

Abstract
Amyotrophic lateral sclerosis (ALS), also known as motor neuron disease (MND), is an adult onset neurodegenerative disorder characterised by the degeneration of cortical and spinal cord motor neurons, resulting in progressive muscular weakness and death. Increasing evidence supports mitochondrial dysfunction and oxidative DNA damage in ALS motor neurons. Several DNA repair enzymes are activated following DNA damage to restore genome integrity, and impairments in DNA repair capabilities could contribute to motor neuron degeneration. After a brief description of the evidence of DNA damage in ALS, this paper focuses on the available data on DNA repair activity in ALS neuronal tissue and disease animal models. Moreover, biochemical and genetic data on DNA repair in ALS are discussed in light of similar findings in other neurodegenerative diseases.
Keywords: Amyotrophic lateral sclerosis, DNA repair, DNA damage, base excision repair, neurodegeneration
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS), also known as motor neuron disease (MND), is an adult onset neurodegenerative disorder characterised by the degeneration of motor neurons of the motor cortex, brainstem, and spinal cord, resulting in progressive weakness and death. The course of the disease is inexorably progressive, with almost 50% of the patients dying within 3 years of onset, generally due to respiratory failure. The typical age at onset is between 50 and 60 years, and the incidence of the disease is similar worldwide ranging from 1 to 3 new cases per 100.000 individuals every year. ALS is predominantly a sporadic disorder (sALS), and only 5 to 10% of the cases have a familial origin (fALS). Studies in families led to the identification of several genes and potential loci responsible for familial ALS cases, whereas the development of the sporadic forms is likely the result of complex gene-environment interactions not yet completely clarified [1–3].
The gene encoding for the cytosolic copper-zinc superoxide dismutase protein (SOD1), on chromosome 21q21, was the first ALS identified gene (SOD1), accounting for autosomal dominant forms. Superoxide dismutases are scavenger enzymes against free radicals mainly produced by mitochondrial respiration, and SOD1 mutations account for approximately 20% of the familial forms. Other common pathogenic ALS mutations have been identified in the 43-KDa TAR DNA-binding protein (TARDBP) and fused in sarcoma (FUS) genes, accounting for approximately 4-5% of fALS cases each. Less common genetic mutations causing fALS include those in alsin (ALS2), senataxin (SETX), spatacsin (SPG11), vesicle-associated membrane protein/synaptobrevin-associated membrane protein B (VAPB), angiogenin (ANG), phosphoinositide phosphatase (FIG4), and optineurin (OPTN) genes, overall accounting for less than 5% of fALS cases [2, 3]. Further, mutations in the dynactin gene (DCTN1) on chromosome 2p13 have been observed in a family with a slowly progressive motor neuron disease, with some clinical features overlapping ALS [4]. Variants in other genes have been suggested to be associated with fALS or found in families with atypical ALS phenotypes, but results are still inconclusive leaving more than 50% of the genetics of fALS yet to be identified [2, 3]. Common polymorphisms in several genes have been hypothesized to be associated with increased risk to develop sporadic ALS forms, including angiogenesis genes (ANG and VEGF), variant sequences of neurofilament proteins (NEFL, NEFM, and NEFH), DNA repair genes (APEX1 and OGG1), paraoxonase genes (PON1, PON2, and PON3), survival motor neuron genes (SMN1 and SMN2), and the haemochromatosis gene (HFE) [5]. The ALSGene database (http://www.alsgene.org/) is a recently available public database containing data on ALS genetic association studies, including genome-wide association studies (GWAS). The database is continuously updated, and meta-analyses are performed for those polymorphisms which have been investigated in at least four independent studies. Accessed on June 2011, the database contained information on 284 polymorphisms in 90 genes, and 21 meta-analyses were available. The available meta-analyses revealed significant associations between four gene polymorphisms (APEX1 rs1130409, GWA_9p21.2 rs2814707, HFE rs1799945, and UNC13A rs12608932) and ALS risk. Moreover, two additional polymorphisms (DPP6 rs10260404 and ATXN2-polyQ-3bpSTR) resulted associated with ALS risk in two independent large-scale association studies each (http://www.alsgene.org/). Concerning ANG, VEGF, and PON genes, recently published meta-analyses [6, 7] and updated meta-analyses (http://www.alsgene.org/) failed to replicate association with disease risk, with the exception of the VEGF-2578AA genotype in males [7].
There is evidence that sALS is more common in men than in women and that gender influences the clinical features of the disease. Generally, the male/female ratio for incidence is greater at younger ages and tends to disappear with increasing age. Moreover, sALS males have a greater likelihood of onset in the spinal regions, and females in the bulbar region [8]. Therefore, it is not surprising that also genetic variants might have a different role in disease risk after stratification by gender.
Concerning environmental factors, the first evidence of an environmental contribution to ALS came from the observed endemic occurrence of ALS with parkinsonism and progressive dementia in regions of Western Pacific that could not be fully explained by genetic factors and was related to the consumption of food made with seeds of the cycad plants [9]. Several other environmental factors have been extensively studied in recent years. The most consistent associations are exposure to pesticides, heavy metals including lead and mercury, an excessive physical activity such as playing professional soccer, head injuries, cigarette smoking, and occupational exposure to electromagnetic fields, even if, to date, no definitive causal link has been consistently established [10–12].
Studies with ALS animal models indicate that the overexpression of mutant SOD1 in mice leads to symptoms similar to those observed in humans [13, 14], suggesting that a toxic gain of function for the mutant gene might be responsible for the disease. Another study indicates that the toxicity of mutant SOD1 might arise from an initial misfolding (gain of function) reducing nuclear protection against oxidative DNA damage compared with the wild-type enzyme [15]. Moreover, neurons obtained from Als2 knock-out mice are more vulnerable to oxidative damage compared to primary cultured neurons derived from controls, suggesting that the loss of the gene predisposes to oxidative damage [16]. More recently, it was observed that the transfection of mutant TAR DNA-binding protein-43 induces mitochondrial dysfunction and oxidative damage in motor neuron-like cells [17]. Moreover, senataxin is a member of the superfamily I DNA/RNA helicases, likely involved in oxidative DNA damage response [18]. Several authors report increased oxidative damage in cortical neurons, spinal cords, and other tissues of each of sALS and fALS patients, and a compromised DNA repair activity in ALS has been largely suggested [19–21]. After a brief description of oxidative DNA damage in either ALS tissues or disease animal models, this paper describes the current knowledge on DNA repair activity in ALS.
