

Abstract
In the title compound, C10H8N2S, the 2-aminobenzothiazole and propyne groups are not coplanar [dihedral angle = 71.51 (1)°]. The crystal structure is stabilized by strong intermolecular N—H⋯N hydrogen bonds and C—H⋯C, C—H⋯π and F-type aromatic–aromatic [centroid–centroid distance = 3.7826 (12) Å] interactions are also observed.
Related literature
For the biological activity of heterocyclic compounds, see: Xuan et al. (2001 ▶) and of benzothiazole and benzimidazole compounds, see: Caroti et al. (1989 ▶); Paget et al. (1969 ▶); Da Settimo et al. (1992 ▶); Johnson et al. (2009 ▶); Kus et al. (1996 ▶). For N—H⋯N hydrogen bonding, see: Mingos & Braga (2004 ▶). For F-type aromatic–aromatic interactions, see: Zhang et al. (2010 ▶). For details of the synthesis, see: Lilienkampf et al. (2009 ▶). For recently reported small crystal structures and their antimicrobial activity, see: Singh, Agarwal & Awasthi (2011 ▶); Singh, Agarwal, Mahawar & Awasthi (2011 ▶); Awasthi et al. (2009 ▶).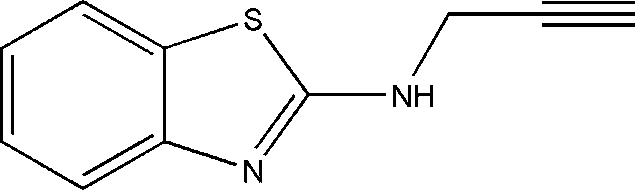
Experimental
Crystal data
C10H8N2S
M r = 188.25
Monoclinic,
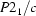
a = 6.8048 (4) Å
b = 8.6071 (5) Å
c = 15.8244 (8) Å
β = 99.445 (5)°
V = 914.26 (9) Å3
Z = 4
Mo Kα radiation
μ = 0.30 mm−1
T = 293 K
0.40 × 0.39 × 0.38 mm
Data collection
Oxford Diffraction Xcalibur Eos diffractometer
Absorption correction: multi-scan (CrysAlis PRO; Oxford Diffraction, 2009 ▶) T min = 0.771, T max = 1.000
4188 measured reflections
2454 independent reflections
1428 reflections with I > 2σ(I)
R int = 0.045
Refinement
R[F 2 > 2σ(F 2)] = 0.043
wR(F 2) = 0.131
S = 0.88
2454 reflections
128 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.17 e Å−3
Δρmin = −0.27 e Å−3
Data collection: CrysAlis PRO (Oxford Diffraction, 2009 ▶); cell refinement: CrysAlis PRO; data reduction: CrysAlis PRO; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: Mercury (Macrae et al., 2006 ▶); software used to prepare material for publication: publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811035136/zj2020sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811035136/zj2020Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811035136/zj2020Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
Cg1 is the centroid of the S1,C1,C6,N1,C7 ring.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N2—HN2⋯N1i | 0.87 (3) | 2.05 (3) | 2.910 (2) | 170 (2) |
| C8—H8A⋯C4ii | 0.98 | 2.86 | 3.756 (3) | 153 (1) |
| C10—H10⋯C1iii | 0.89 | 2.87 | 3.687 (3) | 153 (1) |
| C10—H10⋯C6iii | 0.89 | 2.89 | 3.776 (3) | 174 (1) |
| C10—H10⋯Cg1iii | 0.89 | 2.74 | 3.548 (3) | 151 |
Symmetry codes: (i) 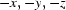 ; (ii)
; (ii) 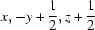 ; (iii)
; (iii) 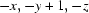 .
.
Acknowledgments
AA and MKS are thankful to the University Grant Commission (scheme No. 34–311/2008), New Delhi, and Banaras Hindu University (BHU), Varanasi, India, respectively, for financial assistance. The authors are highly thankful to the department of Chemistry, Banaras Hindu University, for providing the single-crystal X-ray data.
