

Abstract
Eotaxin-3 (CCL-26), a potent chemokine for eosinophil recruitment and contributing significantly to the pathogenesis of asthma, is secreted by lung epithelial cells in response to T helper 2 cytokines including interleukin 13 (IL-13). Here we showed that vitamin E forms, but not their metabolites, differentially inhibited IL-13-stimulated generation of eotaxin-3 in human lung epithelial A549 cells. The relative inhibitory potency was γ-tocotrienol (γ-TE) (IC50 ~15 μM) > γ-tocopherol, δ-tocopherol (IC50 ~25-50 μM) > α-tocopherol. Consistent with suppression of eotaxin, γ-TE treatment impaired IL-13-induced phosphorylation of STAT6, the key transcription factor for activation of eotaxin expression, and consequently blocked IL-13 stimulated DNA-binding activity of STAT6. In search of the upstream target of γTE by using inhibitor and siRNA approaches, we discovered that the atypical protein kinase C (aPKC) signaling, instead of classical PKC, p38 MAPK, JNK or ERK, played a critical role in IL-13-stimulated eotaxin generation and STAT6 activation. While showing no obvious effect on aPKC expression or phosphorylation, γ-TE treatment resulted in increased expression of PAR4, an endogenous negative regulator of aPKCs. Importantly, γ-TE treatment led to enhanced formation of aPKC/PAR4 complex that is known to reduce aPKC activity via protein-protein crosstalk. Our study demonstrated that γ-TE inhibited IL-13/STAT6-activated eotaxin secretion via up-regulation of PAR4 expression and enhancement of aPKC-PAR-4 complex formation. These results support the notion that specific vitamin E forms may be useful anti-asthmatic agents.
Keywords: tocopherol, tocotrienol, asthma, inflammation
Natural forms of vitamin E are eight structurally-related lipophilic antioxidants, which include α-, β-, γ- and δ-tocopherol (α-, β-, γ- and δ-T) and α-, β-, γ-and δ-tocotrienol (α-, β-, γ-and δ-TE). Research on vitamin E including potential protective effects on asthma has mostly focused on αT, the predominant form of vitamin E in tissues. Although severe αT deficiency appears to modulate airway allergic inflammation [1], inconsistent outcomes have been reported in clinical and animal studies regarding the role of α-T supplementation in airway diseases [2-6]. On the other hand, studies by us and others strongly suggest that other forms of vitamin E including γT, the major form of vitamin E in US diet, appear to have unique properties that are import to disease prevention or treatment [7, 8]. We have shown that γ-T, δ-T and γ-TE as well as their metabolites are more potent than α-T in inhibition of cyclooxygenase- and 5-lipoxygenase-catalyzed eicosanoids in cell-based studies and in an inflammation model in rats [9-13]. γ-T is also better than α-T in scavenging reactive nitrogen species and attenuating inflammation-related damage [11, 14, 15]. γT administered by nebulization is shown to improve pulmonary function in sheep with burn and smoke inhalation injury [16]. Recently, we have demonstrated that γ-T supplementation inhibited ovalbumin-induced airway inflammation in an asthma and allergic rhinitis model, respectively, in Brown Norway rats [17, 18]. In these studies, γ-T supplementation led to marked decrease of airway eosinophil infiltration and reduced proinflammatory cytokines [17, 18]. Despite these exciting findings, the mechanism(s) underlying γ-T-exerted inhibition of eosinophilia was not fully understood.
It is well recognized that interleukin-13 (IL-13), a key cytokine secreted by T helper 2 (Th2) lymphocytes, plays critical roles in the pathogenesis of allergic asthma [19]. IL-13 has been shown to regulate eosinophilic inflammation and mucus production, and promote epithelial damage and airway hyper-responsiveness [19-21]. As a central effector cytokine in the lung, IL-13 stimulates lung epithelial cells to release eotaxins-3 (CCL26) and other members of the eotaxin family including CCL11 and CCL24. Eotaxins are potent chemoattractants for eosinophils and cause airway eosinophilia, a hallmark of asthma. Emerging evidence suggests that CCL26 (or eotaxin-3), but not CCL11 or CCL24, likely accounts for eosinophil recruitment to asthmatic airways following allergen challenge in subjects with mild asthma [22]. Based on the observation that γT suppressed eosinophilia in the asthma model in rats [17, 18], we hypothesize that vitamin E forms including γT may modulate the secretion of eotaxin. Here we investigate the effect and mechanism of different forms of vitamin E and their metabolites on IL-13-stimulated eotaxin-3 secretion in human lung epithelial A549 cells.
