

Abstract
Protein kinase A (PKA) is an evolutionarily conserved negative regulator of the hedgehog (Hh) signal transduction pathway. PKA is known to be required for the proteolytic processing event that generates the repressor forms of the Ci and Gli transcription factors that keep target genes off in the absence of Hh. Here, we show that complete loss of PKA activity in the mouse leads to midgestation lethality and a completely ventralized neural tube, demonstrating that PKA is as strong a negative regulator of the sonic hedgehog (Shh) pathway as patched 1 (Ptch1) or suppressor of fused (Sufu). Genetic analysis shows that although PKA is important for production of the repressor form of Gli3, the principal function of PKA in the Shh pathway in neural development is to restrain activation of Gli2. Activation of the Hh pathway in PKA mutants depends on cilia, and the catalytic and regulatory subunits of PKA are localized to a compartment at the base of the primary cilia, just proximal to the basal body. The data show that PKA does not affect cilia length or trafficking of smoothened (Smo) in the cilium. Instead, we find that there is a significant increase in the level of Gli2 at the tips of cilia of PKA-null cells. The data suggest a model in which PKA acts at the base of the cilium after Gli proteins have transited the primary cilium; in this model the sequential movement of Gli proteins between compartments in the cilium and at its base controls accessibility of Gli proteins to PKA, which determines the fates of Gli proteins and the activity of the Shh pathway.
Keywords: Gli2, Gli3, Hedgehog, PKA, Cilia, Neural patterning, Mouse
INTRODUCTION
The hedgehog (Hh) family of secreted signaling proteins control proliferation, patterning and morphogenesis of nearly every tissue in both Drosophila and vertebrate embryos (Ingham and McMahon, 2001; Jiang and Hui, 2008; Ingham et al., 2011). The activity of the hedgehog pathway is also important in the development of many types of tumor (Teglund and Toftgard, 2010; Theunissen and de Sauvage, 2009; Toftgard, 2000). Despite its significance, important aspects of vertebrate hedgehog signaling have diverged from the Drosophila pathway and are not well understood. For example, Suppressor of fused (Sufu) is dispensable in Drosophila but has a crucial negative regulatory role in Hh signaling in vertebrates that is under active investigation (Tukachinsky et al., 2010; Humke et al., 2010; Wang et al., 2010; Jia et al., 2009). Vertebrate Hh signaling requires the primary cilium for transduction of Hh signals (Goetz and Anderson, 2010; Huangfu et al., 2003), but the relationships between cilia and the core components of the Hh pathway remain unclear.
cAMP-dependent protein kinase A (PKA) is an evolutionarily conserved negative regulator of Hh signaling (Li et al., 1995; Jiang and Struhl, 1995; Hammerschmidt et al., 1996). In Drosophila, genetic and biochemical studies have shown that a central function of Drosophila PKA is to control the formation of the repressor form of the Ci transcription factor. PKA phosphorylates a cluster of serine residues in the C-terminal domain of Ci and phosphorylation of these sites is required for partial proteolysis of Ci by the proteasome to generate a transcriptional repressor that keeps target genes off in the absence of ligand (Chen et al., 1999; Chen et al., 1998; Price and Kalderon, 1999). Consistent with this function, Drosophila mutants that lack catalytic activity of PKA show a gain of pathway activity (Chen et al., 1998; Jia et al., 2005; Jiang and Struhl, 1998; Smelkinson and Kalderon, 2006). In addition to this well-studied role, Drosophila PKA has additional functions in the pathway. PKA negatively regulates the Hh pathway through phosphorylation of full-length Ci, which limits its activity as a transcriptional activator (Wang et al., 1999). PKA also has a positive role in the Drosophila pathway through phosphorylation of the transmembrane protein Smoothened (Smo) (Apionishev et al., 2005; Jia et al., 2004; Ohlmeyer and Kalderon, 1997; Zhang et al., 2004).
As in Drosophila, the mouse homologues of Ci, the Gli proteins, can undergo PKA-dependent proteolytic processing to generate transcriptional repressor forms (Dai et al., 1999). Gli3 is the major transcriptional repressor and, in the absence of ligand, the majority of Gli3 protein undergoes PKA-dependent proteolytic processing to generate the repressor form (Wang et al., 2000). Gli2 is the major transcriptional activator regulated by Hh, although a small fraction of Gli2 can experience PKA-dependent proteolytic processing to a repressor form (Pan et al., 2006; Pan et al., 2009). Decreased activity of PKA in vertebrate embryos leads to a gain of Hh pathway activity, which is due, at least in part, to regulation of Gli3 processing (Concordet et al., 1996; Epstein et al., 1996; Hammerschmidt et al., 1996; Huang et al., 2002; Tiecke et al., 2007; Ungar and Moon, 1996). The PKA phosphorylation sites in Smo are not conserved, suggesting that Smo is not a target of PKA in the vertebrate pathway; recent work suggests that the C-terminal tail of vertebrate Smo is, instead, phosphorylated by CK1 and GRK2 (Chen et al., 2011).
The PKA holoenzyme consists of two regulatory and two catalytic subunits. In the mouse genome, two genes encode PKA catalytic subunits, Cα and Cβ (Prkaca and Prkacb). Mutants that lack either one of these genes survive past birth (Howe et al., 2002; Skalhegg et al., 2002). By contrast, embryos that lack three of the four alleles of Prkaca and Prkacb (referred to as PKA-deficient) die during gestation with neural tube defects associated with an expansion of Shh-dependent ventral neural cell types (Huang et al., 2002), which is consistent with a requirement for PKA in the production of Gli3 repressor. However, the expansion of Shh-dependent cell types in PKA-deficient embryos is much greater than that of Gli3-null mutants (Persson et al., 2002), which suggests that PKA has additional roles in the vertebrate Shh pathway.
Here, we use genetic and cell biological approaches to define the roles of PKA in the Shh pathway in the neural tube of the developing mouse embryo. We find that complete loss of PKA catalytic activity in Cα–/– Cβ–/– double mutant embryos causes a complete ventralization of the neural tube, indicating that the Shh pathway is maximally activated in all neural progenitors in the absence of PKA. As expected, we find that PKA is important for the formation of Gli3 repressor. However, genetic experiments indicate that the major function of PKA in the neural plate is to prevent Gli2 from activating the targets of the pathway.
It has long been recognized that PKA is enriched at centrosomes, including the centrosomes that act as the basal bodies that template primary cilia (Nigg et al., 1985; De Camilli et al., 1986; Barzi et al., 2010). We find that PKA catalytic subunits localize to a specific region adjacent to the basal bodies of cilia in neural progenitors and in embryonic fibroblasts. Our data show that PKA does not regulate cilia length or localization of smoothened (Smo) to cilia in response to Shh in the embryo. However, PKA does regulate the level of Gli2 that accumulates at cilia tips. The data suggest that vertebrate Hh signaling depends on two spatially separated, cilia-associated compartments: a compartment at the base of the cilium where PKA prevents Gli activation in the absence of ligand; and a compartment within the cilium where Gli proteins are modified to allow processing in the absence of ligand or activation in response to ligand.
MATERIALS AND METHODS
Mouse strains and generation of compound mutants
Mutant strains were as follows: Prkcatm1Gsm (Skalhegg et al., 2002), Prkcbtm2Gsm (Howe et al., 2002), Ift172wim (Huangfu et al., 2003), Dynch2h1mmi (Liem et al., 2009), Gli2tm1Alj (Matise et al., 1998) and Gli3Xt (Hui and Joyner, 1993). All alleles were bred onto the C3H genetic background for at least two generations. PKA-deficient embryos were obtained by crossing previously generated knockout strains of the two mouse PKA catalytic subunit genes (Prkaca and Prkacb). In our analysis, we used the same Cα knockout strain described in the original report, Prkcatm1Gsm, that eliminates expression of both Cα1 and Cα2 isoforms (Skalhegg et al., 2002), and a different Cβ mutant, Prkcbtm2Gsm, that disrupts all Cβ isoforms (Cβ1, Cβ2 and Cβ3) (Howe et al., 2002). Mutant embryos that were both PKA deficient and lacked Gli2, Gli3 or Ift172 were obtained from intercrosses between triple heterozygous animals (Prkca+/tm1Gsm; Prkcb+/tm2Gsm, plus Gli2+/tm1Alj, Gli3+/Xt or Ift172+/wim, respectively). PKA double homozygous embryos were obtained from intercrosses between double heterozygous (Prkca+/tm1Gsm; Prkcb+/tm2Gsm) animals. In all the crosses, littermates were used as wild-type controls.