supplementary crystallographic information
Comment
Heterocyclic compounds containing nitrogen, sulfur, oxygen etc have immense importance especially in pharmaceutical industry. Most of the modern drugs contain one or more heteroatom in their scaffold. Further, oxidation of nitrogen in heterocycle plays key role in bioactivity of these scaffolds (Xuan, et al., 2001). It is well documented that benzothiazole and benzimidazole derivatives show wide range of biological activities including antilipidemic (Caroti, et al., 1989), antimicrobial (Kus, et al., 1996), antiviral (Paget, et al., 1969) anti-inflammatory and analgesic properties (Da Settimo, et al., 1992). Moreover, 2-aminobenzimidazole/benzothiazole derivatives are common intermediate for the synthesis of various drugs. Anticancer properties of benzothiazole derivatives in cell based assays are also well documented (Johnson, et al., 2009).Our research interest involves the antimicrobial activities of small molecule (Awasthi, et al., 2009). Recently, we have reported several small crystal structures (Singh, Agarwal & Awasthi 2011, Singh, Agarwal, Mahawar et al., 2011). We report here the crystal structure of N-(prop-2-yn-1-yl)-1,3-benzothiazol-2-amine (Figure 1).
In the title compound, the C7—N2 single bond (1.342 Å) is shorter than normal C—N bond (1.47 Å) suggesting a delocalized double bond in benzothiazole moiety. Further, N2—C8 bond (1.438 Å) is also shorter than a standard C—N bond distance due to delocalization of electrons. Again, it is evident from the crystal structure that the title compound is stabilized by strong intermolecular N—H···N hydrogen bonding as well as C—H···π interactions and aromatic π···π stacking interaction resulting in the formation of supramolecular arrangement in the cystal as seen in the crystal packing along b axis (Figure 2, Table 1). The intermolecular hydrogen bond distance between N2···N1 (2.91 Å) is shorter than N···N average bond range 3.15 Å, suggesting strong hydrogen bonding (Mingos & Braga, 2004).
In the cystal packing two benzothiazole skeletons are arranged in an antiparallel fashion by F-type aromatic–aromatic interactions and form a dimer, the ring A and C of an benzothiazole skeleton stacks with the ring C and A of another adjacent benzothiazole skeleton, respectively. The distance of CgA and CgC is 3.783 Å, where CgA and CgC are the center of ring A and C, respectively and the centroid - centroid distance between two adjacent benzothiazole ring is 3.879Å (Zhang et al., 2010). 2-Aminobenzothiazole and propyne group are not co-planar with a dihedral angle of 71.51°. The torsion angles of C7—N2—C8—C9 and C10—C9—C8—N2 are found 91.1 (3) and 44 (7)° respectively. The CCDC No. of the crystal is 806158.
Experimental
The synthesis of the title compound was carried out according to the published procedure (Lilienkampf, et al., 2009). Briefly, to a solution of 2-aminobenzothaizole (0.90 g, 6 mmol) in dry acetone was added anhydrous K2CO3 (4.97 g m, 32 mmol) and reaction mixture was further refluxed for 15–30 minutes. Subsequently, KI (0.50 g m, 3 mmol) and propargyl bromide (0.64 ml, 7.2 mmol) were added and further refluxed the reaction mixture for 18 hrs. The reaction mixture was cooled, filtered, and the filtrate was evaporated in vacuo to give the product which was purified by column chromatography using hexane and dichloromethane (65:35) as eluent. The product was crystallized from hexane:dichloromethane (1:1). The light pink colored crystals were obtained by slow evaporation of solvent at room temperature in several days. Yield = 20%. MS (Macromass G) m/z = 188 (M+), Rf = 0.59 (98:2, CH2Cl2: MeOH), m.p.= 215°, Elemental analysis (Perkin –Elmer 240 C elemental analyzer) Calculated for: C10H8N2S (%) C– 63.8, H-4.2, N -14.9, S-17.1, found C-63.9, H-4.5, N -14.7, S-16.9. 1H NMR (CDCl3), 8.35 (s, 1H, NH), 7.71–7.68 (m, 1H), 7.44–7.42(m, 1H), 7.26–7.21 (m, 1H), 7.07–7.02(m, 1H), 4.18–4.17(m, 2H, CH2) 3.21 (s, 1H, CH).
Refinement
All H atoms were located from difference Fourier map (range of C—H = 0.87 - 0.98 Å and N–H = 0.87 Å) and allowed to refine freely.
Figures
Fig. 1.

ORTEP diagram of the molecule with thermal ellipsoids drawn at 50% probability level Color code: White: C; yellow: S; blue: N; white: H.
Fig. 2.

Packing diagram of molecule viewed through b plane showing supramolecular arrangement, Intermolecular N—H···N hydrogen bonding, C—H···π interactions and F-type aromatic-aromatic interaction.
Fig. 3.

The formation of the title compound.