2. DNA REPAIR: THE DNA BASE EXCISION REPAIR PATHWAY
It is estimated that every day each single cell of the human body is subjected to an average of approximately one million DNA lesions, including apurinic/apyrimidinic (AP) sites (abasic sites), DNA adducts, single-strand breaks (SSBs), double-strand breaks (DSBs), insertion/deletion mismatches, and DNA-protein crosslinks, all arising as a consequence of either endogenous or exogenous factors. Several DNA repair mechanisms are therefore operative in human cells to counteract the daily amount of DNA damage, including the DNA base excision repair (BER) pathway that primarily removes DNA bases modified by oxidation, alkylation, and deamination, the nucleotide excision repair pathway (NER) that operates on UV photoproducts, DNA crosslinks, and DNA bulky lesions, mismatch repair (MMR) that corrects base mismatches and small insertions or deletions during replication, and DSBs repair mechanisms such as homologous recombination (HR) and nonhomologous end joining (NHEJ) that repair DNA DSBs such as those caused by ionizing radiations [22, 23].
The DNA base excision repair (BER) pathway deserves a detailed description in order to understand the following sections of this paper since it is believed to be the major pathway for repairing DNA base modifications caused by oxidation and has been largely investigated in ALS tissues and animal models (Table 1).
Table 1.
Levels and activity of DNA repair proteins in motor neurons of ALS patients and disease animal models.
| Protein | Function | Activity in ALS | References |
|---|---|---|---|
| ↓ in ALS motor cortex | [20] | ||
| Apurinic/apyrimidinic endonuclease 1 (APEX1) | DNA endonuclease (BER pathway) | ↑ in ALS motor cortex and spinal cord motor neurons | [24] |
| ↓ in spinal cord motor neurons of SOD1 transgenic mice | [25] | ||
|
| |||
| ↓ mitochondrial activity in ALS motor neurons | [21] | ||
| 8-Oxoguanine DNA glycosylase (OGG1) | DNA glycosylase that specifically removes 8-oxoguanine from the DNA (BER pathway) | ↑ nuclear levels in spinal cord motor neurons of SOD1 transgenic mice | [26] |
| = unchanged mitochondrial levels in spinal cord motor neurons of SOD1 transgenic mice | [26] | ||
|
| |||
| DNA polymerase γ (Pol γ) | Mitochondrial DNA polymerase (BER pathway) | ↓ in spinal cord motor neurons of SOD1 transgenic mice | [26] |
|
| |||
| ↑ expression in astrocytes of the spinal cord of ALS patients | [27] | ||
| Poly-ADP-ribose polymerase-1 (PARP1) | Recognition of DNA damage (DNA repair) | ↓ expression in motor neurons of the spinal cord of ALS patients | [28] |
| ↑ expression in motor cortex, parietal cortex, and cerebellum of ALS patients | [28] | ||
|
| |||
| DNA excision repair protein ERCC-1 | Repair protein primarily involved in NER pathway | Errc1 knock-out mice showed age-related motor neuron degeneration | [29] |
The first step in BER is the recognition of a damaged base by an appropriate DNA glycosylase that catalyzes the cleavage of the N-glycosylic bond between the damaged base and the sugar moiety; after the cleavage, the damaged base is released resulting in the formation of an abasic site which is then cleaved by an AP lyase activity (an activity present in some DNA glycosylases) or by the major mammalian apurinic/apyrimidinic endonuclease (APEX1). Repair can then proceed through short- or long-patch BER. In short-patch BER, which is the most common subpathway, a single nucleotide is incorporated into the gap by DNA polymerase β (Pol β), the same polymerase also removes the 5′-sugar phosphate, and the DNA ligase III/X-ray repair cross-complementing group 1 (XRCC1) complex seals the DNA ends. Thus, Pol β carries out two distinct reactions in BER; it uses its DNA polymerase activity to fill in the one-nucleotide gap, and it also uses its 5′-deoxyribophosphatase activity to cleave the 5′ phosphate to allow for efficient ligation. However, if the 5′-deoxyribophosphate (5′dRP) is reduced or oxidised, and therefore resistant to the action of Pol β, then an additional synthesis of DNA is required to displace the modified 5′-sugar phosphate as part of a flap, which is then removed by flap endonuclease (FEN1). Therefore, in long-patch BER, several nucleotides (two to seven-eight) are incorporated, followed by cleavage of the resulting 5′ flap structure and ligation. It has been suggested that after Pol β adds the first nucleotide into the gap, it is substituted by Pol δ/ε which continues long-patch BER. DNA ligase I completes the long-patch pathway. Several other proteins include the proliferating cell nuclear antigen (PCNA), the RPA protein, and FEN-1 participate in long-patch BER [30, 31]. A diagram illustrating the BER pathway is shown in Figure 1.
Figure 1.
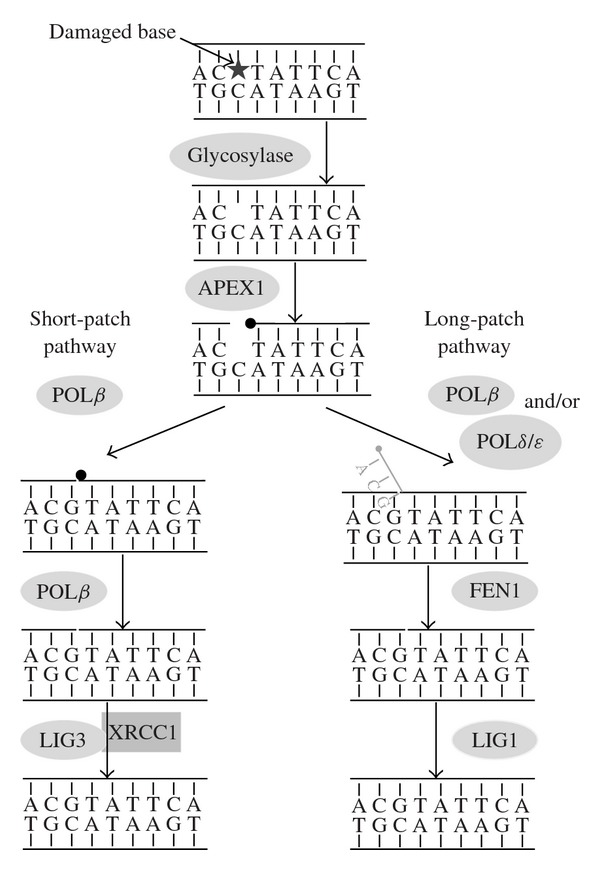
Diagram illustrating the DNA base excision repair (BER) pathway. In this pathway, a DNA glycosylase catalyzes the removal of a damaged base, creating an abasic (AP) site. The APEX1 endonuclease catalyzes the incision of the DNA backbone leaving behind a 5′-deoxyribose phosphate (5′dRP, indicated with a black circle). In short-patch BER, polymerase β (Pol β) displaces the AP site and polymerizes DNA to fill in the gap. Pol β then catalyzes the removal of the displaced AP site, and the ligase III/XRCC1 complex seals the ends. If the 5′dRP is refractory to the action of Pol β, then an additional synthesis of DNA (long-patch BER) is required to displace the modified 5′-sugar phosphate as part of a flap (in grey), which is then removed by flap endonuclease (FEN1). Pol β adds the first nucleotide into the gap and is substituted by polymerase δ/ε which continues long-patch BER. DNA ligase I (LIG1) completes the long-patch pathway.