Immunohistochemistry
Dorsal-ventral patterning of the neural tube was performed by immunostaining of neural tube transverse sections as previously described (Eggenschwiler et al., 2001) with the following antibodies: α-Shh (5E1), α-FoxA2 (4C7), α-Nkx2.2 (74.5A5), α-Nkx6.1 (F55A10), α-HB9 (81.5C10), α-Isl1 (39.4D5), α-Pax6 (PAX6) [developed by Drs A. Kawakami (Tokyo Institute of Technology, Japan), and T. Jessell and S. Brenner-Morton (Columbia University, New York, USA), and obtained through the Developmental Studies Hybridoma Bank]; rabbit polyclonal α-FoxA2 antibody (Abcam) and rabbit polyclonal α-Olig2 antibody (Millipore). Other antibodies used in immunohistochemistry and immunocytochemistry were: anti-Gli2 (guinea pig polyclonal) [a kind gift from J. T. Eggenschwiler (Princeton University, Princeton, NJ, USA)]; anti-Arl13b (rabbit polyclonal); anti-Smo (rabbit polyclonal) [raised by Pocono Rabbit Farm and Laboratory using antigens and procedures described previously (Rohatgi et al., 2007)]; anti-pericentrin (rabbit polyclonal) (Covance); anti-acetylated tubulin (6-11B-1, mouse monoclonal) and anti-γ-tubulin (GTU-88, mouse monoclonal) (Sigma); the anti-PKA-catalytic antibody (which recognizes the products of both the Cα and Cβ genes) (5B) and anti-PKARIIb (all mouse monoclonals) (BD Biosciences); and anti-ninein (rabbit polyclonal) [a kind gift from James E. Sillibourne (Institut Curie, Paris, France)]. Confocal microscopy was performed by using an upright Leica TCS SP2 AOBS laser-scanning microscope. Images were taken with a 63× objective and 2× zoom. Extended views of confocal datasets were processed using the Volocity software (PerkinElmer). Cilia images in MEFs were obtained with a DeltaVision image restoration microscope (Applied Precision/Olympus) equipped with a CoolSnap QE cooled CCD camera (Photometrics). Z-stack images sets were obtained with an Olympus 100×/1.4 NA, UPLS Apo oil immersion objective and deconvolved using the SoftWoRx software (Applied Precision/DeltaVision).
Derivation of MEFs, cell culture and western blotting
Mouse embryonic fibroblasts (MEFs) were prepared from E9.0-9.5 embryos and cultured in medium (high-glucose DMEM, 0.05 mg/ml penicillin, 0.05 mg/ml streptomycin, 2 mM glutamax, 1 mM sodium pyruvate, 0.1 mM MEM nonessential amino acid supplement) containing 10% FBS (Gemini BioProducts). MEFs were grown to confluence and serum starved (in medium containing 0.5% FBS for 24 hours prior to the experiments). MEFs from PKA-null (Cα–/– Cβ–/–) double mutant embryos could not be maintained after a few passages and were assayed after two passages in culture. Activation of the Hh pathway was obtained with the addition of 100 nM SAG (Calbiochem) for 24 hours before fixation and staining. To activate adenylate cyclase, cells were treated with 10 μM forskolin (Sigma) for 12 hours before fixation and staining. Cells were fixed with 2% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 20 minutes at room temperature and subsequently in 100% methanol for 5 minutes at –20°C to preserve centrosomes.
For western blot analysis, whole (E9.0 and E10.5) embryos extracts were prepared in lysis buffer [25 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40 (v/v), 1% deoxycholate (w/v), 0.1% SDS, a protease inhibitor cocktail (1× EDTA-free protease inhibitors from Roche) and phosphatase inhibitor cocktail 1 from Sigma] and processed using standard methods. Antibodies used in western blots were anti-Gli2 (goat polyclonal) from R&D Systems (catalog number AF3635); anti-Gli3 (goat polyclonal) from R&D Systems (catalog number AF3690); anti-γ-tubulin (GTU-88, mouse monoclonal) from Sigma; peroxidase-conjugated AffiniPure rabbit anti-goat IgG (H+L) from Jackson ImmunoResearch Laboratories; and HRP-goat anti-mouse IgG (H+L) conjugate from ZyMax/Invitrogen. The relative intensity of full-length Gli2 and Gli3, and of processed Gli3 was quantified using ImageJ software (NIH).
For qPCR experiments, total RNA was extracted from E9.5 wild-type and PKA-null embryos using the RNeasy Mini Kit (QIAGEN). Taqman Gene Expression assays (Applied Biosystems) were performed for mouse Gli2 (Mm01293111_m1) and mouse Gli3 (Mm00492333_m1) genes. At least three replicates assays were analyzed per embryo sample and gene. Relative expression values were normalized to HPRT gene expression.
Scanning electron microscopy
E8.0 and E9.0 embryos were dissected in PBS at room temperature and fixed in 2.5% glutaraldehyde in PBS (pH 7.4) for 24 hours at 4°C and processed as described previously (Huangfu et al., 2003). Scanning electron microscopy (SEM) was performed using a Zeiss SUPRA 25 FESEM. The ImageJ software (NIH) was used to quantify cilia length in SEM 12,000× images of the embryonic node.
Alcian Blue/Alizarin Red staining of cartilage and bone
Alcian Blue and Alizarin Red were used to stain cartilage and bone, as previously described (Nagy et al., 2003).
RESULTS
Absence of all PKA catalytic activity fully activates the Shh pathway
It was shown previously that PKA-deficient mouse embryos (Cα–/– Cβ–/+ and Cα–/+ Cβ–/–) show an expansion of Shh-dependent ventral cell types in the developing neural tube (Huang et al., 2002). Those studies used a Cβ allele that did not eliminate all splice isoforms of the gene. We therefore analyzed neural patterning using a null Cβ allele. We found that the neural patterning phenotype of E10.5 PKA-deficient embryos carrying the null allele was indistinguishable from the previously described phenotype: ventral progenitors in the thoracic and lumbar neural tube were expanded, associated with neural tube closure defects in the head and lumbar spinal cord (supplementary material Fig. S1) (Huang et al., 2002).
It has previously been noted that embryos that lacked both genes encoding PKA catalytic subunits arrested early in development (Huang et al., 2002), but the double mutant phenotype was not analyzed. We found that embryos that lack all four catalytic subunit alleles (Cα–/– Cβ–/–; we designate this genotype as PKA-null) arrested at E9.0 with a striking abnormal morphology similar to that of Ptch1- and Sufu-null embryos (Cooper et al., 2005; Goodrich et al., 1997; Svard et al., 2006), in which the Shh pathway is fully activated in all cells. Like Ptch1 and Sufu mutants, PKA-null embryos arrested at E9.0, with an open neural tube and a characteristic small head, and failed to undergo embryonic turning (Fig. 1A-D; supplementary material Fig. S2).
Fig. 1.
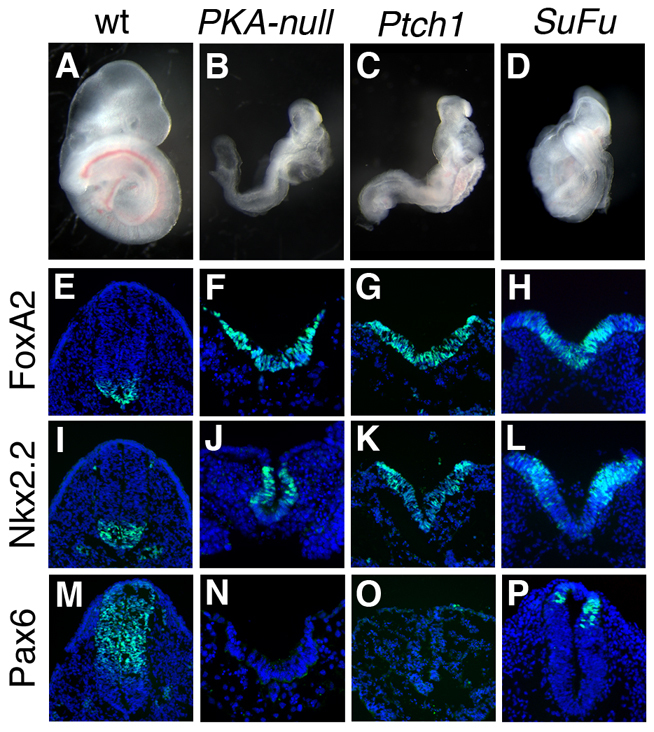
Absence of PKA activity causes maximal activation of the Shh pathway. (A-D) Embryos that lack all four alleles encoding PKA catalytic subunits arrest at E9.0 with a morphology resembling that of Ptch1–/– and Sufu–/– embryos. (E-P) Cross-sections through the open neural plate (thoracic) region of E9.0 wild-type, PKA-null, Ptch1–/– and Sufu–/– embryos, showing a strong ventralization of the neural tube. The expression of FoxA2 (floor plate) (E-H) and Nkx2.2 (V3 progenitors) (I-L) is expanded at all levels of the neural tube, and expression of Pax6 is absent (M-P) (all markers are green). The Sufu–/– embryos express Pax6 in the neural tube, indicating that phenotype is slightly weaker than that of PKA-null and Ptch1–/–. DAPI is blue.