MATERIALS AND METHODS
Materials
α-T (99%), γ-T (97-99%) and δ-T (97%) were purchased from Sigma (St Louis, MO). 2-(β-Carboxyethyl)-7, 8-dimethyl-6-hydroxychroman (γ-CEHC) was from Cayman Chemical (Ann Arbor, MI). γ-TE was a gift from BASF (Germany). Tissue culture reagents were from Invitrogen (Rockville, MD). Recombinant human IL-13 was purchased from R&D Systems Minneapolis, MN. Human interleukin-4 (IL-4) was from Atlanta Biologicals, Inc (Lawrenceville, GA). Inhibitors for PKCs (Gö-6983), MEK (U0126), p38 MAPK (SB202190), NFkB (Parthenolide), and JAK inhibitor I were from Calbiochem (La Jolla, CA). Highly specific inhibitors for classic PKCs (cPKC) and atypical PKCs (aPKC), i.e., myristoylated cPKC and aPKC pseudosubstrates (for cPKC: N-Myristoyl-Phe-Ala-Arg-Lys-Gly-Ala-Leu-Arg-Gln-OH; for aPKC: N-Myristoyl-Ser-Ile-Tyr-Arg-Arg-Gly-Ala-Arg-Arg-Trp-Arg-Lys-Leu-OH), were purchased from Enzo Life Sci (Plymouth Meeting, PA). JNK inhibitor (SP600125) was from Biomol (Plymouth Meeting, PA). [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] (MTT), inhibitor of JAK/STAT6 (Leflunomide), and all other chemicals were from Sigma (St Louis, MO).
Eotaxin-3 generation by IL-13-stimulated A549 cells
Human lung A549 cells were obtained from American Type Culture Collection (Manassas, VA) and were routinely cultured in RPMI-1640 with 10% fetal bovine serum (FBS). Cells (3 × 105 per well) were seeded and allowed to attach overnight in a 24-well plate. Vitamin E stock solutions were initially made in DMSO and then diluted in 10 mg/mL of fatty acid-free bovine serum albumin. Confluent cells were pre-incubated with vitamin E forms for 14-16 h in DMEM containing 1% FBS and 0.05% DMSO (solvent), and then stimulated by 10 ng/ml of IL-13 for 24 h. Eotaxin-3 accumulation in the media was measured using a Quantikine Human Eotaxin-3/CCL26 Immunoassay kit (R&D Systems, Minneapolis, MN).
Evaluation of cellular dehydrogenase/reductase activity by MTT assays
The cellular metabolic status was evaluated by estimation of dehydrogenase/reductase activity that reduces MTT to form an insoluble purple product which was dissolved in DMSO and measured at 570 nm [23].
Western Blot
Cells were lysed in Tris-EDTA, 1% SDS, 1 mM DTT and 2 mM sodium vanadate with protease inhibitor cocktails (Sigma). The resulting solution was heated at 95°C for 5 min. Proteins (25-50 μg) were loaded on 10% pre-cast SDS-PAGE gels.
Resolved proteins were transferred onto a PVDF membrane (Millipore, Billerica, MA) and probed by antibodies. Membranes were exposed to chemiluminescent reagent (Perkin Elmer, Waltham, WA) and visualized on a Kodak film.
Electrophoretic Mobility Shift Assay (EMSA)
Based on the functional STAT6-binding element in the promoter of human eotaxin-3 [24], biotin 3’ end-labeled DNA duplex of sequence (5’-GATCACAGAAttctctggaaTTGTTT-3’) was synthesized and labeled by Integrated DNA Technologies (IDT, Coralville, IA). EMSA was performed using the LightShift™ Chemiluminescent EMSA kit” (Pierce, Rockford, IL). Briefly, 8 μg of nuclear extracts isolated by NE-PER nuclear kit (Pierce, Rockford, IL) were incubated with 50 ng/μL Poly (dI•dC) and 40 fmol of biotin-labeled probes in 1X-binding buffer at room temperature for 20 min. Samples were separated on a 5% nondenaturing polyacrylamide gel (Bio-Rad, Hercules, CA), semi-transferred and cross-linked to Biodyne® Nylon transfer membranes (Pall Life Sciences, Ann Arbor, MI). The STAT6-probe complex was detected by a chemiluminescent nucleic acid detection module (Pierce, Rockford, IL) according to the manufacturer’s instructions. The STAT6-DNA complex was confirmed by competition experiments with 200-fold excess of unlabeled probes and super-shift with an anti-STAT6 antibody.