Several different DNA glycosylases, each specific for certain lesions, operate in human cells. However, individual glycosylases may recognize more than one type of damage, and each specific lesion may be recognized by more than one type of glycosylase, allowing some degrees of redundancy in the process. Moreover, monofunctional glycosylases such as the uracil DNA glycosylase (UNG) have only glycosylase activity and require APEX1 for the incision of the resulting sugar residue. In contrast, bifunctional glycosylases such as oxoguanine DNA glycosylase (OGG1) have both glycosylase and AP lyase activity. The diagram illustrated in Figure 1 should be considered only as indicative of the process, since there is indication of interaction and cooperation between the enzymes at successive steps in BER. For example, XRCC1 acts as a scaffold protein in short-patch BER, regulating and coordinating the whole process. XRCC1 recruits DNA Pol β and DNA ligase III required for filling and sealing the damaged strand. Moreover, it also interacts with DNA glycosylases and APEX1, mediating their exchange at the damaged site. There is a specific interaction between APEX1 and Pol β that assembles the polymerase onto the AP site in DNA. XRCC1 also interacts with poly-ADP-ribose polymerase-1 (PARP-1), which is one of the cellular sensors of DNA SSBs and DSBs. Long-patch BER requires the nuclear protein PCNA that promotes this pathway via its ability to interact with and coordinate the activity of a DNA polymerase and FEN1. Moreover, the decision to proceed via the long-patch or short-patch BER is still poorly understood and might depend on the availability of ATP and/or by the state of the 5′-dRP intermediate [30].
BER takes places either in nuclei and mitochondria, and mitochondria have independent BER machinery encoded by nuclear genes. Indeed, several BER enzymes have been identified which have both nuclear and mitochondrial forms. For example, among DNA glycosylases, nuclear and mitochondrial adenine-DNA-glycosylases are generated by alternative spliced forms of the MYH gene. Similarly nuclear and mitochondrial uracil DNA glycosylases are encoded by alternative splicing and transcription of the UNG gene, and two isoforms of the human oxoguanine DNA glycosylase operate in human cells, the alpha and the beta forms, arising as alternative splice products from the OGG1 gene. The beta form operates in mitochondria, while the alpha form operates in the nucleus as well as in mitochondria; however, it seems that the alpha form is responsible for the base excision activity also in mitochondria, whilst the beta form lacks DNA glycosylase activity, and its function remains unclear. The gaps generated by the action of AP endonucleases/lyases are filled in by the DNA polymerase γ (Pol γ) in the mitochondria, and ligation is mediated by ligase III. There is also some recent evidence of long-patch BER in mitochondria [32].
3. OXIDATIVE DNA DAMAGE IN AMYOTROPHIC LATERAL SCLEROSIS
A wide variety of reactive oxygen species (ROS) are produced in biological systems during the course of the normal metabolism, and they have several important physiological functions. However, the accumulation of ROS beyond the needs of the cell can potentially damage several biomolecules, including lipids, proteins, and nucleic acids. Mammalian cells have an intricate network of defence mechanisms, such as superoxide dismutases, to neutralize an excessive accumulation of ROS and, under physiological conditions, are able to cope with the flux of ROS. Oxidative stress describes a condition in which cellular antioxidant defences are insufficient to keep the levels of ROS below a toxic threshold. Some tissues, including neuronal and muscular tissues, are much more vulnerable to oxidative stress because of their elevated consumption of oxygen required to produce energy and the consequent generation of large amounts of ROS. Evidence of oxidative damage to lipids, proteins, and nucleic acids has been largely detected in spinal cords, cerebrospinal fluid (CSF), and other tissues of both sALS and fALS patients [33].
The type of DNA damage that most likely occurs in neuronal cells is oxidative damage, which is primarily removed by the DNA base excision repair (BER) pathway [34]. Oxidative DNA damage accumulates in the DNA of the neurons over time, especially in mitochondrial DNA (mtDNA), and is supposed to play a critical role in the pathogenesis of several neurodegenerative diseases, including ALS [19, 35, 36]. The mtDNA is believed to be particularly sensitive to oxidative damage due to its proximity to the inner mitochondrial membrane, where oxidants are formed. Several of the mtDNA genes encode for subunits of the mitochondrial respiratory chain; thus, oxidative mtDNA damage, if not correctly repaired, could result in mutations and deletions disrupting the function of genes involved in the production of ATP and leading to mitochondrial dysfunctions, increased production of ROS, and cellular death. Therefore, a decline in one or more BER enzymes may result in the accumulation of nDNA and mtDNA mutations ultimately leading to the production of altered proteins and possible neurodegeneration [31].