Analysis of markers of neural patterning showed that the PKA-null embryos exhibited a strong ventralization of the neural plate. The ventral neural cell types that require the highest level of activity of the Shh pathway were expanded to fill the entire neural plate: FoxA2-expressing floor-plate cells and V3 interneuron progenitors (marked by expression of Nkx2.2) were intermingled at all positions of the open neural plate (Fig. 1E-L). The strong expansion of ventral progenitors correlated with the absence of dorsal interneuron progenitors, shown by the lack of Pax6 expression (Fig. 1M-P). This ventralization was identical to the phenotype of Ptch1-null embryos, and slightly stronger than seen in Sufu-null embryos. Thus, in the absence of PKA, the Shh pathway is maximally activated in all cells of the neural plate.
Gli2 is the major target of PKA in neural patterning
To understand how loss of PKA activity leads to activation of the Shh pathway in the neural plate, we generated compound mutants that were PKA deficient and lacked the Gli transcription factors that implement Shh activity. In the developing neural tube, Gli3 acts primarily as a transcriptional repressor that keeps target genes off in the absence of Shh. Genetic and biochemical studies have established that phosphorylation of Gli3 by PKA is required for its proteolytic processing into the transcriptional repressor form (Pan et al., 2006; Pan et al., 2009; Wang and Li, 2006). Gli3 single mutants have only subtle defects in neural patterning (Persson et al., 2002). In the caudal neural tube, where PKA-deficient embryos show a strong phenotype, removal of Gli3 did not modify the phenotype: in PKA-deficient embryos that also lacked Gli3 (Cα–/– Cβ–/+ Gli3–/– and Cα–/+ Cβ–/– Gli3–/–; we designate these embryos as PKA-deficient Gli3) the floor-plate and V3 progenitors were dorsally expanded and the motoneuron domain was shifted into the dorsal neural tube (Fig. 2). These results are consistent with the hypothesis that Gli3 repressor is not made in the caudal neural tube of PKA-deficient embryos. In the more rostral neural tube and in the limbs, where the PKA-deficient phenotype is mild, presumably owing to the activity of the one remaining wild-type allele, decreased PKA and loss of Gli3 had additive effects on patterning (supplementary material Fig. S3).
Fig. 2.
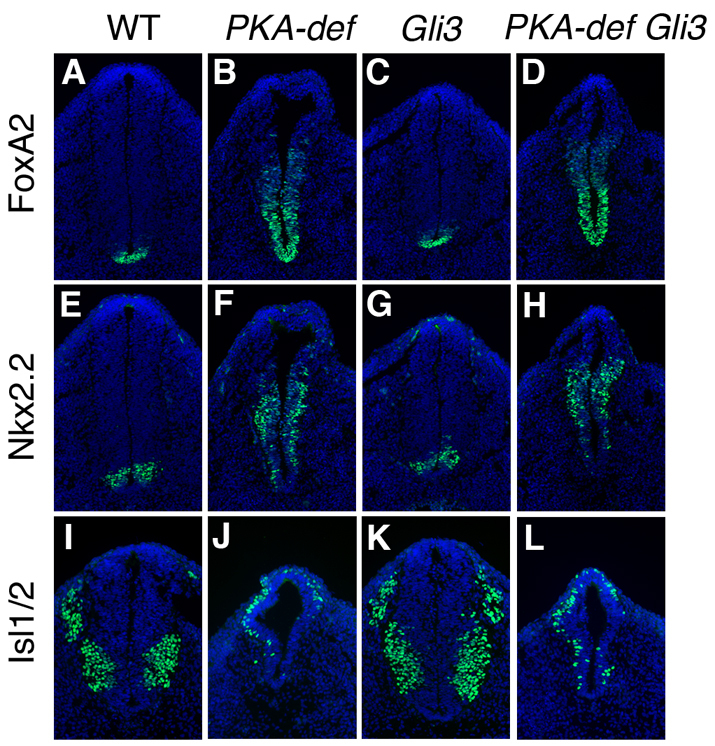
The neural tube of PKA-deficient Gli3 compound mutants is ventralized. (A-L) Expression of markers of floor plate (FoxA2) (A-D), V3 interneuron progenitors (Nkx2.2) (E-H) and motoneurons (Isl1) (I-L) in cross-sections at the level of the hindlimb of E10.5 wild-type, PKA-deficient, Gli3 single mutant and PKA-deficient Gli3 compound mutant embryos. Markers are in green; DAPI is blue. Whereas Gli3 mutant shows a subtle defects in the caudal neural tube patterning, the floor plate and V3 progenitor domains are expanded, and the motoneuron domain is shifted dorsally in the PKA-deficient Gli3 compound mutants.
Gli2 is required for normal specification of the ventral neural cell types that require highest level Shh signaling, the floor plate and V3 interneurons (Ding et al., 1998; Matise et al., 1998), and plays a role in the specification of motoneurons, which require intermediate levels of Shh signaling (Bai et al., 2004; Motoyama et al., 2003; Lei et al., 2004). Because these ventral neural types were expanded in PKA-deficient embryos, we analyzed neural patterning in E10.5 embryos that were both PKA deficient and lacked Gli2 (Cα–/– Cβ+/– Gli2–/– and Cα+/– Cβ–/– Gli2–/–; designated as PKA-deficient Gli2). Neural tube patterning in PKA-deficient Gli2 compound mutant embryos was very similar to that in Gli2 single mutants. In both PKA-deficient Gli2 and Gli2 homozygous embryos, expression of floor-plate markers was absent or strongly reduced (Fig. 3A-D), Nkx2.2-expressing V3 interneuron progenitors were located at the ventral midline (Fig. 3E-H) and the domain of mature motoneurons was not expanded dorsally (Fig. 3I-L). This indicates that most of the expansion of Shh-dependent cell types in PKA-deficient embryos is the result of inappropriate activation of Gli2. The phenotype of the PKA-deficient Gli2 mutants was not identical to that of Gli2 single mutants, as the domain of expression of the motoneuron progenitor marker Olig2 was expanded dorsally in the compound mutants (supplementary material Fig. S4), which is consistent with the role of PKA in the production of Gli3 repressor.
Fig. 3.
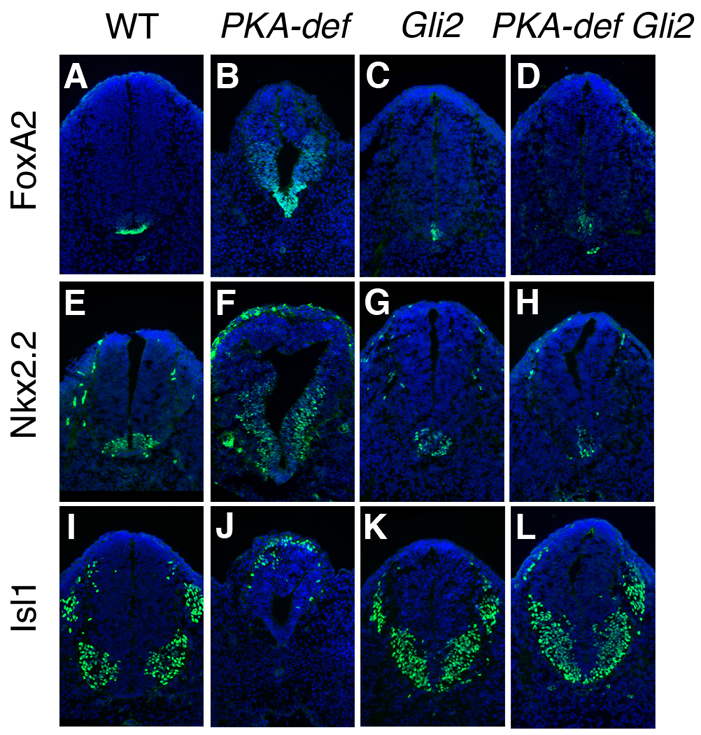
The neural tube phenotype of PKA-deficient Gli2 compound mutant embryos resembles that of Gli2 single mutants. (A-L) Expression of markers of floor plate (FoxA2), V3 interneuron progenitors (Nkx2.2) and motoneurons (Isl1) in cross-sections through at the level of the hindlimb of E10.5 wild-type, PKA-deficient, Gli2 single mutant and PKA-deficient Gli2 compound mutant embryos. Markers are in green; DAPI is blue. In PKA-deficient Gli2 compound mutants, expression of the floor-plate marker FoxA2 is strongly reduced (A-D), Nkx2.2-expressing V3 interneuron progenitors are located in the ventral midline (E,F) and motoneurons span the ventral midline (I-L), as in Gli2 homozygous embryos.