siRNA transfection
Human PKCλ/ι (s11110) and PKC ζ siRNA (s11128) were purchased from Ambion Applied Biosystems(Ambion, Carlsbad, CA). Transfection was performed with lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen Carlsbad, CA). Sixty-four hours after transfection, cells were stimulated with IL-13 (10 ng/ml) for 10min, collected and analyzed by Western blotting.
PKC activity
PKCζ kinase activity was measured using HTScan® PKCζ Kinase Assay Kit according to the manufacture’s instructions (Cell Signaling, Danvers, MA). Briefly, tested compounds including γTE at 20 μM and 50 μM were incubated with 50 ng of recombinant PKCζ in 25 mM Tris-HCl (pH 7.5) with10 mM MgCl2, 5 mM β-glycerophosphate, 0.1 mM Na3VO4, and 2 mM DTT at room temperature for 5 min. The reaction mixture was then added with the substrate peptide (1.5 μM) and ATP (200 μM) and incubated at room temperature 30 min. The reaction was stopped with 50 mM EDTA (pH 8). Phosphorylation of the substrate peptide was quantified by colorimetric ELISA with Phospho-PKA Substrate antibody.
Immunoprecipitation (IP)
Fresh cell pellets were lysed with 100 μL of lysis buffer containing 50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, protease inhibitors and 2 mM sodium vanadate. After lysis, 100 μL of Tris-NaCl buffer was added to dilute NP-40 to 0.5%. Samples were incubated with PAR-4 antibody (Cell Signaling, Danvers, MA) on ice for 1.5 h with vortexing every 30 min and then rocking at 4 °C overnight. Samples were then incubated with 100 μL of Immobilized Protein G Slurry (Pierce, Rockford, IL) by gently rocking at 4°C for 2 h. After being thoroughly washed, the beads were mixed with loading buffer and incubated at 95 °C for 5 min. The supernatant was analyzed by Western blot.
Statistical analysis
One-way ANOVA and post hoc Tukey’s multiple comparisons were used in statistical analyses.
RESULTS
Vitamin E forms, but not their metabolites, differentially inhibited IL-13-induced eotaxin-3 secretion in human lung epithelial A549 cells
In the early stage of the study, we found that IL-4 and IL-13, at 2 to 10 ng/ml, similarly stimulated eotaxin-3 secretion in a dose-dependent manner in human lung epithelial A549 cells (Data not shown). Compared with baseline controls in which less than 40 pg/106 cells of eotaxin-3 was detected, IL-13 (10 ng/ml) treatment led to a marked increase of eotaxin-3 secretion to 1300-5500 pg/106 cells. On the other hand, IL-1β (at 10 ng/ml) failed to cause any significant increase of eotaxin-3, despite being reported to induce eotaxin in a previous study [25]. In the subsequent studies, IL-13 was used to stimulate eotaxin-3.
We observed that vitamin E forms differentially inhibited IL-13-induced eotaxin-3 formation. At 25-50 μM, γ-T and δ-T decreased 35-50% secretion of eotaxin-3, while α-T showed 20-25% inhibition (Figure 1A). γ-TE was the strongest among all the vitamin E forms tested, with an IC50 of ~15 μM (Figure 1B). Besides lipophilic vitamin E forms, we also examined hydrophilic phenolic compounds including γ-CEHC, the terminal metabolite of γ-T, and resveratrol, a polyphenolic compound mainly found in grapes. γ-CEHC at 10 to 50 μM did not show significant effects on IL-13-induced eotaxin-3 (Figure 1C). Resveratrol inhibited 35% of eotaxin-3 production at 10 μM, but its inhibitory effect was not evident at 20 μM (Figure 1C).
Figure 1. Effects of vitamin E forms, γ-CEHC and resveratrol on IL-13-induced eotaxin-3 secretion.