In case of oxidative DNA damage, one of the major alterations in the DNA is the formation of 8-hydroxy-2′-deoxyguanosine (8-OHdG), and increased levels of 8-OHdG have been identified in the neuronal DNA of the motor cortex of sALS patients, and in both sALS and fALS spinal cords [19]. The analysis of plasma, urine, and CSF of ALS patients revealed increased levels of 8-OHdG. Moreover, plasma and urine 8-OHdG levels increased significantly with time in the ALS group, and the rate of increase in urine 8-OHdG levels with time was significantly correlated with disease severity [35]. Another study aimed at evaluating oxidative damage in blood and CSF of each of sALS and fALS patients showed that blood concentrations of hydroxyl radicals and CSF values of 8-OHdG and ascorbate free radical were higher in both sALS and fALS patients compared to controls [37]. Increased oxidative damage to the nuclear DNA, measured as the content of 8-OHdG, has been also observed in the G93A-SOD1 transgenic mouse model of fALS [36]. The analysis of 8-OHdG levels in the spinal cord of transgenic mice harbouring a mutant SOD1 gene showed oxidative damage to mtDNA in spinal motor neurons from very early stage of the disease [38]. Others observed that in G93A-SOD1 mutant mice, motor neuron degeneration is characterized by somal and mitochondrial swelling and formation of DNA single-strand breaks prior to double-strand breaks occurring in nuclear and mitochondrial DNA. Single-strand breaks of nuclear DNA and mtDNA were evident in motor neurons of 6-week old mice, and double-strand breaks appeared by 9 weeks of age and progressively increased thereafter [39]. It was also demonstrated that, in sporadic ALS, there is a selective decrease in the activity of the mtDNA-encoded enzyme cytochrome c oxidase in human spinal cord motor neurons, and authors proposed that this could well be caused by oxidative damage to mtDNA leading to the accumulation of mtDNA mutations [40]. Murata et al. [41] measured the concentrations of the reduced and oxidized forms of coenzyme Q10 (CoQ10) and 8-OHdG in the CSF of sALS patients and age-matched healthy controls, observing increased levels of both oxidized CoQ10 and 8-OHdG in the CSF of sALS patients more than in the CSF of controls. Moreover, the percentage of oxidized CoQ10 was correlated with the concentrations of 8-OHdG in the CSF of sALS patients, suggesting that both mitochondrial oxidative damage and oxidative DNA damage play important roles in the pathogenesis of sALS [41]. Significantly higher levels of oxidative DNA damage and DNA strand breaks, increased p53 activity, and a greater percentage of apoptotic cells were observed in SH-SY5Y neuroblastoma cells overexpressing the mutant G93A-SOD1 protein when compared to cells overexpressing wild-type SOD1 and untransfected cells [42]. Collectively, the available data suggest increased nuclear and mitochondrial DNA damage in ALS, likely contributing to mitochondrial dysfunction and death of motor neurons.
4. DNA REPAIR IN AMYOTROPHIC LATERAL SCLEROSIS
Almost 30 years ago, Bradley and Krasin advanced the hypothesis that the primary abnormality in ALS is the accumulation of abnormal DNA, which is unable to undertake normal transcription in motor neurons, and suggested that this abnormal DNA may arise from a deficiency of an isozyme of one of the DNA repair enzymes [43]. A few studies performed over 20 years ago revealed that skin fibroblasts obtained from sALS patients were relatively deficient in the repair of alkylation damage, possibly of apurinic/apyrimidinic sites [44, 45]. This has driven researchers to investigate the activity of the apurinic/apyrimidinic enzyme in ALS tissues, and the frontal cortical levels and the activity of APEX1 (alternatively called APE1 or APE/Ref-1) were determined in 11 sALS patients and 6 age-matched controls, resulting significantly reduced in sALS patients than in controls [20]. Others observed an increased APEX1 activity in ALS motor neurons and suggested that DNA damage could be an upstream mechanism for motor neuron degeneration in ALS and that mechanisms for DNA repair are activated in this disease [24]. APEX1 expression was also examined in the spinal cord of transgenic mice expressing an ALS-linked mutant SOD1. Immunoblotting and immunocytochemical analyses showed that most spinal motor neurons lost the immunoreactivity for APEX1 in the early presymptomatic stage that preceded significant loss of the neurons, suggesting that an early impairment of DNA repair in the spinal motor neurons may account for the mutant SOD1-mediated motor neuronal death in this model [25].
OGG1 is the DNA glycosylase that specifically removes oxidized guanine from DNA. Increased levels of 8-OHdG and an impaired mitochondrial OGG1 activity were observed in the spinal motor neurons of sALS individuals compared with control subjects, indicating that oxidative DNA damage accumulates in the mtDNA and that OGG1 does not repair it efficiently [21]. Another study showed that nuclear ogg1 levels were increased, while mitochondrial ogg1 remained unchanged, and mitochondrial DNA polymerase γ was downregulated in spinal cord motor neurons of SOD1 transgenic mice [26]. More recently, an age-related motor neuron degeneration was observed in DNA repair-deficient Ercc1 mice, that is, mice that are impaired in several DNA repair systems, including nucleotide excision repair (NER), interstrand crosslink repair, and double-strand break repair, supporting the notion that accumulation of DNA damage and genotoxic stress may contribute to neuronal aging and motor neuron vulnerability in human neuromuscular disorders [29].
PARP-1 is a zinc-finger DNA-binding protein that is activated by single-strand breaks or double-strand breaks in DNA. The primary function of PARP-1 is in DNA repair processes through the detection of DNA damage and the prevention of chromatid exchanges. PARP-1 poly-ADP-ribosylates several proteins involved in DNA repair including histones, thus inducing local relaxation of the chromatin structure and facilitating the access of repair proteins to damaged DNA. However, PARP-1 activation can also result in cell death. Indeed, cells with extensive DNA damage have a widespread PARP-1 activation with a subsequent depletion of its substrate NAD+, and then ATP, leading to energy failure and cell death. Using immunohistochemical analyses of postmortem tissue, Kim et al. [27] demonstrated that PARP-1 expression is increased in astrocytes but decreased in motor neurons in the spinal cord of sporadic ALS patients. The same group observed that in ALS brain, PARP-1 expression was increased in the motor cortex, parietal cortex, and cerebellum. PARP-1 immunostaining in the motor cortex was increased in ALS neurons and subcortical glia and macrophages. Moreover, there was widespread increased PARP-1 expression in neurons in the parietal cortex and cerebellum, regions that are typically clinically unaffected in ALS, suggesting widespread oxidative stress [28]. A summary of DNA repair activity in ALS is shown in Table 1. Collectively, the available data suggest that DNA repair mechanisms are activated following oxidative DNA damage in ALS tissues, and that impairments in DNA repair activities might contribute to the accumulation of DNA damage critical for motor neuron survival.
The results of studies attempting to evaluate the association between variants and polymorphisms of BER genes and sALS risk have been conflicting and inconsistent. Common BER polymorphisms, including APEX1 Asp148Glu (rs1130409), OGG1 Ser326Cys (rs1052133), XRCC1 Arg194Trp (rs1799782), XRCC1 Arg280His (rs25489), and XRCC1 Arg399Gln (rs25487), have been investigated as candidate sALS risk factors [46–53]. However, despite some positive associations in initial studies [47, 49, 51, 52], subsequent research has failed to replicate those findings [48, 50, 53]. Additionally, all the studies were largely limited by the sample size of case-control groups [46–53]. Also a reanalysis of GWAS data failed to confirm association between XRCC1 polymorphisms and sALS risk [53].