PKA is important for processing of Gli3 and for stability of full-length Gli2 and Gli3
As the genetic experiments showed that PKA regulates both Gli3 and Gli2, we analyzed both proteins in PKA-deficient and PKA-null embryo extracts on western blots. There was a reduction in the relative amount of processed Gli3 in extracts of whole E10.5 PKA-deficient embryos: the ratio of full-length to processed Gli3 was 0.12 in wild-type and 0.21 in the mutant embryos (Fig. 4C), which is consistent with a role of PKA in promoting processing of Gli3. In PKA-null embryos, the amounts of both processed and full-length Gli3 were reduced to 10-20% of the wild-type levels (Fig. 4D), which prevented accurate determination of the ratio of processed/full-length Gli3. We did, however, detect some processed Gli3 in PKA-null embryos. The level of Gli2 was normal in E10.5 PKA-deficient embryos, but Gli2 was reduced in extracts of E9.0 PKA-null embryos (Fig. 4A,B). It has been shown that activated full-length Gli proteins are unstable (Humke et al., 2010; Wen et al., 2010; Chen et al., 2009; Jia et al., 2009; Wang et al., 2010), so the reduction in the level of full-length Gli2 and Gli3 in PKA-null embryos suggests that the absence of PKA activity leads to activation of the Gli proteins, consistent with the strongly ventralized neural plate of the mutants.
Fig. 4.

PKA is required for the stability and processing of Gli proteins. (A,C) Levels of full-length Gli2 and Gli3 are not affected by the partial loss of PKA activity in PKA-deficient embryos, whereas there is a moderate decrease in the level of Gli3R (C). (B,D) Total loss of PKA activity in PKA-null embryos drastically reduces the levels of full-length Gli2 and Gli3, which are almost undetectable, suggesting that PKA is important for the stability of activated full-length Gli2 and Gli3. qPCR experiments showed that RNA levels for Gli3 and Gli2 are reduced 2.2±0.1-fold and 1.7±0.1-fold, respectively, in PKA-null embryos, which is not sufficient to explain the decrease in level of the full-length Gli proteins.
The primary cilium is required for the ectopic activation of the Hh pathway in PKA-deficient embryos
We have shown previously that a dominant-negative form of PKA activates the Hh pathway in wild-type mouse embryo fibroblasts (MEFs), but not in MEFs that lack cilia (Ocbina and Anderson, 2008), which argued that cilia are required for the effects of decreased PKA. To investigate the relationship between cilia and PKA-dependent control of Gli activity more directly, we generated PKA-deficient mutants that also lacked Ift172 (Cα–/– Cβ–/+ Ift172–/– and Cα–/+ Cβ–/– Ift172–/–; designated as PKA-deficient Ift172). Ift172 mutant embryos lack cilia and fail to specify the ventral neural cell types that depend on Shh signaling (Huangfu et al., 2003). PKA-deficient Ift172 compound mutants showed the same morphology (Fig. 5A-D) and the same absence of Shh-dependent ventral neural cell types in the neural tube as Ift172 single mutants: floor-plate (FoxA2) and V3 interneuron progenitors (Nkx2.2) were absent (Fig. 5E-L), whereas the Pax6 expression domain expanded ventrally (Fig. 5M-P). Thus, the activation of the Shh pathway caused by partial loss of PKA can only occur in the presence of the primary cilium.
Fig. 5.
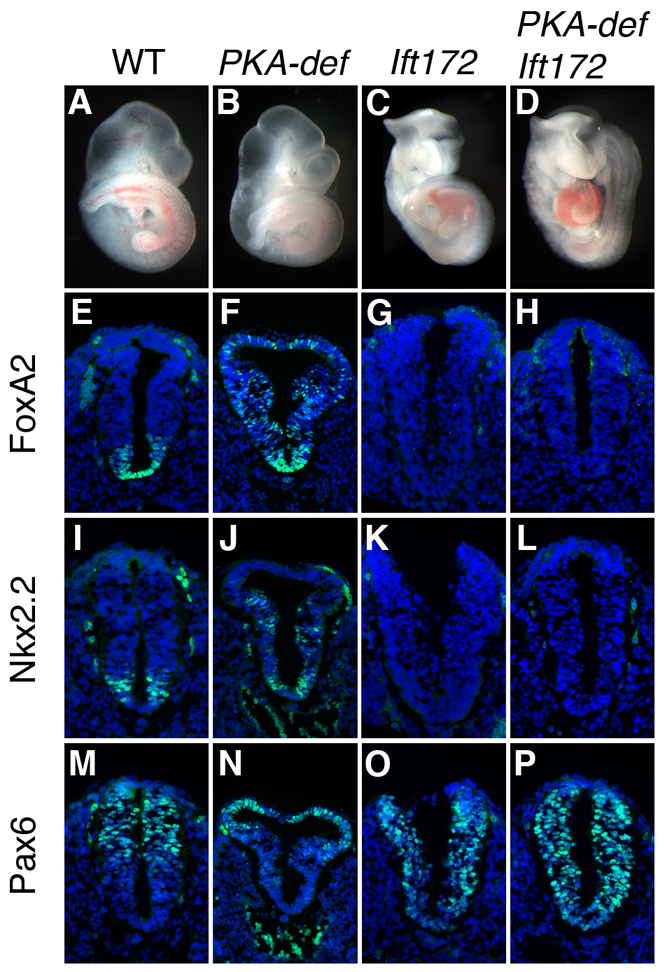
The primary cilium is required for the effect of PKA mutations on the neural tube. (A-D) PKA-deficient Ift172 compound mutants resemble Ift172 single mutants. (E-P) Expression of neural tube patterning markers FoxA2, Nkx2.2 and Pax6 (in green) in cross-sections through the posterior neural tube, at the position of where the hindlimb is beginning to develop, of E9.5 wild-type, PKA-deficient, Ift172 single and PKA-deficient Ift172 compound mutant embryos. Markers in green; DAPI is blue. PKA-deficient Ift172 compound mutants show the same lack of Hh signaling in the neural tube as Ift172 mutants (compare H with G, L with K, and P with O), indicating that the expansion of ventral neural cell types in PKA-deficient mutants depends on cilia.
PKA is localized near the base of the cilium in the Shh-responsive cells
As PKA is associated with centrosomes in cultured cells (Nigg et al., 1985; De Camilli et al., 1986; Barzi et al., 2010), we examined the localization of PKA subunits in vivo. Immunofluorescent staining of transverse sections of E9.5 and E10.5 wild-type embryos with an antibody that recognizes both PKA catalytic subunits (PKAC) showed that PKAC colocalized with the pericentriolar marker pericentrin (Pcnt) in both the neural tube and mesenchymal cells (Fig. 6A-D); the regulatory subunit PKARIIβ also localized to the same region (supplementary material Fig. S5A-E). In MEFs, PKAC and the regulatory subunit PKARIIβ were highly enriched in a domain at the base of the primary cilium, and were also detected in the Golgi (Fig. 6E,F; supplementary material Fig. S5F,G). High-resolution imaging in MEFs showed that PKA at the base of the cilium was proximal to γ-tubulin (which marks the centrosome), on the opposite side of the centriole from the cilium and transition zone (Fig. 6E,F). Activation of the Hh pathway with the Smo agonist SAG did not detectably modify the intensity or localization of the signal of either PKAC and PKARIIβ at the base of the cilium (Fig. 6E,F; supplementary material Fig. S5F,G).
Fig. 6.

PKA localizes to the basal side of the centrosome in wild-type embryos and MEFs. (A-C) PKA catalytic subunits (PKAC, green) are apically enriched in the E9.5 wild-type neural tube and colocalize with pericentrin (Pcnt) (red). (D) PKA catalytic subunits are localized to the base of cilia (visualized by the cilia marker Arl13b, red) in the mesenchymal cells adjacent to the neural tube of the E10.5 embryo. (E) In MEFs, PKAC (green) is enriched at the base of the primary cilium (Arl13b, pink). PKAC is adjacent to, but does not overlap with, the centrosome marker γ-tubulin (red). (F) PKAC localization to the base of the cilia does not change when Shh pathway is activated in the wild-type MEFs treated with 100 nM SAG for 24 hours. (G,H) PKAC does not accumulate in cilia (Arl13b) of MEFs where retrograde trafficking is blocked by mutation of the retrograde motor Dync2h1mmi. Scale bars: 20 μm in A-C; 10 μm in D; 5 μm in E-H.
Despite the strong signal at the base of the cilium, we did not detect any PKAC or PKARIIβ within the primary cilium. To test whether low undetectable levels of PKAC traffic through the cilium, we removed the retrograde ciliary motor. Inactivation of Dync2h1, which encodes the heavy chain of the IFT-dynein motor, leads to accumulation of Smo, Gli2, Ptch1 and IFT proteins to high levels in the cilium (Ocbina et al., 2011). However, even in Dync2h1 mutant MEFs, we detected PKAC only at the base of the cilium (Fig. 6G,H). This provides strong evidence that PKAC acts at the base of the cilium and not in the axoneme, although it does not rule out the possibility that a small fraction of PKA could enter the cilium.
