrat H, Brandon BA, Olcott HS. 1947. The reaction of formaldehyde with proteins; participation of indole groups; gramicidin. J Biol Chem. 168:99–118 [PubMed] [Google Scholar]
- Fraenkel-Conrat H, Mecham DK. 1949. The reaction of formaldehyde with proteins: demonstration of intermolecular cross-linking by means of osmotic pressure measurements. J Biol Chem. 177:477–486 [PubMed] [Google Scholar]
- Fraenkel-Conrat H, Ollcott HS. 1948. Reaction of formaldehyde with proteins: cross-linking of amino groups with phenol, imidazole, or indole groups. J Biol Chem. 174:827–843 [PubMed] [Google Scholar]
- Freire E, van Osdol WW, Mayorga OL, Sanchez-Ruiz JM. 1990. Calorimetrically determined dynamics of complex unfolding transitions in proteins. Annu Rev Biophys Chem. 19:159–188 [DOI] [PubMed] [Google Scholar]
- Homouz D, Stagg L, Wittung-Stafshede P, Cheung MS. 2009. Macromolecular crowding modulates folding mechanism of alpha/beta protein apoflavodoxin. Biophys J. 92:671–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irace G, Bismuto E, Savy F, Colonna G. 1986. Unfolding pathway of myoglobin: molecular properties of intermediate forms. Arch Biochem Biophys. 244:459–469 [DOI] [PubMed] [Google Scholar]
- Jackson WM, Brandts JF. 1970. Thermodynamics of protein denaturation: a calorimetric study of the reversible denaturation of chymotrypsinogen and conclusions regarding the accuracy of the two-state approximation. Biochemistry. 9:2294–2301 [DOI] [PubMed] [Google Scholar]
- Jasani B, Rhodes A. 2001. The role and mechanism of high-temperature antigen retrieval in diagnostic pathology. Curr Diagn Pathol. 7:153–160 [Google Scholar]
- Kakimoto K, Takekoshi S, Miyajima K, Osamura RY. 2008. Hypothesis for the mechanism for heat-induced antigen retrieval occurring on fresh frozen sections without formalin-fixation in immunohistochemistry. J Mol Histol. 39:389–399 [DOI] [PubMed] [Google Scholar]
- Kamatari YO, Konno T, Kataoka M, Akasaka K. 1998. The methanol-induced transition and the expanded helical conformation in hen lysozyme. Protein Sci. 7:681–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S, Sundd M, Jagannadham MV. 2002. Alcohol and temperature induced conformational transitions in ervatamin B: sequential unfolding of domains. J Biochem Mol Biol. 35:155–164 [DOI] [PubMed] [Google Scholar]
- Kunkel GR, Mehrabian M, Martinson HG. 1981. Contact-site cross-linking agents. Mol Cell Biochem. 34:3–13 [DOI] [PubMed] [Google Scholar]
- Le Botlan DJ, Mechin BG, Martin GJ. 1983. Proton and carbon-13 nuclear magnetic resonance spectrometry of formaldehyde in water. Anal Chem. 55:587–591 [Google Scholar]
- Lobo MV, Alonso FJ, Arenas MI, Caso E, Fraile B, del Rio RM. 2002. Ultrastructural staining with sodium metaperiodate and sodium borohydride. J Histochem Cytochem. 50:11–19 [DOI] [PubMed] [Google Scholar]
- Mason JT, O’Leary TJ. 1991. Effects of formaldehyde fixation on protein secondary structure: a calorimetric and infrared spectroscopic investigation. J Histochem Cytochem. 39:225–229 [DOI] [PubMed] [Google Scholar]
- Mehl AF, Crawford MA, Zhang L. 2009. Determination of myoglobin stability by circular dichroism spectroscopy: classic and modern data analysis. J Chem Educ. 86:600–602 [Google Scholar]
- Metz B, Jiskoot W, Hennink WE, Crommelin DJ, Kersten GF. 2003. Physicochemical and immunochemical techniques predict the quality of diphtheria toxoid vaccines. Vaccine. 22:156–167 [DOI] [PubMed] [Google Scholar]
- Metz B, Kersten GFA, Baart GJ, de Jong A, Meiring H, ten Hove J, van Steenbergen MJ, Hennink WE, Crommelin DJ, Jiskoot W. 2006. Identification of formaldehyde-induced modifications in proteins: reactions with insulin. Bioconjug Chem. 17:815–822 [DOI] [PubMed] [Google Scholar]