5. DISCUSSION AND CONCLUSIONS
The present paper describes the currently available literature concerning DNA repair in ALS. Overall, mitochondrial dysfunction and oxidative DNA damage have been largely detected in motor neurons of either ALS patients or disease animal models [19, 35–41], and a widespread PARP-1 activation was observed in several ALS brain regions [28], consistent with an increase in DNA damage. The activity of APEX1 resulted increased in ALS spinal cord motor neurons [24] and decreased in ALS cortical neurons or in spinal cord motor neurons of SOD1 transgenic mice [20, 25]. The activity of other BER enzymes resulted increased, unchanged, or reduced, depending on the enzyme or the model analysed (Table 1). These apparently contradictory findings have been observed also in other major neurodegenerative diseases such as Alzheimer's disease (AD) and Parkinson's disease (PD). Overall, increased oxidative DNA damage, a widespread PARP-1 activation, and an increased expression of BER enzymes were observed in neurons of AD, PD, and ALS individuals suggesting that DNA repair mechanisms could be activated following oxidative DNA attack in early stages of the neurodegenerative process (for a review, see [23]). However, several investigators reported reduced/impaired BER activity in AD-, PD-, or ALS-affected tissues [23], suggesting that following a persistent condition of oxidative stress in neurons, DNA repair proteins, activated to counteract the oxidative insult, could be themselves the subject of ROS attack resulting in protein modification and degradation, and ultimately leading to a reduced DNA repair activity. For example, there is some indication that OGG1 activity might be impaired by posttranslational oxidative modifications and degradation in AD, suggesting that increased oxidative stress in AD brain neurons could cause BER impairments [54, 55].
Another interesting question is whether or not inherited DNA repair impairments, such as those caused by inherited gene variants, could contribute to the neurodegenerative process. Hereditary spinocerebellar ataxias deficient in DNA damage responses represent the most robust set of data linking mutations in DNA repair genes to neurodegeneration (reviewed in [56]). Spinocerebellar ataxia with axonal neuropathy 1 (SCAN1) is caused by autosomal recessive mutations in the gene encoding tyrosyl-DNA phosphodiesterase 1 (TDP1), a protein required for the repair of DNA single-strand breaks that arise independent of DNA replication from abortive topoisomerase 1 activity or oxidative stress. Ataxia-telangiectasia (AT), ataxia-telangiectasia-like disorder (ATLD), ataxia oculomotor apraxia type 1 (AOA1), and ataxia oculomotor apraxia type 2A (AOA2) are a subgroup of the autosomal recessive spinocerebellar ataxias characterized by cerebellar atrophy and oculomotor apraxia. The progressive neurodegeneration described in AT and ATLD is due to mutations in genes encoding for ATM and Mre11, respectively. ATM recognizes and signals DNA double-strand breaks to the cell cycle checkpoints and the DNA repair machinery. The Mre11 DNA repair complex, composed of Rad50, Mre11, and Nbs1 proteins, is involved in DNA damage recognition, DNA repair, and initiating cell cycle checkpoints. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. AOA1 is caused by mutations in the gene encoding aprataxin (APTX), a nuclear protein that interacts with several DNA repair proteins, including XRCC1, DNA polymerase β, DNA ligase III, PARP-1, and p53. It functions in the end processing of DNA single-strand breaks removing 3′-phosphate, 5′-phosphate, and 3′-phosphoglycolate ends. AOA2 is caused by autosomal recessive mutations in the gene encoding senataxin (SETX) [56]. SETX is a member of the superfamily I DNA/RNA helicases, likely involved in oxidative DNA damage response, and, as discussed in the introduction section of this paper, SETX mutations have been also linked to familial ALS [1, 18].
Parkin is an E3 ubiquitin ligase that acts on a variety of substrates, resulting in polyubiquitination and degradation by the proteasome or monoubiquitination and regulation of biological activity. Mutation of parkin is one of the most prevalent causes of autosomal recessive familial PD, and a recent study has shown that parkin is essential for optimal repair of DNA damage. Particularly, DNA damage induces nuclear translocation of parkin leading to interactions with nuclear proteins involved in DNA repair [57]. Moreover, parkin protects mitochondrial genome integrity and supports mtDNA repair [58]. Missense mutations in the gene encoding mitochondrial DNA polymerase γ (POLG1) cosegregate with a phenotype that includes progressive external ophthalmoplegia and parkinsonism and have been reported in case studies, in which parkinsonism was part of the clinical symptoms [59, 60]. In addition, a POLG1 polymorphism (a polymorphic polyglutamine tract in the N-terminal region of the protein) has been associated with risk of sporadic idiopathic PD [61].
To date, there is no clear evidence linking mutations in DNA repair genes to AD. Novel OGG1 mutations leading to a serious impairment in protein activity have been observed in nuclear DNA from postmortem brain specimens of 4 AD subjects, but no causal link with the disease has been yet proven [62].
Concerning ALS, inconsistent and/or contradictory findings have been observed when attempting to evaluate common BER gene polymorphisms as candidate sALS risk factors [46–53].
Similarly, common BER gene polymorphisms, namely, OGG1 Ser326Cys, APEX1 Asp148Glu, XRCC1 Arg194Trp, XRCC1 Arg280His, and XRCC1 Arg399Gln, are not significantly associated with increased AD risk [63–66], and the OGG1 Ser326Cys polymorphism does not affect PD risk or age at onset [67]. There is however evidence that PARP-1 haplotypes could contribute to both AD and PD risk [68–70]. Interestingly, there is also evidence of BER involvement in Huntington's disease (HD), a progressive neurodegenerative disorder resulting in cognitive impairment, choreiform movements, and death which usually occurs 15–20 years after the onset of the symptoms. HD is an autosomal dominant disorder caused by a CAG repeat expansion within exon 1 of the gene encoding for huntingtin (IT15). Disease age at onset is inversely correlated with the number of CAG repeats, and the OGG1 protein has been involved in somatic expansion of the CAG repeat tract in HD cells, occurring during the repair of oxidative DNA damage in the CAG repeat tract and likely contributing to disease age at onset [71].
The recent observation of age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice [29] supports the notion that the impairment of additional DNA repair mechanisms, such as the nucleotide excision repair pathway, might contribute to motor neuron vulnerability. NER involves at least 20–30 proteins or complexes of proteins and is required for the removal of a wide variety of forms of DNA damage, including UV-induced photoproducts, DNA crosslinks, and other bulky DNA lesions. Xeroderma pigmentosum (XP), Cockayne's syndrome (CS), and trichothiodystrophy (TTD) represent a clinically heterogeneous group of progeroid syndromes characterized by defects in NER proteins. A subset of these patients exhibit neurological dysfunction and neurodegeneration, and several studies performed in mice, as well as in cell cultures, suggest that neurodegeneration in XP and CS patients might arise as a consequence of impaired repair of DNA lesions caused by mutations of NER genes (reviewed in [23]). More recently, it has been suggested that polymorphisms of NER genes might contribute to the risk of multiple sclerosis, an inflammatory demyelinating disease of the central nervous system characterized by a significant neurodegenerative component [72].