Absence of PKA does not affect cilia length and ciliary trafficking of Smo
Several recent reports have suggested that treatment of cells with forskolin, which should increase the level of cAMP and therefore activate PKA, can affect cilia length (Besschetnova et al., 2010; Low et al., 1998). We therefore examined how loss of PKA affected cilia and ciliary protein localization. No defects in cilia length, Gli2 localization or Smo localization were seen in PKA-deficient embryos or MEFs (supplementary material Fig. S6). In E8.0 PKA-null embryos, the node cilia were of normal length and morphology, as assayed by scanning electron microscopy (SEM) (Fig. 7A,B). At E9.0, primary cilia were present in normal numbers on the apical side of cells in the neural tube in PKA-null embryos, as assayed by staining of the cilia marker Arl13b (Fig. 7E,F; supplementary material Fig. S5D). In MEFs derived from PKA-null mutants, cilia were normal in length, as assayed by staining of the axonemal marker acetylated α-tubulin (Fig. 7G-N). Thus, absence of PKA does not have a detectable effect on cilia length in either the midgestation embryo or in cultured embryonic fibroblasts.
Fig. 7.

PKA is not required for cilia structure or for localization of Smo, but does affect Gli2 localization to cilia. (A,B) SEM shows that cilia are the same length in the node of E8.0 wild-type (wt) and PKA-null embryos. Wild-type cilia were 3.5±0.5 μm long (n=37); PKA-null cilia were 3.6±0.6 μm long (n=27). (C-F) Localization of Smo and Gli2 in sections of PKA-null embryos. (C,D) Smo (green) localizes to the short cilia of the E9.5 neural plate in wild-type (C) and PKA-null mutant (D) embryos. The base of cilia is marked by γ-tubulin. (E,F) Gli2 is enriched in the cilia of mesenchymal cells surrounding the neural tube in the E9.5 wild-type (E) and PKA-null (F) embryos. Cilia are marked by Arl13b, and the base of cilium is marked by γ-tubulin. (G-N) Localization of Hh pathway protein in wild-type and mutant MEFs. (G-J) Smo localization. Smo is cytoplasmic in unstimulated wild-type MEFs (G) and localizes to the cilia after 24 hours of SAG treatment (H). Smo is not localized to cilia in unstimulated PKA-null MEFs (I) and moves into cilia after 24 hours of SAG treatment (J). (K-N) Gli2 localization. Gli2 is present at wild-type cilia tips in the absence of SAG (K) and is further enriched in cilia tips after SAG treatment. (M,N) Gli2 is enriched in cilia tips in PKA-null MEFs regardless of SAG stimulation. Basal bodies are marked by γ-tubulin and cilia are marked by acetylated α-tubulin. (O) Percentage of Smo+ and Gli2+ cilia in experiments shown in G-N. Smo localization is not affected in PKA-null MEFs, whereas significantly more Gli2 is present in the cilia of PKA-null MEFs than in wild type, in the absence of SAG stimulation (P<0.05). Data are mean±s.e.m. Scale bars: 2 μm in A,B; 5 μm in C-N.
Smo moves into cilia in response to Shh (Corbit et al., 2005). Treatment of cells with forskolin promotes Smo movement into cilia in the absence of ligand (Milenkovic et al., 2009; Wen et al., 2010; Wilson et al., 2009). This finding predicts that Smo would fail to move into cilia in PKA-null cells, if forskolin affected Smo by activating PKA. By contrast, we found that when the Hh pathway was inactive, Smo was absent from cilia of both wild-type and PKA-null MEFs (Fig. 7G,I) and that Smo moved normally into the ciliary membrane in response to SAG in PKA-null cells (Fig. 7H,J,O). Smo was also present in cilia of the E9.0 PKA-null neural plate (Fig. 7C,D; supplementary material Fig. S5E). Thus, Smo trafficking into cilia is not regulated by PKA.
Gli2 is enriched at cilia tips in the absence of PKA
In contrast to the normal trafficking of Smo in the absence of PKA, we found that Gli2 was enriched at the tips of cilia in cultured PKA-null cells, both in the absence and presence of SAG (Fig. 7M,N). While 25% of cilia tips in wild-type cells had detectable Gli2 staining, 66% of PKA-null cilia had Gli2 staining in the absence of SAG (Fig. 7O). The number of cilia with detectable Gli2 and the level of Gli2 at cilia tips in untreated PKA-null cells was approximately the same as that in wild-type cells treated with SAG and was not further enhanced by treatment with SAG (Fig. 7K-N). In both wild-type and PKA-null embryos, Gli2 was detected at cilia tips in embryo sections (Fig. 7E,F), although the variable level of Gli2 expression precluded a direct comparison with wild type. Based on the MEF data, we conclude that although cilia length and Smo trafficking are unchanged in the absence of PKA, the amount of Gli2 at cilia tips is increased in the absence of PKA.
DISCUSSION
Absence of PKA leads to full activation of the Shh pathway
Complete loss of PKA activity in the mouse embryo leads to arrest at E9.0, associated with unrestrained activity of the Shh pathway in the neural plate, such that all neural cells acquire the most ventral neural fates, floor plate and V3 progenitors. The neural patterning phenotype of PKA-null embryos is as strong as that in Ptch1-null embryos. Although PKA has many different cellular functions (Willis et al., 2011), the strikingly similar morphologies of PKA- and Ptch1-null embryos suggests that the primary function of PKA in the early embryo is to restrain the activity of the Hh pathway.
The strong activation of the Shh pathway in PKA-null, Sufu and Ptch1 mutants indicates that each of these three components is essential to prevent full activation of Gli2 in the absence of ligand, but each appears to act at a different step in the pathway. Ptch1 prevents ciliary location of Smo, which is essential for Gli activation; Sufu binds Gli proteins and prevents their activation. As described below, our data suggest that PKA acts on Gli proteins at a step independent of both Smo ciliary localization and Sufu.
The primary role of PKA in the neural plate is to block the production of Gli2 activator
Biochemical and cell-based experiments have shown that PKA phosphorylates a cluster of sites in the C-terminal domain of both Gli2 and Gli3, which targets the proteins for ubiquitylation and partial proteolysis by the proteasome to form the Gli repressors (Wang et al., 2000; Pan et al., 2009). We find that the amount of Gli3 repressor is reduced in PKA-deficient embryos and that removal of Gli3 does not enhance or suppress the gain of Hh phenotype seen in the caudal neural tube of PKA-deficient embryos, consistent with the biochemical data that show PKA regulates Gli3 processing. We were, however, surprised to find that there was some processed Gli3 in the PKA-null embryos. This result suggests that there may be a PKA-independent mode of Gli3 processing. It has previously been noted that although Gli3 processing depends on cilia, there is some residual Gli3 repressor in mutants that lack cilia (Huangfu and Anderson, 2005; Liu et al., 2005). The results raise the possibility that there is a PKA-independent, cilia-independent mechanism for formation of Gli3 repressor.
Although there is a clear role for PKA in the formation of Gli repressors, that role is not sufficient to account for the very strong ventralization of the neural tube seen in PKA-null embryos. A knock-in allele of Gli2 that carries mutations in the four PKA phosphorylation sites required for processing causes mild ectopic activation of Shh targets in the neural tube (Pan et al., 2009), and the combined absence of Gli3 and Gli2 repressor could contribute to the PKA-deficient and PKA-null phenotypes. However, our genetic experiments show clearly that ectopic expression of ventral neural markers in PKA-deficient embryos depends on the Gli2 activator: PKA-deficient Gli2 compound mutants show the same loss of floor plate and V3 progenitors seen in Gli2 single mutants. A role for PKA in negative regulation of full-length Gli is also supported by the observation that constitutively active PKA can inhibit the activity of Gli1, which does not undergo proteolytic processing (Ruiz i Altaba, 1999). Our findings also agree well with recent studies showing that the activity of human full-length Gli1 expressed in Drosophila is inhibited by PKA through direct phosphorylation of specific residues in Gli1 (Marks and Kalderon, 2011). Our studies focused on the neural tube, where Gli activators have a central role in patterning; it is likely that the primary role of PKA in tissues such as the limb, where Gli repressors play the central role in patterning, would be in the regulation of Gli3 processing.
PKA acts at the basal body
Our genetic analysis shows that the inappropriate activation of the Shh pathway caused by decreased PKA activity can only occur in the presence of the primary cilium. Although we have not analyzed triple mutants that lack both genes encoding PKA catalytic subunits and also lack cilia, it is very likely that the strong activation of Shh signaling in the PKA-null phenotype is also cilia dependent. We find that the PKA catalytic and regulatory subunits localize to a region proximal to the basal body that overlaps with the pericentriolar material marked by pericentrin in both embryonic cells and in MEFs. Although PKA is important for motility of flagella and cilia (Wirschell et al., 2011), our findings indicate that PKA is not present in primary cilia, even when retrograde trafficking is blocked, which causes accumulation of proteins that traffic through cilia (Ocbina et al., 2011). It is interesting to note that centrosomes are not present in interphase cells in Drosophila (Rogers et al., 2008), which do not use cilia for Hh signaling. Thus, in vertebrates, but not in Drosophila, PKA is localized at the base of the primary cilium, where it can regulate Hh signaling.