- Metz B, Kersten GFA, Hoogerhout P, Brugghe HF, Timmermans HA, de Jong A, Meiring H, ten Hove J, Hennink WM, Crommelin DJA, et al. 2004. Identification of formaldehyde-induced modifications in proteins: reactions with model peptides. J Biol Chem. 279:6235–6243 [DOI] [PubMed] [Google Scholar]
- Miller RT, Swanson PE, Wick MR. 2000. Fixation and epitope retrieval in diagnostic immunohistochemistry: a concise review with practical considerations. Appl Immunohistochem Mol Morphol. 8:228–235 [DOI] [PubMed] [Google Scholar]
- Minton AP. 1997. Influence of excluded volume upon macromolecular structure and associations in ‘crowded’ media. Curr Opin Biotechnol. 1:65–69 [DOI] [PubMed] [Google Scholar]
- Minton AP. 2000. Effect of a concentrated “inert” macromolecular cosolute on the stability of a globular protein with respect to denaturation by heat and by chaotropes: a statistical-thermodynamic model. Biophys J. 78:101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JM, Navabi H, Jasani B. 1997. Role of calcium chelation in high-temperature antigen retrieval at different pH values. J Pathol. 182:233–237 [DOI] [PubMed] [Google Scholar]
- Morgan JM, Navabi H, Schmid KW, Jasani B. 1994. Possible role of tissue-bound calcium ions in citrate-mediated high-temperature antigen retrieval. J Pathol. 174:301–307 [DOI] [PubMed] [Google Scholar]
- Naseem F, Khan RH. 2003. Effect of ethylene glycol and polyethylene glycol on the acid-unfolded state of trypsinogen. J Protein Chem. 22:677–682 [DOI] [PubMed] [Google Scholar]
- Pace CN, Trevino S, Prabhakaran E, Scholtz JM. 2004. Protein structure, stability and solubility in water and other solvents. Philos Trans R Soc B Biol Sci. 359:1225–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchtler H, Meloan SN. 1985. On the chemistry of formaldehyde fixation and its effects on immunohistochemical reactions. Histochemistry. 82:201–204 [DOI] [PubMed] [Google Scholar]
- Rait VK, O’Leary TJ, Mason JT. 2004. Modeling formalin fixation and antigen retrieval with bovine pancreatic ribonuclease A: I-structural and functional alterations. Lab Invest. 84:292–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rait VK, Xu L, O’Leary TJ, Mason JT. 2004. Modeling formalin fixation and antigen retrieval with bovine pancreatic RNase A: II. Interrelationship of cross-linking, immunoreactivity, and heat treatment. Lab Invest. 84:300–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JM, Vandre DD. 2001. Antigen retrieval in cells and tissues: enhancement with sodium dodecyl sulfate. Histochem Cell Biol. 116:119–130 [DOI] [PubMed] [Google Scholar]
- Rojo F, Tabernero J, Albanell J, Van CE, Ohtsu A, Doi T, Koizumi W, Shirao K, Takiuchi H, Ramon y Cajal S, et al. 2006. Pharmacodynamic studies of gefitinib in tumor biopsy specimens from patients with advanced gastric carcinoma. J Clin Oncol. 24:4309–4316 [DOI] [PubMed] [Google Scholar]
- Roth J. 1986. Post-embedding cytochemistry with gold-labelled reagents: a review. J Microsc. 143:125–137 [DOI] [PubMed] [Google Scholar]
- Rott GM, Poverennyi AM. 1982. Initiation by formaldehyde of conformational changes from helix to globule in the myoglobin molecule. Molekulyarnaya Biologiya (Mosk). 16:998–1003 [PubMed] [Google Scholar]
- Shi SR, Cote RJ, Taylor CR. 1997. Antigen retrieval immunohistochemistry: past, present, and future. J Histochem Cytochem. 45:327–343 [DOI] [PubMed] [Google Scholar]
- Shi SR, Cote RJ, Taylor CR. 2001. Antigen retrieval techniques: current perspectives. J Histochem Cytochem. 49:931–937 [DOI] [PubMed] [Google Scholar]
- Shi SR, Cote RJ, Yang C, Chen C, Xu HJ, Benedict WF, Taylor CR. 1996. Development of an optimal protocol for antigen retrieval: a ‘test battery’ approach exemplified with reference to the staining of retinoblastoma protein (pRB) in formalin-fixed paraffin sections. J Pathol. 179:347–352 [DOI] [PubMed] [Google Scholar]