In summary, increasing evidence supports a role for DNA repair mechanisms in neurodegenerative diseases, and an impaired DNA repair activity, either of genetic origin or environmentally/oxidatively induced, is likely to contribute to the onset and progression of neurodegenerative disorders [23], making this field a timely and exciting research topic in neuropathological studies. Concerning ALS, additional studies are required to elucidate the molecular mechanisms leading to impaired DNA repair activity in motor neurons.
ACKNOWLEDGMENT
The author acknowledges all the patients and controls that participated in research studies [50–52, 63, 67].
References
- 1.Migliore L, Coppedè F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutation Research. 2009;667(1-2):82–97. doi: 10.1016/j.mrfmmm.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 2.Ticozzi N, Tiloca C, Morelli C, et al. Genetics of familial amyotrophic lateral sclerosis. Archives Italiennes de Biologie. 2011;149:65–82. doi: 10.4449/aib.v149i1.1262. [DOI] [PubMed] [Google Scholar]
- 3.Traub R, Mitsumoto H, Rowland LP. Research advances in amyotrophic lateral sclerosis, 2009 to 2010. Current Neurology and Neuroscience Reports. 2011;11:67–77. doi: 10.1007/s11910-010-0160-0. [DOI] [PubMed] [Google Scholar]
- 4.Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nature Genetics. 2003;33(4):455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 5.Schymick JC, Talbot K, Traynor BJ. Genetics of sporadic amyotrophic lateral sclerosis. Human Molecular Genetics. 2007;16:R233–R242. doi: 10.1093/hmg/ddm215. [DOI] [PubMed] [Google Scholar]
- 6.Wills AM, Cronin S, Slowik A, et al. A large-scale international meta-analysis of paraoxonase gene polymorphisms in sporadic ALS. Neurology. 2009;73(1):16–24. doi: 10.1212/WNL.0b013e3181a18674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambrechts D, Poesen K, Fernández-Santiago R, et al. Meta-analysis of vascular endothelial growth factor variations in amyotrophic lateral sclerosis: increased susceptibility in male carriers of the -2578AA genotype. Journal of Medical Genetics. 2009;46(12):840–846. doi: 10.1136/jmg.2008.058222. [DOI] [PubMed] [Google Scholar]
- 8.McCombe PA, Henderson RD. Effects of gender in amyotrophic lateral sclerosis. Gender Medicine. 2010;7(6):557–570. doi: 10.1016/j.genm.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Kisby GE, Ellison M, Spencer PS. Content of the neurotoxins cycasin (methylazoxymethanol β-D-glucoside) and BMAA (β-N-methylamino-L-alanine) in cycad flour prepared by Guam Chamorros. Neurology. 1992;42(7):1336–1340. doi: 10.1212/wnl.42.7.1336. [DOI] [PubMed] [Google Scholar]
- 10.Johnson FO, Atchison WD. The role of environmental mercury, lead and pesticide exposure in development of amyotrophic lateral sclerosis. NeuroToxicology. 2009;30(5):761–765. doi: 10.1016/j.neuro.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutedja NA, Veldink JH, Fischer K, et al. Exposure to chemicals and metals and risk of amyotrophic lateral sclerosis: a systematic review. Amyotrophic Lateral Sclerosis. 2009;10(5-6):302–309. doi: 10.3109/17482960802455416. [DOI] [PubMed] [Google Scholar]
- 12.Sutedja NA, Fischer K, Veldink JH, et al. What we truly know about occupation as a risk factor for ALS: a critical and systematic review. Amyotrophic Lateral Sclerosis. 2009;10(5-6):295–301. doi: 10.3109/17482960802430799. [DOI] [PubMed] [Google Scholar]
- 13.Gurney ME. The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. Journal of the Neurological Sciences. 1997;152(1):S67–S73. doi: 10.1016/s0022-510x(97)00247-5. [DOI] [PubMed] [Google Scholar]
- 14.Shibata N. Transgenic mouse model for familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation. Neuropathology. 2001;21(1):82–92. doi: 10.1046/j.1440-1789.2001.00361.x. [DOI] [PubMed] [Google Scholar]
- 15.Sau D, De Biasi S, Vitellaro-Zuccarello L, et al. Mutation of SOD1 in ALS: a gain of a loss of function. Human Molecular Genetics. 2007;16(13):1604–1618. doi: 10.1093/hmg/ddm110. [DOI] [PubMed] [Google Scholar]
- 16.Cai H, Lin X, Xie C, et al. Loss of ALS2 function is insufficient to trigger motor neuron degeneration in knock-out mice but predisposes neurons to oxidative stress. Journal of Neuroscience. 2005;25(33):7567–7574. doi: 10.1523/JNEUROSCI.1645-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duan W, Li X, Shi J, Guo Y, Li Z, Li C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience. 2010;169(4):1621–1629. doi: 10.1016/j.neuroscience.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 18.Suraweera A, Becherel OJ, Chen P, et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. Journal of Cell Biology. 2007;177(6):969–979. doi: 10.1083/jcb.200701042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrante RJ, Browne SE, Shinobu LA, et al. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. Journal of Neurochemistry. 1997;69(5):2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- 20.Kisby GE, Milne J, Sweatt C. Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. NeuroReport. 1997;8(6):1337–1340. doi: 10.1097/00001756-199704140-00004. [DOI] [PubMed] [Google Scholar]
- 21.Kikuchi H, Furuta A, Nishioka KI, Suzuki SO, Nakabeppu Y, Iwaki T. Impairment of mitochondrial DNA repair enzymes against accumulation of 8-oxo-guanine in the spinal motor neurons of amyotrophic lateral sclerosis. Acta Neuropathologica. 2002;103(4):408–414. doi: 10.1007/s00401-001-0480-x. [DOI] [PubMed] [Google Scholar]
- 22.Weissman L, de Souza-Pinto NC, Stevnsner T, Bohr VA. DNA repair, mitochondria, and neurodegeneration. Neuroscience. 2007;145(4):1318–1329. doi: 10.1016/j.neuroscience.2006.08.061. [DOI] [PubMed] [Google Scholar]
- 23.Coppedè F, Migliore L. Dna repair in premature aging disorders and neurodegeneration. Current Aging Science. 2010;3(1):3–19. doi: 10.2174/1874609811003010003. [DOI] [PubMed] [Google Scholar]