Recent studies in cerebellar granule neuron precursors (CGNPs) suggested that the subcellular localization of PKA can be controlled by the activity of the Hh pathway (Barzi et al., 2010). CGNPs can be maintained only in the presence of Shh and PKA is localized to the base of the cilia under these conditions. It was observed that removal of Shh from CGNP cultures triggered the dispersion of PKA from the centrosome, accompanied by activation of the enzyme (Barzi et al., 2010). By contrast, we did not detect any change in the localization of PKA at the base of the primary cilium in MEFs after serum starvation or after activation of the Shh pathway by SAG. In the embryo, the catalytic subunits of PKA were highly enriched at the base of the cilium in cell types both near the source of Shh and at a distance from the source. Thus, in the embryo and in MEFs, there was no detectable change in the localization of PKA in response to activity of the Shh pathway.
PKA is not required for normal cilia length or Smo trafficking
The proximity of PKA to the primary cilium has raised a number of hypotheses about the relationships between PKA activity, ciliary trafficking and hedgehog signaling that have been tested in pharmacological experiments in cultured cells. For example, treatment with forskolin, which activates adenylate cyclase and thereby increases the level of cellular cyclic AMP (cAMP) and should activate PKA, increases the length of primary cilia in mammalian epithelial (MDCK and IMCD) and primary bone mesenchyme cells (Besschetnova et al., 2010; Low et al., 1998). Cilia length is determined in part by the balance of anterograde and retrograde IFT (Engel et al., 2009), and an increase in the rate of anterograde IFT was observed after exposure to forskolin that could account for the longer cilia (Besschetnova et al., 2010). By contrast, we find that decreased or absent PKA activity did not affect cilia length in the neural tube, mesenchyme, embryonic node and MEFs. Thus, we conclude that PKA activity is not required to control cilia length or the balance of anterograde and retrograde trafficking within the primary cilium in embryonic tissues or MEFs.
Several recent findings suggested that PKA activity could regulate trafficking of Smo within the primary cilium. It has been reported that treatment of NIH3T3 cells with forskolin induced translocation of Smo to cilia (Milenkovic et al., 2009; Wen et al., 2010; Wilson et al., 2009). Such an activity is counter-intuitive, as the function of PKA is to keep the pathway off in the absence of ligand, and entry of Smo into the cilium should activate the pathway. In contrast to the pharmacological experiments, we find that Smo traffics normally in PKA-null cells: Smo is not detected in the cilium in the absence of pathway activation and Smo moves into the cilium in response to SAG. We also observed that forskolin promotes Smo translocation into the cilia of wild-type MEFs and we find that effect is blocked in PKA-null cells. This indicates that forskolin causes hyperactivation of PKA that drives Smo into cilia (supplementary material Fig. S7A). However, our findings show that Smo trafficking is independent of PKA activity under physiological conditions.
It has also been reported that forskolin blocks the entry of Gli proteins into cilia (Wen et al., 2010; Tukachinsky et al., 2010). However, we found that forskolin also blocked the accumulation of Gli2 at cilia tips in PKA-null cells (supplementary material Fig. S7B), indicating that this effect of forskolin is independent of PKA. The differences between our genetic findings on the role of PKA in control of cilia length and trafficking and the pharmacological experiments suggest that forskolin can affect aspects of cilia biology that are independent of PKA. It is known, for example, that cAMP activates EPAC proteins, guanine nucleotide exchange factors for Rap1 and Rap2 (Murray, 2008; Borland et al., 2009), that cAMP gated channels are important in specialized cilia (Bradley et al., 2005) and that specific channels in kidney primary cilia are directly stimulated by forskolin and inhibited by PKAi (Raychowdhury et al., 2009). Thus, experiments based only on the effects of small molecule modulators of cAMP levels in cilia should be interpreted with caution.
Regulation of Gli activation at the base of the cilium by PKA
Our genetic and cell biological experiments indicate that PKA acts at the base of the cilium and regulates activity of the Shh pathway by promoting the formation of Gli repressors and preventing the formation of Gli activator in the absence of ligand. We therefore consider two models that could account for the action of Gli that differ in whether PKA acts on Gli proteins before or after they enter the cilium (Fig. 8).
Fig. 8.

Two two-compartment models for cilia-dependent regulation of Gli activity. PKA is localized to the base of the cilia, where its function is to prevent activation of the pathway in the absence of ligand, and crucial modifications of Gli protein complexes take place within the cilium. (A) In this model, PKA phosphorylates Gli proteins before they enter the cilium and PKA activity is regulated by Smo. In the absence of ligand, phosphorylation of Gli/Sufu complexes by PKA limits trafficking of the complexes. Phosphorylation of Gli proteins by PKA is also required for processing of Gli repressors by proteasome after they exit the cilium. In the presence of ligand, PKA activity is turned off, perhaps by active Smo in the cilium. Unphosphorylated Gli/Sufu then enters the cilium where the complex dissociates. Full-length free Gli proteins are stabilized and become transcription activators. (B) In this model, PKA phosphorylation is determined by the appropriate Gli substrate, which becomes available only when Gli is complexed with Sufu and only after it has transited the primary cilium. In the absence of Shh, unphosphorylated Gli-Sufu complexes enter the cilium and are modified within the cilium (red sunburst), either via covalent modification or a change in the composition of the complex. After exiting the cilium, Gli2 complexed with Sufu is recognized and phosphorylated by PKA, which stabilizes Gli2/Sufu complex and thereby prevents Gli2 activation and recycling of Gli2 back into the cilium. Phosphorylation by PKA targets Gli3/Sufu complex to the SCF E3 ubiquitin ligase and proteasome for processing into Gli3 repressor. In the presence of Shh ligand, active Smo localizes to the cilia and promotes Gli-Sufu dissociation in the cilium; free Gli proteins are modified in the cilia and are no longer substrates for PKA at the base of cilia. Full-length unphosphorylated Gli proteins can also recycle into the cilium. Smo could, in principle, also have a second activity that turns down PKA activity when the pathway is activated, even if PKA acts on Gli proteins only after they exit the cilium.
Recent reports have argued that dissociation of Gli/Sufu complexes in cilia is a central step in the activation of the vertebrate Hh pathway (Humke et al., 2010; Tukachinsky et al., 2010). These groups showed that pathway activation causes dissociation of the Gli-Sufu complexes, that dissociation depends on the cilium and that forskolin blocked dissociation of the Gli-Sufu complex (Humke et al., 2010; Tukachinsky et al., 2010). These results were used to suggest that PKA phosphorylates Gli proteins before they enter the cilium, and active Smo turns off PKA activity to allow dissociation of the Gli-Sufu complexes in the cilium (Fig. 8A). This model is, however, largely based on the effect of forskolin, which might not reflect physiologically relevant activities of PKA, as described above.
An alternative model is based on the colocalization of PKA with the proteasome and the components of the SCF (Skp1-Cul1-F-box protein) E3 ubiquitin ligase complex that are required for Gli3 processing (Fabunmi et al., 2000; Freed et al., 1999) (Fig. 8B). In the absence of ligand, the cilium is required for formation of Gli3 repressor (Huangfu and Anderson, 2005; May et al., 2005), so it is attractive to propose that phosphorylation of Gli proteins by PKA and Gli processing occur in a concerted process after Gli proteins transit the cilium. In this model, in the absence of pathway activation, the Gli/Sufu complex enters the cilium, is modified in the cilium, either by covalent modification or through interactions with other proteins such as Kif7, but does not dissociate in the cilium. After exit from the cilium, the modified Gli/Sufu complex has become a substrate for PKA. Phosphorylation of the Gli3/Sufu complex by PKA would target Gli3 for immediate processing by the proteasome. Phosphorylation of the Gli2/Sufu complex by PKA would lead to processing of a small amount of Gli2, but the major action of PKA would be to stabilize the Gli2/Sufu complex and prevent release of full-length Gli2, as suggested by the data of Humke et al. (Humke et al., 2010). When the pathway is activated by Hh ligand, activated Smo would promote dissociation of Gli/Sufu complexes in the cilium; free Gli3 and Gli2 proteins would not be substrates for PKA and would therefore not be targeted for processing or degradation at the base of the cilium. In this model, PKA activity is controlled by the availability of appropriate substrates.