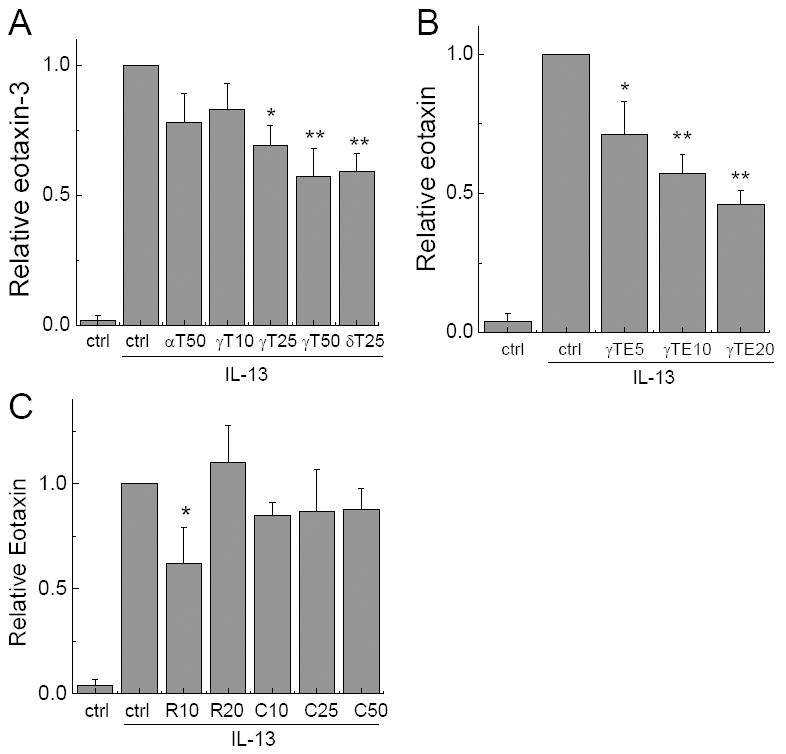
After preincubated with tocopherols (Panel A) and γTE (Panel B) for 14-18 h, or γ-CEHC/resveratrol (Panel C) for 1 h, A549 cells were activated by IL-13 (10 ng/mL) for 24 h and media were collected to measure eotaxin-3 secretion. Results are the ratio of eotaxin secreted by cells treated with tested compounds to that of solvent controls. αT50, γT10, γT25, γT50, δT25, γTE5, γTE10 and γTE20 stand for the corresponding vitamin E forms at indicated concentrations (μM). R10 and R20 are resveratrol at 10 and 20 μM, respectively. C10, C25 and C50 are γ-CEHC at indicated concentrations (μM). Data are shown as mean values ± SD. *P<0.05 and **P<0.01 are significant difference between the treated and control cells. n >2 per bar.
Because A549 cells have been shown to metabolize vitamin E forms to long-chain carboxychromanols that are potent inhibitors of cyclooxygenases [12, 26], we examined whether these long-chain metabolites contribute to the inhibitory effect on eotaxin-3. Coincubation of vitamin E forms with sesamin (1 or 5 μM), which inhibits tocopherol hydroxylase-mediated vitamin E metabolism and prevents the accumulation of long-chain metabolites in the cultured media [12], did not significantly affect the inhibitory effect on eotaxin-3 by γ-T or γ-TE (not shown). This strongly suggests that the un-metabolized vitamins, rather than their metabolites, are responsible for the observed inhibitory effects.
Since previous studies have reported that treatment of certain vitamin E forms impairs A549 cell viability when cells are cultured under sub-confluent conditions [27], it is important to examine whether the tested compounds affect cell viability in this study. Under the current experimental conditions where confluent A549 cells were used, none of these tested compounds had any significant effects on cell morphology based on microscopic examination. MTT assays showed slight decrease (10-15%) of cellular dehydrogenase/reductase activity in the cells treated with γ-T at 50 μM, and γ-TE or resveratrol at 20 μM for 36 hrs or longer.