Crystal data
| C10H8N2S | F(000) = 392 |
| Mr = 188.25 | Dx = 1.368 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 6.8048 (4) Å | Cell parameters from 1697 reflections |
| b = 8.6071 (5) Å | θ = 3.5–29.1° |
| c = 15.8244 (8) Å | µ = 0.30 mm−1 |
| β = 99.445 (5)° | T = 293 K |
| V = 914.26 (9) Å3 | Block, light pink |
| Z = 4 | 0.40 × 0.39 × 0.38 mm |
Data collection
| Oxford Diffraction Xcalibur Eos diffractometer | 2454 independent reflections |
| Radiation source: fine-focus sealed tube | 1428 reflections with I > 2σ(I) |
| graphite | Rint = 0.045 |
| ω scans | θmax = 29.1°, θmin = 3.5° |
| Absorption correction: multi-scan (CrysAlis PRO; Oxford Diffraction, 2009) | h = −9→5 |
| Tmin = 0.771, Tmax = 1.000 | k = −10→9 |
| 4188 measured reflections | l = −19→19 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.043 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.131 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.88 | w = 1/[σ2(Fo2) + (0.0804P)2] where P = (Fo2 + 2Fc2)/3 |
| 2454 reflections | (Δ/σ)max = 0.05 |
| 128 parameters | Δρmax = 0.17 e Å−3 |
| 0 restraints | Δρmin = −0.27 e Å−3 |
Special details
| Geometry. All s.u.'s (except the s.u. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell s.u.'s are taken into account individually in the estimation of s.u.'s in distances, angles and torsion angles; correlations between s.u.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell s.u.'s is used for estimating s.u.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| HN2 | −0.004 (4) | 0.093 (3) | 0.0660 (17) | 0.085 (8)* | |
| S1 | 0.42981 (8) | 0.29415 (6) | 0.04155 (3) | 0.0554 (2) | |
| N1 | 0.2087 (2) | 0.08399 (18) | −0.04719 (10) | 0.0464 (4) | |
| N2 | 0.0971 (3) | 0.1551 (2) | 0.07821 (11) | 0.0548 (5) | |
| C7 | 0.2251 (3) | 0.1665 (2) | 0.02239 (12) | 0.0444 (5) | |
| C6 | 0.3657 (3) | 0.1159 (2) | −0.09078 (12) | 0.0457 (5) | |
| C9 | −0.0017 (3) | 0.3891 (3) | 0.14573 (12) | 0.0505 (5) | |
| C1 | 0.5014 (3) | 0.2280 (2) | −0.05259 (13) | 0.0497 (5) | |
| C8 | 0.1095 (3) | 0.2429 (3) | 0.15623 (14) | 0.0541 (5) | |
| H8A | 0.250 (2) | 0.2658 (4) | 0.1781 (3) | 0.065* | |
| H8B | 0.0582 (7) | 0.1790 (9) | 0.1993 (6) | 0.065* | |
| C2 | 0.6662 (4) | 0.2711 (3) | −0.08909 (17) | 0.0638 (7) | |
| H2 | 0.750 (2) | 0.3410 (19) | −0.0650 (7) | 0.077* | |
| C5 | 0.3948 (3) | 0.0473 (3) | −0.16713 (14) | 0.0577 (6) | |
| H5 | 0.306 (2) | −0.0263 (18) | −0.1935 (7) | 0.069* | |
| C3 | 0.6923 (4) | 0.2005 (3) | −0.16390 (17) | 0.0705 (7) | |
| H3 | 0.805 (3) | 0.2274 (8) | −0.1895 (7) | 0.085* | |
| C4 | 0.5588 (4) | 0.0906 (3) | −0.20341 (16) | 0.0674 (7) | |
| H4 | 0.5794 (6) | 0.0458 (12) | −0.2544 (14) | 0.081* | |
| C10 | −0.0941 (4) | 0.5029 (3) | 0.13609 (15) | 0.0737 (7) | |