In unstimulated PKA-null cells, we observed a two- to threefold increase in the fraction of cells that accumulate Gli2 at cilia tips, as well as an increase in the intensity of the Gli2 signal at cilia tips. The increase in Gli2 in cilia in PKA-null cells in the absence of ligand would appear to be more consistent with models in which PKA negatively regulates the entry of Gli proteins into the cilium (Tukachinsky et al., 2010) than the model proposed above (Fig. 8B) in which PKA acts after Gli proteins exit the cilium. However, cilia localization of Gli2 and Gli3 is not affected by the phosphorylation of the major PKA sites (Zeng et al., 2010), and it is not clear how a two- to threefold fold increase in the amount of Gli2 at the cilia tip would lead to full activation of the pathway. Instead, we propose that the increase in ciliary Gli2 is secondary to the primary role for PKA in control of fate of Gli complexes after they exit the cilium. The data could be explained, for example, if unphosphorylated Gli proteins at the base of the cilium can recycle back into the axoneme (Fig. 8), as has been proposed for IFT proteins (Engel et al., 2009), but phosphorylated Gli proteins cannot enter the cilium.
No matter whether PKA acts before or after Gli proteins traffic through cilia, the data suggest that the activity of the Shh pathway depends on the sequential movement of Gli proteins between two spatially separated, cilia-associated compartments, one in the cilium and one at its base, that control accessibility of Gli proteins to PKA. We suggest that the structure of the primary cilium, and the localization of PKA to base of the cilium, provides the spatial scaffold that creates separate compartments required for Hh signaling in vertebrates.
Supplementary Material
Acknowledgments
We thank Stanley McKnight (University of Washington) for mice carrying the null allele of Cβ and for the Cα mice; Jonathan Eggenschwiler, Sarah Goetz and Hisham Bazzi for comments on the manuscript; and Bryan Tsou for helpful discussions. We thank Yevgeniy Romin and Sho Fujisawa (MSKCC Molecular Cytology Core) for assistance with confocal microscopy and Metamorph analysis, and Shawn Galdeen (Rockefeller University Bio-Imaging Resource Center) for assistance with Deltavision microscopy.
Footnotes
Funding
The work was supported by The National Institutes of Health [R01NS044385 to K.V.A.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.070805/-/DC1
References
- Apionishev S., Katanayeva N. M., Marks S. A., Kalderon D., Tomlinson A. (2005). Drosophila Smoothened phosphorylation sites essential for Hedgehog signal transduction. Nat. Cell Biol. 7, 86–92 [DOI] [PubMed] [Google Scholar]
- Bai C. B., Stephen D., Joyner A. L. (2004). All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev. Cell 6, 103–115 [DOI] [PubMed] [Google Scholar]
- Barzi M., Berenguer J., Menendez A., Alvarez-Rodriguez R., Pons S. (2010). Sonic-hedgehog-mediated proliferation requires the localization of PKA to the cilium base. J. Cell Sci. 123, 62–69 [DOI] [PubMed] [Google Scholar]
- Besschetnova T. Y., Kolpakova-Hart E., Guan Y., Zhou J., Olsen B. R., Shah J. V. (2010). Identification of signaling pathways regulating primary cilium length and flow-mediated adaptation. Curr. Biol. 20, 182–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borland G., Smith B. O., Yarwood S. J. (2009). EPAC proteins transduce diverse cellular actions of cAMP. Br. J. Pharmacol. 158, 70–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J., Reisert J., Frings S. (2005). Regulation of cyclic nucleotide-gated channels. Curr. Opin. Neurobiol. 15, 343–349 [DOI] [PubMed] [Google Scholar]
- Concordet J. P., Lewis K. E., Moore J. W., Goodrich L. V., Johnson R. L., Scott M. P., Ingham P. W. (1996). Spatial regulation of a zebrafish patched homologue reflects the roles of sonic hedgehog and protein kinase A in neural tube and somite patterning. Development 122, 2835–2846 [DOI] [PubMed] [Google Scholar]
- Cooper A. F., Yu K. P., Brueckner M., Brailey L. L., Johnson L., McGrath J. M., Bale A. E. (2005). Cardiac and CNS defects in a mouse with targeted disruption of suppressor of fused. Development 132, 4407–4417 [DOI] [PubMed] [Google Scholar]
- Chen M. H., Wilson C. W., Li Y. J., Law K. K., Lu C. S., Gacayan R., Zhang X., Hui C. C., Chuang P. T. (2009). Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 23, 1910–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Gallaher N., Goodman R. H., Smolik S. M. (1998). Protein kinase A directly regulates the activity and proteolysis of cubitus interruptus. Proc. Natl. Acad. Sci. USA 95, 2349–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Cardinaux J. R., Goodman R. H., Smolik S. M. (1999). Mutants of cubitus interruptus that are independent of PKA regulation are independent of hedgehog signaling. Development 126, 3607–3616 [DOI] [PubMed] [Google Scholar]
- Chen Y., Sasai N., Ma G., Yue T., Jia J., Briscoe J., Jiang J. (2011). Sonic Hedgehog dependent phosphorylation by CK1α and GRK2 is required for ciliary accumulation and activation of Smoothened. PLoS Biol. 9, e1001083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y., Reiter J. F. (2005). Vertebrate Smoothened functions at the primary cilium. Nature 13, 1018–1021 [DOI] [PubMed] [Google Scholar]
- Dai P., Akimaru H., Tanaka Y., Maekawa T., Nakafuku M., Ishii S. (1999). Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem. 274, 8143–8152 [DOI] [PubMed] [Google Scholar]
- De Camilli P., Moretti M., Donini S. D., Walter U., Lohmann S. M. (1986). Heterogeneous distribution of the cAMP receptor protein RII in the nervous system: evidence for its intracellular accumulation on microtubules, microtubule-organizing centers, and in the area of the Golgi complex. J. Cell Biol. 103, 189–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q., Motoyama J., Gasca S., Mo R., Sasaki H., Rossant J., Hui C. C. (1998). Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 125, 2533–2543 [DOI] [PubMed] [Google Scholar]
- Eggenschwiler J. T., Espinoza E., Anderson K. V. (2001). Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature 412, 194–198 [DOI] [PubMed] [Google Scholar]
- Engel B. D., Ludington W. B., Marshall W. F. (2009). Intraflagellar transport particle size scales inversely with flagellar length: revisiting the balance-point length control model. J. Cell Biol. 187, 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein D. J., Marti E., Scott M. P., McMahon A. P. (1996). Antagonizing cAMP-dependent protein kinase A in the dorsal CNS activates a conserved Sonic hedgehog signaling pathway. Development 122, 2885–9284 [DOI] [PubMed] [Google Scholar]
- Fabunmi R. P., Wigley W. C., Thomas P. J., DeMartino G. N. (2000). Activity and regulation of the centrosome-associated proteasome. J. Biol. Chem. 275, 409–413 [DOI] [PubMed] [Google Scholar]
- Freed E., Lacey K. R., Huie P., Lyapina S. A., Deshaies R. J., Stearns T., Jackson P. K. (1999). Components of an SCF ubiquitin ligase localize to the centrosome and regulate the centrosome duplication cycle. Genes Dev. 13, 2242–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz S. C., Anderson K. V. (2010). The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich L. V., Milenkovic L., Higgins K. M., Scott M. P. (1997). Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113 [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M., Bitgood M. J., McMahon A. P. (1996). Protein kinase A is a common negative regulator of Hedgehog signaling in the vertebrate embryo. Genes Dev. 10, 647–658 [DOI] [PubMed] [Google Scholar]
- Howe D. G., Wiley J. C., McKnight G. S. (2002). Molecular and behavioral effects of a null mutation in all PKA C beta isoforms. Mol. Cell. Neurosci. 20, 515–524 [DOI] [PubMed] [Google Scholar]
- Huang Y., Roelink H., McKnight G. S. (2002). Protein kinase A deficiency causes axially localized neural tube defects in mice. J. Biol. Chem. 277, 19889–19896 [DOI] [PubMed] [Google Scholar]
- Huangfu D., Anderson K. V. (2005). Signaling from Smo to Ci/Gli: conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development 133, 3–14 [DOI] [PubMed] [Google Scholar]
- Huangfu D., Liu A., Rakeman A. S., Murcia N. S., Niswander L., Anderson K. V. (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87 [DOI] [PubMed] [Google Scholar]
- Hui C. C., Joyner A. L. (1993). A mouse model of greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat. Genet. 3, 241–246 [DOI] [PubMed] [Google Scholar]
- Humke E. W., Dorn K. V., Milenkovic L., Scott M. P., Rohatgi R. (2010). The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 24, 670–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham P. W., McMahon A. P. (2001). Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087 [DOI] [PubMed] [Google Scholar]
- Ingham P. W., Nakano Y., Seger C. (2011). Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet. 12, 393–406 [DOI] [PubMed] [Google Scholar]
- Jia J., Tong C., Wang B., Luo L., Jiang J. (2004). Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432, 1045–1050 [DOI] [PubMed] [Google Scholar]
- Jia J., Zhang L., Zhang Q., Tong C., Wang B., Hou F., Amanai K., Jiang J. (2005). Phosphorylation by double-time/CKIepsilon and CKIalpha targets cubitus interruptus for Slimb/beta-TRCP-mediated proteolytic processing. Dev. Cell 9, 819–830 [DOI] [PubMed] [Google Scholar]