STAT6, but not NFκB, was responsible for IL-13-stimulated eotaxin formation, and γ-TE inhibited IL-13-induced STAT6 phosphorylation and IL-13-stimulated DNA binding activity of STAT6
Both NFκB and STAT have previously been reported to regulate eotaxin-3 expression in various cell types [24, 25, 28]. Here we found that leflunomide, which has been shown to inhibit JAK-STAT6 signaling [28, 29], but not parthnolide, an inhibitor of NFκB [30], dose-dependently decreased IL-13 stimulated eotaxin secretion (Figure 2A). In addition, JAK inhibitor I that specifically inhibits JAK-STAT also markedly decreased eotaxin (data not shown). These results indicated that JAK-STAT but not NFkB is responsible for IL-13-induced eotaxin formation in A549 cells. Consistently, immunoblotting with anti-phospho-Tyr-641-STAT6 antibody revealed that IL-13 treatment caused a strong induction of STAT6 phosphorylation (Figure 2B), whereas no activation of STAT3 was observed (not shown). Interestingly, γ-TE treatment inhibited IL-13-induced STAT6 phosphorylation (Figure 2B). To test whether γTE affects STAT6 binding to the eotaxin-3 promoter, EMSA experiments were performed. Nuclear extracts were prepared from IL-13 treated cells in the presence or absence of γTE and incubated with biotin-labeled nucleotide probes containing STAT6-binding elements. As shown in Figure 2C, IL-13 induced formation of a STAT6-DNA complex, which was verified by the effective competition with unlabeled STAT6-recognition sequence and supershift with an antibody to STAT6. Importantly, consistent with inhibition of STAT6 phosphorylation and reduction of eotaxin-3, γTE suppressed IL-13-induced DNA binding activity of STAT6.
Figure 2. Panel A - The inhibitor for JAK-Stat6 (Leflunomide, LF) but not NFkB (Parthnolide, P) inhibits IL-13-induced eotaxin secretion.
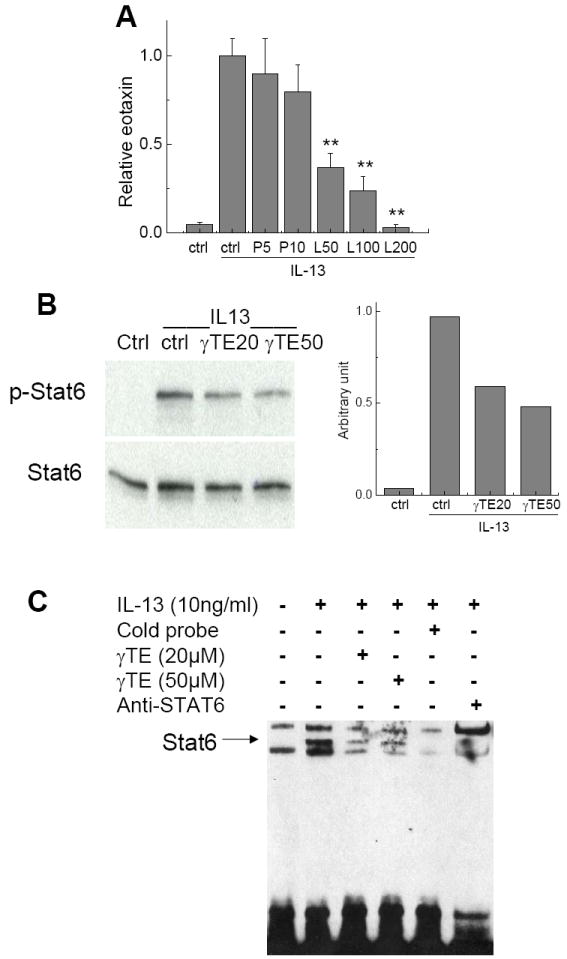
A549 cells were preincubated with LF and P at indicated concentrations (μM) for 1 h and stimulated by IL-13 for 24 h. Eotaxin formation was measured by ELISA. Data are shown as mean values ± SD. *P<0.05 and **P<0.01 are difference between treated and control cells. n >2 per bar. Panel B -γ-TE treatment partially impaired IL-13-stimulated STAT6 phosphorylation. Cells were preincubated with γ-TE at indicated concentrations (μM) or DMSO (0.05%) for 14-16 h, and stimulated with IL-13 (10ng/ml) for 20 min. Immunoblotting was performed with anti-phosphor-Tyr-641-STAT6 or anti-STAT6 and the densitometry was shown (right). Panel C – γ-TE inhibited IL-13 stimulated STAT6 DNA binding activity. Cells were pretreatment with γTE for 18h and stimulation with IL-13 for 30 min. Nuclear extracts were isolated and EMSA was conducted as described in Methods and Materials.