| H10 | −0.165 (2) | 0.591 (2) | 0.1287 (3) | 0.088* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0584 (4) | 0.0468 (4) | 0.0591 (4) | −0.0137 (2) | 0.0038 (3) | −0.0032 (2) |
| N1 | 0.0444 (9) | 0.0428 (9) | 0.0508 (9) | −0.0020 (7) | 0.0046 (7) | −0.0031 (7) |
| N2 | 0.0561 (12) | 0.0534 (11) | 0.0553 (10) | −0.0136 (9) | 0.0101 (9) | −0.0129 (9) |
| C7 | 0.0454 (11) | 0.0370 (10) | 0.0484 (10) | −0.0012 (9) | 0.0009 (8) | 0.0009 (8) |
| C6 | 0.0435 (11) | 0.0409 (11) | 0.0506 (11) | 0.0074 (9) | 0.0010 (9) | 0.0062 (9) |
| C9 | 0.0494 (12) | 0.0545 (13) | 0.0476 (11) | −0.0088 (10) | 0.0081 (9) | −0.0043 (9) |
| C1 | 0.0482 (11) | 0.0429 (12) | 0.0567 (12) | 0.0018 (9) | 0.0045 (9) | 0.0114 (9) |
| C8 | 0.0607 (13) | 0.0532 (13) | 0.0480 (11) | −0.0043 (11) | 0.0078 (10) | −0.0035 (10) |
| C2 | 0.0572 (14) | 0.0577 (14) | 0.0773 (17) | −0.0066 (11) | 0.0130 (12) | 0.0125 (12) |
| C5 | 0.0582 (13) | 0.0584 (14) | 0.0544 (12) | 0.0092 (11) | 0.0030 (10) | 0.0013 (10) |
| C3 | 0.0617 (15) | 0.0776 (19) | 0.0761 (17) | 0.0052 (13) | 0.0227 (13) | 0.0242 (14) |
| C4 | 0.0686 (16) | 0.0786 (17) | 0.0572 (13) | 0.0226 (14) | 0.0164 (12) | 0.0117 (12) |
| C10 | 0.0827 (17) | 0.0700 (17) | 0.0664 (15) | 0.0148 (15) | 0.0062 (13) | −0.0025 (13) |
Geometric parameters (Å, °)
| S1—C1 | 1.738 (2) | C1—C2 | 1.394 (3) |
| S1—C7 | 1.7612 (19) | C8—H8A | 0.9843 |
| N1—C7 | 1.299 (2) | C8—H8B | 0.9843 |
| N1—C6 | 1.391 (2) | C2—C3 | 1.368 (3) |
| N2—C7 | 1.342 (3) | C2—H2 | 0.8728 |
| N2—C8 | 1.438 (3) | C5—C4 | 1.388 (3) |
| N2—HN2 | 0.87 (3) | C5—H5 | 0.9257 |
| C6—C5 | 1.388 (3) | C3—C4 | 1.387 (3) |
| C6—C1 | 1.402 (3) | C3—H3 | 0.9510 |
| C9—C10 | 1.160 (3) | C4—H4 | 0.9255 |
| C9—C8 | 1.464 (3) | C10—H10 | 0.8942 |
| C1—S1—C7 | 88.46 (10) | C9—C8—H8A | 108.9 |
| C7—N1—C6 | 110.27 (17) | N2—C8—H8B | 108.9 |
| C7—N2—C8 | 125.02 (19) | C9—C8—H8B | 108.9 |
| C7—N2—HN2 | 118.2 (16) | H8A—C8—H8B | 107.7 |
| C8—N2—HN2 | 116.7 (16) | C3—C2—C1 | 117.9 (2) |
| N1—C7—N2 | 122.92 (18) | C3—C2—H2 | 121.1 |
| N1—C7—S1 | 116.23 (15) | C1—C2—H2 | 121.1 |
| N2—C7—S1 | 120.83 (15) | C6—C5—C4 | 119.0 (2) |
| C5—C6—N1 | 125.34 (19) | C6—C5—H5 | 120.5 |
| C5—C6—C1 | 119.37 (19) | C4—C5—H5 | 120.5 |
| N1—C6—C1 | 115.29 (17) | C2—C3—C4 | 121.7 (2) |
| C10—C9—C8 | 178.2 (2) | C2—C3—H3 | 119.2 |
| C2—C1—C6 | 121.5 (2) | C4—C3—H3 | 119.2 |
| C2—C1—S1 | 128.74 (19) | C3—C4—C5 | 120.6 (2) |
| C6—C1—S1 | 109.74 (14) | C3—C4—H4 | 119.7 |
| N2—C8—C9 | 113.45 (18) | C5—C4—H4 | 119.7 |
| N2—C8—H8A | 108.9 | C9—C10—H10 | 180.0 |
| C6—N1—C7—N2 | −177.76 (18) | C7—S1—C1—C2 | −179.2 (2) |
| C6—N1—C7—S1 | 1.1 (2) | C7—S1—C1—C6 | 0.15 (15) |
| C8—N2—C7—N1 | 179.58 (19) | C7—N2—C8—C9 | 91.1 (3) |
| C8—N2—C7—S1 | 0.7 (3) | C10—C9—C8—N2 | 44 (7) |
| C1—S1—C7—N1 | −0.77 (16) | C6—C1—C2—C3 | 0.0 (3) |
| C1—S1—C7—N2 | 178.15 (17) | S1—C1—C2—C3 | 179.24 (17) |