- Jia J., Kolterud A., Zeng H., Hoover A., Teglund S., Toftgard R., Liu A. (2009). Suppressor of Fused inhibits mammalian Hedgehog signaling in the absence of cilia. Dev. Biol. 330, 452–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J., Struhl G. (1995). Protein kinase A and hedgehog signaling in Drosophila limb development. Cell 80, 563–572 [DOI] [PubMed] [Google Scholar]
- Jiang J., Struhl G. (1998). Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature 391, 493–496 [DOI] [PubMed] [Google Scholar]
- Jiang J., Hui C. C. (2008). Hedgehog signaling in development and cancer. Dev. Cell 15, 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Q., Zelman A. K., Kuang E., Li S., Matise M. P. (2004). Transduction of graded Hedgehog signaling by a combination of Gli2 and Gli3 activator functions in the developing spinal cord. Development 131, 3593–3604 [DOI] [PubMed] [Google Scholar]
- Li W., Ohlmeyer J. T., Lane M. E., Kaldero D. (1995). Function of protein kinase A in hedgehog signal transduction and Drosophila imaginal disc development. Cell 80, 553–562 [DOI] [PubMed] [Google Scholar]
- Liem K. F., Jr, He M., Ocbina P. J., Anderson K. V. (2009). Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA 106, 13377–13382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A., Wang B., Niswander L. A. (2005). Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development 132, 3103–3111 [DOI] [PubMed] [Google Scholar]
- Low S. H., Roche P. A., Anderson H. A., van Ijzendoorn S. C., Zhang M., Mostov K. E., Weimbs T. (1998). Targeting of SNAP-23 and SNAP-25 in polarized epithelial cells. J. Biol. Chem. 273, 3422–3430 [DOI] [PubMed] [Google Scholar]
- Marks S. A., Kalderon D. (2011). Regulation of mammalian Gli proteins by Costal 2 and PKA in Drosophila reveals Hedgehog pathway conservation. Development 138, 2533–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise M. P., Epstein D. J., Park H. L., Platt K. A., Joyner A. L. (1998). Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development 125, 2759–2770 [DOI] [PubMed] [Google Scholar]
- May S. R., Ashique A. M., Karlen M., Wang B., Shen Y., Zarbalis K., Reiter J., Ericson J., Peterson A. S. (2005). Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 287, 378–389 [DOI] [PubMed] [Google Scholar]
- Milenkovic L., Scott M. P. (2010). Not lost in space: trafficking in the hedgehog signaling pathway. Sci. Signal. 13, pe14 [DOI] [PubMed] [Google Scholar]
- Milenkovic L., Scott M. P., Rohatgi R. (2009). Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J. Cell Biol. 187, 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama J., Milenkovic L., Iwama M., Shikata Y., Scott M. P., Hui C. C. (2003). Differential requirement for Gli2 and Gli3 in ventral neural cell fate specification. Dev. Biol. 259, 150–161 [DOI] [PubMed] [Google Scholar]
- Murray A. J. (2008). Pharmacological PKA inhibition: all may not be what it seems. Sci. Signal. 1, re4 [DOI] [PubMed] [Google Scholar]
- Nagy A., Gertsenstein M., Vintersten K., Behringer R. (2003). Manipulating the Mouse Embryo: A Laboratory Manual (Third Edition). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Nigg E. A., Schafer G., Hilz H., Eppenberger H. M. (1985). Cyclic-AMP-dependent protein kinase type II is associated with the Golgi complex and with centrosomes. Cell 41, 1039–1051 [DOI] [PubMed] [Google Scholar]
- Ocbina P. J., Anderson K. V. (2008). Intraflagellar transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Dev. Dyn. 237, 2030–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocbina P. J., Eggenschwiler J. T., Moskowitz I., Anderson K. V. (2011). Complex interactions between genes controlling trafficking in primary cilia. Nat. Genet. 43, 547–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlmeyer J. T., Kalderon D. (1997). Dual pathways for induction of wingless expression by protein kinase A and Hedgehog in Drosophila embryos. Genes Dev. 11, 2250–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y., Bai C. B., Joyner A. L., Wang B. (2006). Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 26, 3365–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y., Wang C., Wang B. (2009). Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog-regulated mouse development. Dev. Biol. 326, 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson M., Stamataki D., te, Welscher P., Andersson E., Bose J., Ruther U., Ericson J., Briscoe J. (2002). Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 16, 2865–2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. A., Kalderon D. (1999). Proteolysis of cubitus interruptus in Drosophila requires phosphorylation by protein kinase A. Development 126, 4331–4339 [DOI] [PubMed] [Google Scholar]
- Raychowdhury M. K., Ramos A. J., Zhang P., McLaughin M., Dai X.-Q., Chen X.-Z., Montalbetti N., del Rocío Cantero M., Ausiello D. A., Cantiello H. F. (2009). Vasopressin receptor-mediated functional signaling pathway in primary cilia of renal epithelial cells. Am. J. Physiol. Renal. Physiol. 296, F87–F97 [DOI] [PubMed] [Google Scholar]
- Rogers G. C., Rusan N. M., Peifer M., Rogers S. L. (2008). A multicomponent assembly pathway contributes to the formation of acentrosomal microtubule arrays in interphase Drosophila cells. Mol. Biol. Cell 19, 3163–3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R., Milenkovic L., Scott M. P. (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372–376 [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A. (1999). Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development 126, 3205–3216 [DOI] [PubMed] [Google Scholar]
- Skalhegg B. S., Huang Y., Su T., Idzerda R. L., McKnight G. S., Burton K. A. (2002). Mutation of the Calpha subunit of PKA leads to growth retardation and sperm dysfunction. Mol. Endocrinol. 16, 630–639 [DOI] [PubMed] [Google Scholar]
- Smelkinson M. G., Kalderon D. (2006). Processing of the Drosophila hedgehog signaling effector Ci-155 to the repressor Ci-75 is mediated by direct binding to the SCF component Slimb. Curr. Biol. 16, 110–116 [DOI] [PubMed] [Google Scholar]
- Svard J., Heby-Henricson K., Persson-Lek M., Rozell B., Lauth M., Bergstrom A., Ericson J., Toftgard R., Teglund S. (2006). Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev. Cell 10, 187–197 [DOI] [PubMed] [Google Scholar]
- Teglund S., Toftgard R. (2010). Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta. 1805, 181–208 [DOI] [PubMed] [Google Scholar]
- Theunissen J. W., de Sauvage F. J. (2009). Paracrine Hedgehog signaling in cancer. Cancer Res. 69, 6007–6010 [DOI] [PubMed] [Google Scholar]
- Tiecke E., Turner R., Sanz-Ezquerro J. J., Warner A., Tickle C. (2007). Manipulations of PKA in chick limb development reveal roles in digit patterning including a positive role in Sonic Hedgehog signaling. Dev. Biol. 305, 312–324 [DOI] [PubMed] [Google Scholar]
- Toftgard R. (2000). Hedgehog signalling in cancer. Cell Mol. Life Sci. 57, 1720–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukachinsky H., Lopez L. V., Salic A. (2010). A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 191, 415–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar A. R., Moon R. T. (1996). Inhibition of protein kinase A phenocopies ectopic expression of hedgehog in the CNS of wild-type and cyclops mutant embryos. Dev. Biol. 178, 186–191 [DOI] [PubMed] [Google Scholar]
- Wang B., Li Y. (2006). Evidence for the direct involvement of βTrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 103, 33–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Fallon J. F., Beachy P. A. (2000). Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434 [DOI] [PubMed] [Google Scholar]
- Wang C., Pan Y., Wang B. (2010). Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 137, 2001–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Wang B., Jiang J. (1999). Protein kinase A antagonizes Hedgehog signaling by regulating both the activator and repressor forms of Cubitus interruptus. Genes Dev. 13, 2828–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X., Lai C. K., Evangelista M., Hongo J. A., de Sauvage F. J., Scales S. J. (2010). Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell. Biol. 30, 1910–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis B. S., Niswender C. M., Su T., Amieux P. S., McKnight G. S. (2011). Cell-type specific expression of a dominant negative PKA mutation in mice. PLoS One 6, e18772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. W., Chen M. H., Chuang P. T. (2009). Smoothened adopts multiple active and inactive conformations capable of trafficking to the primary cilium. PLoS One 4, e5182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirschell M., Yamamoto R., Alford L., Gokhale A., Gaillard A., Sale W. S. (2011). Regulation of ciliary motility: conserved protein kinases and phosphatases are targeted and anchored in the ciliary axoneme. Arch. Biochem. Biophys. 510, 93–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H., Jia J., Liu A. (2010). Coordinated translocation of mammalian Gli proteins and suppressor of fused to the primary cilium. PLoS One 5, e15900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Williams E. H., Guo Y., Lum L., Beachy P. A. (2004). Extensive phosphorylation of Smoothened in Hedgehog pathway activation. Proc. Natl. Acad. Sci. USA 101, 17900–17907 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}