- 24.Shaikh AY, Martin LJ. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. NeuroMolecular Medicine. 2002;2(1):47–60. doi: 10.1007/s12017-002-0038-7. [DOI] [PubMed] [Google Scholar]
- 25.Manabe Y, Warita H, Murakami T, et al. Early decrease of redox factor-1 in spinal motor neurons of presymptomatic transgenic mice with a mutant SOD1 gene. Brain Research. 2001;915(1):104–107. doi: 10.1016/s0006-8993(01)02870-0. [DOI] [PubMed] [Google Scholar]
- 26.Murakami T, Nagai M, Miyazaki K, et al. Early decrease of mitochondrial DNA repair enzymes in spinal motor neurons of presymptomatic transgenic mice carrying a mutant SOD1 gene. Brain Research. 2007;1150(1):182–189. doi: 10.1016/j.brainres.2007.02.057. [DOI] [PubMed] [Google Scholar]
- 27.Kim SH, Henkel JS, Beers DR, et al. PARP expression is increased in astrocytes but decreased in motor neurons in the spinal cord of sporadic ALS patients. Journal of Neuropathology and Experimental Neurology. 2003;62(1):88–103. doi: 10.1093/jnen/62.1.88. [DOI] [PubMed] [Google Scholar]
- 28.Kim SH, Engelhardt JI, Henkel JS, et al. Widespread increased expression of the DNA repair enzyme PARP in brain in ALS. Neurology. 2004;62(2):319–322. doi: 10.1212/01.wnl.0000103291.04985.dc. [DOI] [PubMed] [Google Scholar]
- 29.de Waard MC, van der Pluijm I, Zuiderveen Borgesius N, et al. Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathologica. 2010;120(4):461–475. doi: 10.1007/s00401-010-0715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robertson AB, Klungland A, Rognes T, Leiros I. Base excision repair: the long and short of it. Cellular and Molecular Life Sciences. 2009;66(6):981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson DM, III, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair. 2007;6(4):544–559. doi: 10.1016/j.dnarep.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 32.Liu P, Demple B. DNA repair in mammalian mitochondria: much more than we thought? Environmental and Molecular Mutagenesis. 2010;51(5):417–426. doi: 10.1002/em.20576. [DOI] [PubMed] [Google Scholar]
- 33.Barber SC, Shaw PJ. Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radical Biology and Medicine. 2010;48(5):629–641. doi: 10.1016/j.freeradbiomed.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 34.Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don’t divide what’s to repair? Mutation Research. 2007;614(1-2):24–36. doi: 10.1016/j.mrfmmm.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 35.Bogdanov M, Brown RH, Matson W, et al. Increased oxidative damage to DNA in ALS patients. Free Radical Biology and Medicine. 2000;29(7):652–658. doi: 10.1016/s0891-5849(00)00349-x. [DOI] [PubMed] [Google Scholar]
- 36.Aguirre N, Beal MF, Matson WR, Bogdanov MB. Increased oxidative damage to DNA in an animal model of amyotrophic lateral sclerosis. Free Radical Research. 2005;39(4):383–388. doi: 10.1080/10715760400027979. [DOI] [PubMed] [Google Scholar]
- 37.Ihara Y, Nobukuni K, Takata H, Hayabara T. Oxidative stress and metal content in blood and cerebrospinal fluid of amyotrophic lateral sclerosis patients with and without a Cu, Zn-superoxide dismutase mutation. Neurological Research. 2005;27(1):105–108. doi: 10.1179/016164105X18430. [DOI] [PubMed] [Google Scholar]
- 38.Warita H, Hayashi T, Murakami T, Manabe Y, Abe K. Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Molecular Brain Research. 2001;89(1-2):147–152. doi: 10.1016/s0169-328x(01)00029-8. [DOI] [PubMed] [Google Scholar]
- 39.Martin LJ, Liu Z, Chen K, et al. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. Journal of Comparative Neurology. 2007;500(1):20–46. doi: 10.1002/cne.21160. [DOI] [PubMed] [Google Scholar]
- 40.Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Annals of Neurology. 1999;46(5):787–790. doi: 10.1002/1531-8249(199911)46:5<787::aid-ana17>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 41.Murata T, Ohtsuka C, Terayama Y. Increased mitochondrial oxidative damage and oxidative DNA damage contributes to the neurodegenerative process in sporadic amyotrophic lateral sclerosis. Free Radical Research. 2008;42(3):221–225. doi: 10.1080/10715760701877262. [DOI] [PubMed] [Google Scholar]
- 42.Barbosa LF, Cerqueira FM, Macedo AF, et al. Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS. Biochimica et Biophysica Acta. 2010;1802:462–471. doi: 10.1016/j.bbadis.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 43.Bradley WG, Krasin F. A new hypothesis of the etiology of amyotrophic lateral sclerosis. The DNA hypothesis. Archives of Neurology. 1982;39(11):677–680. doi: 10.1001/archneur.1982.00510230003001. [DOI] [PubMed] [Google Scholar]
- 44.Tandan R, Robison SH, Munzer JS, Bradley WG. Deficient DNA repair in amyotrophic lateral sclerosis cells. Journal of the Neurological Sciences. 1987;79(1-2):189–203. doi: 10.1016/0022-510x(87)90272-3. [DOI] [PubMed] [Google Scholar]
- 45.Bradley WG, Robison SH, Tandan R. Deficient repair of alkylation damage of DNA in Alzheimer’s disease and amyotrophic lateral sclerosis cells. Advances in Experimental Medicine and Biology. 1987;209:3–6. doi: 10.1007/978-1-4684-5302-7_1. [DOI] [PubMed] [Google Scholar]
- 46.Olkowski ZL. Mutant AP endonuclease in patients with amyotrophic lateral sclerosis. NeuroReport. 1998;9(2):239–242. doi: 10.1097/00001756-199801260-00012. [DOI] [PubMed] [Google Scholar]
- 47.Hayward C, Colville S, Swingler RJ, Brock DJH. Molecular genetic analysis of the APEX nuclease gene in amyotrophic lateral sclerosis. Neurology. 1999;52(9):1899–1901. doi: 10.1212/wnl.52.9.1899. [DOI] [PubMed] [Google Scholar]
- 48.Tomkins J, Dempster S, Banner SJ, Cookson MR, Shaw PJ. Screening of AP endonuclease as a candidate gene for amyotrophic lateral sclerosis (ALS) NeuroReport. 2000;11(8):1695–1697. doi: 10.1097/00001756-200006050-00020. [DOI] [PubMed] [Google Scholar]