The atypical Protein kinase C (aPKC) signaling, but not p38 MAPK, MEK1/2, JNK or classic PKCs, was critical to IL-13-triggered STAT6 activation in A549 cells
The inhibition of STAT6 phosphorylation suggests that γTE likely affect upstream signaling(s) for STAT6 activation. Therefore, we searched for potential signaling that regulates both eotaxin formation and STAT6 phosphorylation. p38 MAPK (mitogen-activated protein kinases) and JNK (c-jun N-terminal kinase) signaling pathways have previously been reported to modulate STAT6-mediated gene expression [31, 32]. However, we found that their respective inhibitors, SB202190 (p38) and SP60012 (JNKs), had no effect on STAT6 phosphorylation (Figure 3A). On the other hand, PKC inhibitor Gö-6983, but not ERK inhibitor (U0126), potently inhibited IL-13-induced STAT6 phosphorylation and eotaxin-3 (Figure 3B and 3C), which are similar to γ-TE-caused effects.
Figure 3. Panel A: Inhibitors for p38 (SB) or JNK (SP) had no effect on IL-13-stimulated STAT6 activation.
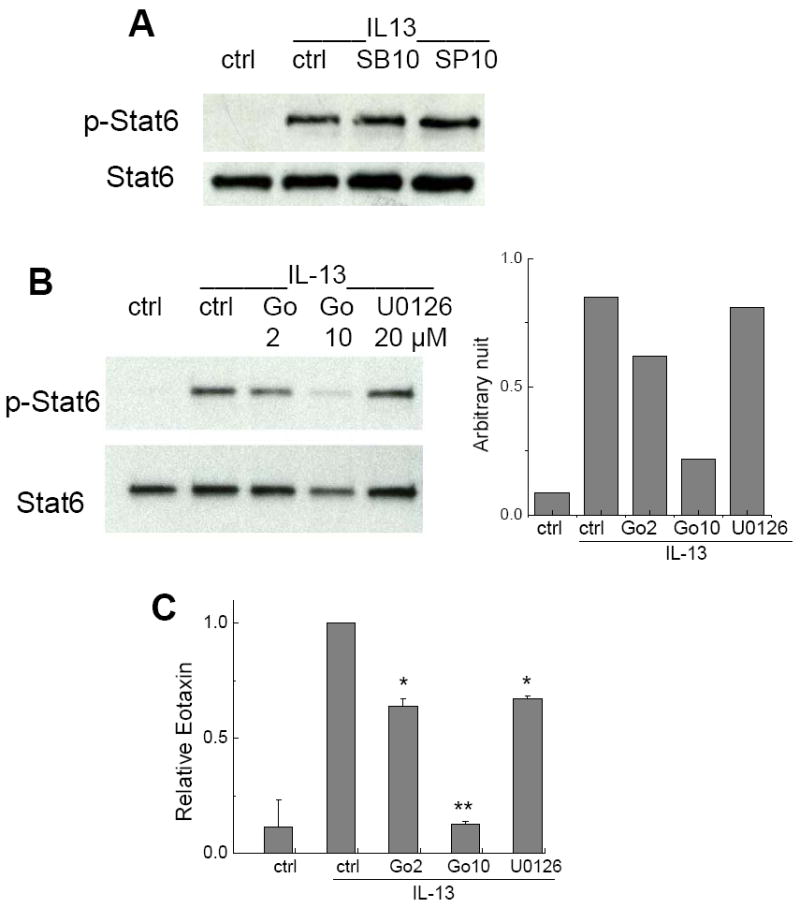
A549 cells were pre-incubated with SB or SP (10 μM) for 1 h, and activated by IL-13 (10 ng/mL) for 10 min. Western blotting was conducted to measure STAT6 phosphorylation. Panel B: Inhibition of PKC but not MEK resulted in blocking IL-13-stimulated STAT6 phosphorylation. A549 cells were preincubated with Gö-6983 (Go, a PKC inhibitor) at 2 or 10 μM, or U0126 (a MEK inhibitor) at 10 μM or vehicle (ctrl) for 1 h, and stimulated by IL-13 for 10 min. Western blotting was conducted to assess STAT6 phosphorylation and its densitometry data were shown (right). Panel C: Inhibitory effects of inhibitors for PKC (Go) and MEK (U0126) on IL-13-induced eotaxin secretion. Gö-6983 at 2 or 10 μM and U0126 at 10 μM were preincubated with A549 cells for 1 h. Cells were then treated by IL-13 (10 ng/mL) for 24 h and eotaxin secretion into the media was measured. Data are shown as mean values ± SD, *P<0.05, **P<0.01: difference between treated and control cells. n=2 or more per bar.