| C7—N1—C6—C5 | 179.50 (18) | N1—C6—C5—C4 | −179.97 (17) |
| C7—N1—C6—C1 | −1.0 (2) | C1—C6—C5—C4 | 0.6 (3) |
| C5—C6—C1—C2 | −0.7 (3) | C1—C2—C3—C4 | 0.8 (4) |
| N1—C6—C1—C2 | 179.82 (18) | C2—C3—C4—C5 | −0.9 (3) |
| C5—C6—C1—S1 | 179.97 (15) | C6—C5—C4—C3 | 0.2 (3) |
| N1—C6—C1—S1 | 0.4 (2) |
Hydrogen-bond geometry (Å, °)
| Cg1 is the centroid of the S1,C1,C6,N1,C7 ring. |
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—HN2···N1i | 0.87 (3) | 2.05 (3) | 2.910 (2) | 170 (2) |
| C8—H8A···C4ii | 0.98 | 2.86 | 3.756 (3) | 153.(1) |
| C10—H10···C1iii | 0.89 | 2.87 | 3.687 (3) | 153 (1) |
| C10—H10···C6iii | 0.89 | 2.89 | 3.776 (3) | 174 (1) |
| C10—H10···Cg1iii | 0.89 | 2.74 | 3.548 (3) | 151 |
Symmetry codes: (i) −x, −y, −z; (ii) x, −y+1/2, z+1/2; (iii) −x, −y+1, −z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: ZJ2020).
References
- Awasthi, S. K., Mishra, N., Kumar, B., Sharma, M., Bhattacharya, A., Mishra, L. C. & Bhasin, V. K. (2009). Med. Chem. Res. 18, 407–420.
- Caroti, P., Ceccotti, C., Settimo, F. D., Primofiore, G., Franzone, J. S., Reboani, M. C. & Cravanzola, C. (1989). Farmaco, 44, 327–355. [PubMed]
- Da Settimo, A., Primofiore, G., Da Settimo, F. & Marini, A. M. (1992). Farmaco, 47, 1293–313. [PubMed]
- Johnson, S. L., Chen, L. H., Barille, E., Emdadim, A., Sabet, M., Yuan, H., Wei, J., Guiney, D. & Palencia, M. (2009). Bioorg. Med. Chem. 17, 3352–68. [DOI] [PMC free article] [PubMed]
- Kus, C., Goker, H., Ayhan, G., Ertan, R., Antanlar, N. & Akin, A. (1996). Farmaco, 51, 413–417. [PubMed]
- Lilienkampf, A., Mao, J., Wan, B., Wang, Y., Franzblau, S. G. & Kozikowski, A. P. (2009). J. Med. Chem. 52, 2109–2118. [DOI] [PubMed]
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst. 39, 453–457.
- Mingos, D. M. P. & Braga, D. (2004). Supramolecular Assembly via Hydrogen Bonds, Vol. 2, pp. 39–41. Berlin: Springer.
- Oxford Diffraction (2009). CrysAlis PRO Oxford Diffraction Ltd, Yarnton, England.
- Paget, C. J., Kisnar, K., Stone, R. L. & De Long, D. C. (1969). J. Med. Chem. 12, 1010–1015. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Singh, M. K., Agarwal, A. & Awasthi, S. K. (2011). Acta Cryst. E67, o1137. [DOI] [PMC free article] [PubMed]
- Singh, M. K., Agarwal, A., Mahawar, C. & Awasthi, S. K. (2011). Acta Cryst. E67, o1382. [DOI] [PMC free article] [PubMed]
- Westrip, S. P. (2010). J Appl Cryst 43, 920–925.
- Xuan, X., Zhiguang, X., Yifang, L. & Weihua, L. (2001). Chin. J. Med. Chem. 11, 317–321.
- Zhang, Z.-T., Gao, M.-X. & He, Q. (2010). J. Chem. Crystallogr. 40, 841–845.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811035136/zj2020sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811035136/zj2020Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811035136/zj2020Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report