- Shi SR, Key ME, Kalra KL. 1991. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem. 39:741–748 [DOI] [PubMed] [Google Scholar]
- Shi SR, Liu C, Balgley BM, Lee C, Taylor CR. 2006. Protein extraction from formalin-fixed, paraffin-embedded tissue sections: quality evaluation by mass spectrometry. J Histochem Cytochem. 54:739–743 [DOI] [PubMed] [Google Scholar]
- Singh BR, DasGupta BR. 1989. Molecular differences between type A botulinum neurotoxin and its toxoid. Toxicon. 27:403–410 [DOI] [PubMed] [Google Scholar]
- Sompuram SR, Kodela V, Ramanathan H, Wescott C, Radcliffe G, Bogen SA. 2002. Synthetic peptides identified from phage-displayed combinatorial libraries as immunodiagnostic assay surrogate quality-control targets. Clin Chem. 48:410–420 [PubMed] [Google Scholar]
- Sompuram SR, Vani K, Bogen SA. 2006. A molecular model of antigen retrieval using a peptide array. Am J Clin Pathol. 125:91–98 [PubMed] [Google Scholar]
- Sompuram SR, Vani K, Hafer LJ, Bogen SA. 2006. Antibodies immunoreactive with formalin-fixed tissue antigens recognize linear protein epitopes. Am J Clin Pathol. 125:82–90 [PubMed] [Google Scholar]
- Sompuram SR, Vani K, Messana E, Bogen SA. 2004. A molecular mechanism of formalin fixation and antigen retrieval. Am J Clin Pathol. 121:190–199 [DOI] [PubMed] [Google Scholar]
- Sreerama N, Venyaminov SY, Woody RW. 2001. Analysis of protein circular dichroism spectra based on the tertiary structure classification. Anal Biochem. 299:271–274 [DOI] [PubMed] [Google Scholar]
- Sreerama N, Woody RW. 2000. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 287:252–260 [DOI] [PubMed] [Google Scholar]
- Stagg L, Zhang SQ, Cheung MS, Wittung-Stafshede P. 2007. Molecular crowding enhances native structure and stability of alpha/beta protein flavodoxin. Proc Natl Acad Sci U S A. 108:18976–18981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suurmeijer AJ, Boon ME. 1993. Notes on the application of microwaves for antigen retrieval in paraffin and plastic tissue sections. Eur J Morphol. 31:144–150 [PubMed] [Google Scholar]
- Swaminathan R, Hoang CP, Verkman AS. 1997. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys J. 72:1900–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CR. 1986. Immunomicroscopy: a diagnostic tool for the surgical pathologist. Philadelphia: Saunders [Google Scholar]
- Tomizawa H, Yamada H, Imoto T. 1994. The mechanism of irreversible inactivation of lysozyme at pH 4 and 100 degrees C. Biochemistry. 33:13032–13037 [DOI] [PubMed] [Google Scholar]
- van der Oord AH, Wesdorp JJ, van Dam AF, Verheij JA. 1969. Occurrence and nature of equine and bovine myoglobin dimers. Eur J Biochem. 10:140–145 [DOI] [PubMed] [Google Scholar]
- Volkin DB, Klibanov A. 1989. Minimizing protein inactivation. In Creighton TE, ed. Protein function: a practical approach. Oxford, UK: Oxford University Press, 1–24 [Google Scholar]
- Wang X, Moore SC, Laszckzak M, Ausio J. 2000. Acetylation increases the alpha-helical content of the histone tails of the nucleosome. J Biol Chem. 275:35013–35020 [DOI] [PubMed] [Google Scholar]
- Woody RW. 1995. Circular dichroism. Methods Enzymol. 246:34–71 [DOI] [PubMed] [Google Scholar]
- Woody RW, Dunker AK. 1996. Aromatic and cysteine sidechain circular dichroism in proteins. In Fasman GD, ed. Circular dichroism and the conformational analysis of biomolecules. New York: Plenum, 109–157 [Google Scholar]
- Zaia J, Annan RS, Biemann K. 1992. The correct molecular weight of myoglobin, a common calibrant for mass spectrometry. Rapid Commun Mass Spectrom. 6:32–36 [DOI] [PubMed] [Google Scholar]
- Zale SE, Klibanov AM. 1986. Why does ribonuclease irreversibly inactivate at high temperatures? Biochemistry. 25:5432–5444 [DOI] [PubMed] [Google Scholar]