Previous studies have demonstrated that the atypical PKCs, i.e., PKCζ and PKCλ/ι, are critically important to the activation of IL-4/STAT6 signaling in T cells [33-35]. Since IL-4 and IL-13 share similar receptors, we determined whether the classic PKCs or aPKCs are necessary for IL-13-triggered STAT6 activation. For this purpose, we used synthetic myristoylated peptides that are designed based on the pseudosubstrate sequence of individual protein kinase Cs. Since there is considerable sequence variability in the pseudosubstrate domains between classic PKCs (PKCα or β) and aPKC (PKCλ/ι or ζ), these pseudosubstrate peptides serve as highly specific inhibitors for these PKC isoforms [36]. We found that the pseudosubstrate peptide for aPKC, but not its analog inhibitor for the classical PKCs, potently suppressed IL-13-stimulated STAT6 activation (Figure 4A). These data are consistent with the observation that Gö-6983, which inhibits cPKCs at nM and aPKCs at low μM, potently reduced IL-13-induced STAT6 activation only at ~10μM concentrations (Figure 3).
Figure 4. Panel A - The pseudosubstrate peptide for the atypical PKCs (aPKC) but not the classic PKCs (cPKC) suppressed STAT6 activation.
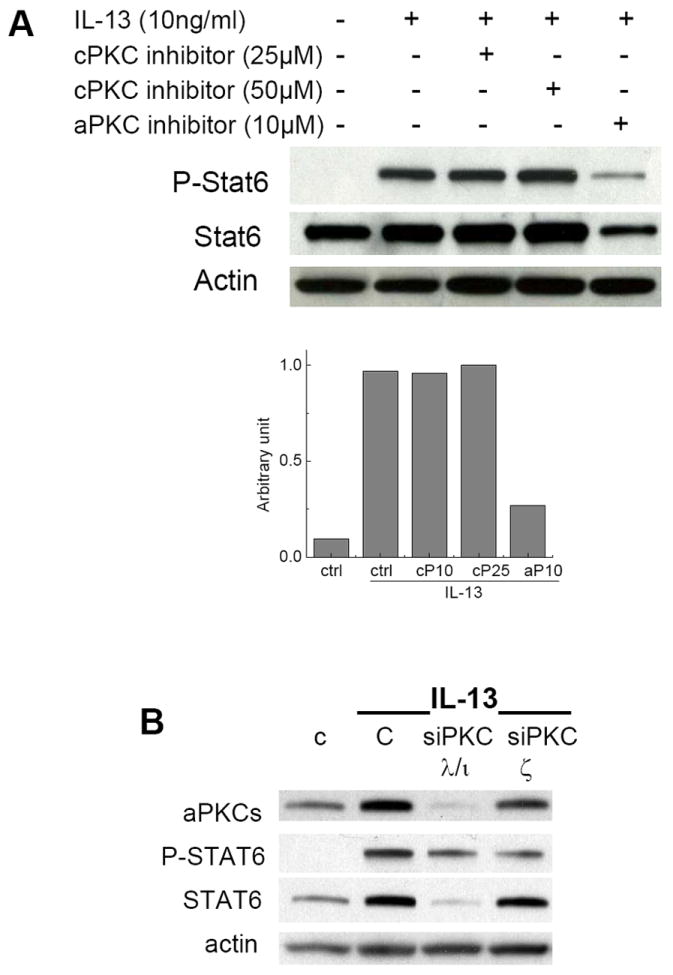
A549 cells were incubated with pseudosubstrate peptide specific for aPKCs or cPKCs at indicated concentrations for 30 min, and then stimulated by IL-13 for 10 min. Western blotting was conducted to probe the effects on STAT6 phosphorylation. Panel B – Downregulation of aPKCs affected STAT6 activation and expression. Cells were transfected with control siRNA (C), siRNAs of PKCλ/ι or PKCζ. Sixty-four hours later, cells were activated by IL13 (10 ng/ml) for 10 min.