- 49.Greenway MJ, Alexander MD, Ennis S, et al. A novel candidate region for ALS on chromosome 14q11.2. Neurology. 2004;63(10):1936–1938. doi: 10.1212/01.wnl.0000144344.39103.f6. [DOI] [PubMed] [Google Scholar]
- 50.Coppedè F, Lo Gerfo A, Carlesi C, et al. Lack of association between the APEX1 Asp148Glu polymorphism and sporadic amyotrophic lateral sclerosis. Neurobiology of Aging. 2010;31(2):353–355. doi: 10.1016/j.neurobiolaging.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 51.Coppedè F, Mancuso M, Lo Gerfo A, et al. Association of the hOGG1 Ser326Cys polymorphism with sporadic amyotrophic lateral sclerosis. Neuroscience Letters. 2007;420(2):163–168. doi: 10.1016/j.neulet.2007.04.067. [DOI] [PubMed] [Google Scholar]
- 52.Coppedè F, Migheli F, Lo Gerfo A, et al. Association study between XRCC1 gene polymorphisms and sporadic amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis. 2010;11(1-2):122–124. doi: 10.3109/17482960903220297. [DOI] [PubMed] [Google Scholar]
- 53.Fang F, Umbach DM, Xu Z, et al. No association between DNA repair gene XRCC1 and amyotrophic lateral sclerosis. doi: 10.1016/j.neurobiolaging.2010.07.004. Neurobiology of Aging. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shao C, Xiong S, Li GM, et al. Altered 8-oxoguanine glycosylase in mild cognitive impairment and late-stage Alzheimer’s disease brain. Free Radical Biology and Medicine. 2008;45(6):813–819. doi: 10.1016/j.freeradbiomed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hill JW, Hu JJ, Evans MK. OGG1 is degraded by calpain following oxidative stress and cisplatin exposure. DNA Repair. 2008;7(4):648–654. doi: 10.1016/j.dnarep.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gueven N, Chen P, Nakamura J, et al. A subgroup of spinocerebellar ataxias defective in DNA damage responses. Neuroscience. 2007;145(4):1418–1425. doi: 10.1016/j.neuroscience.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 57.Kao SY. Regulation of DNA repair by Parkin. Biochemical and Biophysical Research Communications. 2009;382:321–325. doi: 10.1016/j.bbrc.2009.03.048. [DOI] [PubMed] [Google Scholar]
- 58.Rothfuss O, Fischer H, Hasegawa T, et al. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Human Molecular Genetics. 2009;18(20):3832–3850. doi: 10.1093/hmg/ddp327. [DOI] [PubMed] [Google Scholar]
- 59.Davidzon G, Greene P, Mancuso M, et al. Early-onset familial parkinsonism due to POLG mutations. Annals of Neurology. 2006;59(5):859–862. doi: 10.1002/ana.20831. [DOI] [PubMed] [Google Scholar]
- 60.Remes AM, Hinttala R, Kärppä M, et al. Parkinsonism associated with the homozygous W748S mutation in the POLG1 gene. Parkinsonism and Related Disorders. 2008;14(8):652–654. doi: 10.1016/j.parkreldis.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 61.Eerola J, Luoma PT, Peuralinna T, et al. POLG1 polyglutamine tract variants associated with Parkinson’s disease. Neuroscience Letters. 2010;477(1):1–5. doi: 10.1016/j.neulet.2010.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mao G, Pan X, Zhu BB, et al. Identification and characterization of OGG1 mutations in patients with Alzheimer’s disease. Nucleic Acids Research. 2007;35(8):2759–2766. doi: 10.1093/nar/gkm189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coppedè F, Mancuso M, Lo Gerfo A, et al. A Ser326Cys polymorphism in the DNA repair gene hOGG1 is not associated with sporadic Alzheimer’s disease. Neuroscience Letters. 2007;414(3):282–285. doi: 10.1016/j.neulet.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 64.Parildar-Karpuzoğlu H, Doğru-Abbasoğlu S, Hanagasi HA, et al. Single nucleotide polymorphisms in base-excision repair genes hOGG1, APE1 and XRCC1 do not alter risk of Alzheimer’s disease. Neuroscience Letters. 2008;442(3):287–291. doi: 10.1016/j.neulet.2008.07.047. [DOI] [PubMed] [Google Scholar]
- 65.Qian Y, Chen W, Wu J, et al. Association of polymorphism of DNA repair gene XRCC1 with sporadic late-onset Alzheimer’s disease and age of onset in elderly Han Chinese. Journal of the Neurological Sciences. 2010;295(1-2):62–65. doi: 10.1016/j.jns.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 66.Coppedè F. Variants and polymorphisms of DNA base excision repair genes and Alzheimer’s disease. Journal of the Neurological Sciences. 2011;300(1-2):200–201. doi: 10.1016/j.jns.2010.09.020. [DOI] [PubMed] [Google Scholar]
- 67.Coppedè F, Ceravolo R, Migheli F, et al. The hOGG1 Ser326Cys polymorphism is not associated with sporadic Parkinson’s disease. Neuroscience Letters. 2010;473(3):248–251. doi: 10.1016/j.neulet.2010.02.059. [DOI] [PubMed] [Google Scholar]
- 68.Infante J, Llorca J, Mateo I, et al. Interaction between poly(ADP-ribose) polymerase 1 and interleukin 1A genes is associated with Alzheimer’s disease risk. Dementia and Geriatric Cognitive Disorders. 2007;23(4):215–218. doi: 10.1159/000099471. [DOI] [PubMed] [Google Scholar]
- 69.Infante J, Sánchez-Juan P, Mateo I, et al. Poly (ADP-ribose) polymerase-1 (PARP-1) genetic variants are protective against Parkinson’s disease. Journal of the Neurological Sciences. 2007;256(1-2):68–70. doi: 10.1016/j.jns.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 70.Liu HP, Lin WY, Wu BT, et al. Evaluation of the poly(ADP-ribose) polymerase-1 gene variants in Alzheimer’s disease. Journal of Clinical Laboratory Analysis. 2010;24(3):182–186. doi: 10.1002/jcla.20379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447(7143):447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Briggs FB, Goldstein BA, McCauley JL, et al. Variation within DNA repair pathway genes and risk of multiple sclerosis. The American Journal of Epidemiology. 2010;172:217–224. doi: 10.1093/aje/kwq086. [DOI] [PMC free article] [PubMed] [Google Scholar]