To further confirm the role of aPKCs in STAT6 signaling, we transfected cells with siRNAs for PKCλ/ι and PKC ζ, respectively. Figure 4B showed that down regulation of PKCλ/ι resulted in >80% reduction of aPKC levels detected with an antibody that presumably recognizes both aPKC isoforms, whereas downregulation of PKCζ only moderately reduced aPKC levels. These observations strongly suggest that compared with PKCζ, PKCλ/ι is predominantly expressed in A549 cells, which was consistent with the reports by Regala et al. [37]. This explains the relatively modest reduction of PKCζ in response to the siRNA treatment. Downregulation of either isoform led to decrease of STAT6 phosphorylation. Interestingly, PKCλ/ι downregulation caused marked reduction of STAT6 protein levels, which was similarly observed when cells were treated with Gö-6983 and pseudosubstrate peptide of aPKC (Fig 3 and Fig 4A). These siRNA experiments together with inhibitor studies demonstrate that aPKCs play critical roles in STAT6 phosphorylation and protein status in A549 cells.
γ-TE treatment enhanced PAR4 expression and PAR4/aPKC interaction
Given that the aPKCs appear to be critical to IL-13-induced STAT6 activation, we next examined whether γ-TE has any effect on aPKC-mediated signaling. In a cell-free assay, γ-TE did not show any effect on the aPKC activity (data not shown). Immunoblotting results indicated that aPKC expression or its phosphorylation was not affected by γ-TE or IL-13 treatment (Figure 5A).
Figure 5. The effects of γ-TE on atypical PKC (aPKC) expression and its phosphorylatd state (p-aPKC) (Panel A), PAR4 expression levels (Panel B) and potential crosstalk between aPKCs and PAR4 by immunoprecipitation (IP) (Panel C).
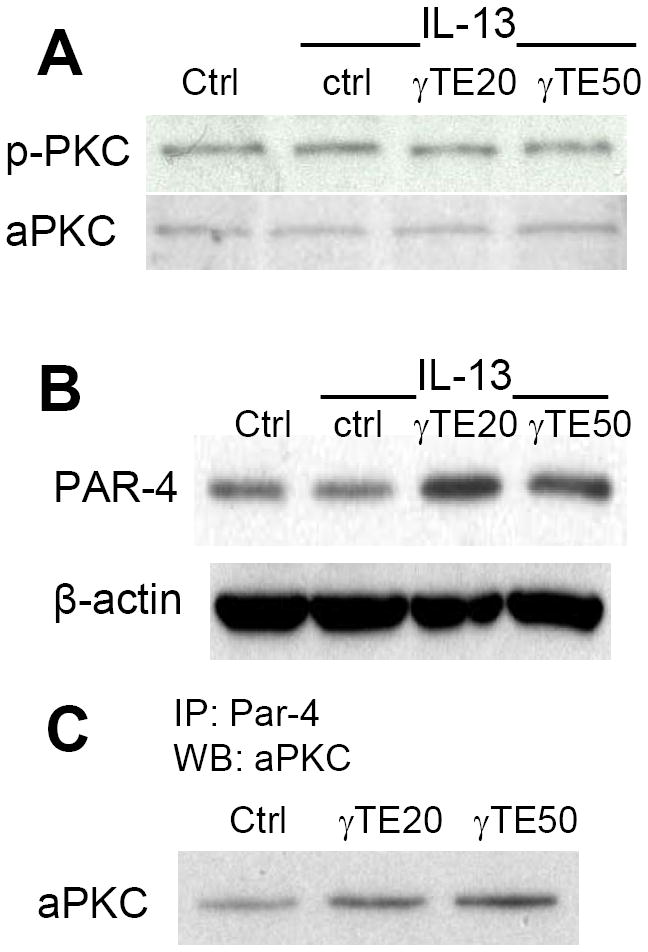
A549 cells were incubated with γ-TE at 20 or 50 μM for 14-16 h, stimulated by IL-13 (10 ng/ml) for 10 min, and collected for Western blotting. In some studies, immunoprecipitation was performed for cells treated with γTE or solvent controls using antibody for PAR4. The precipitated proteins were analyzed by Western blot using antibody for aPKCs.
Besides phosphorylation, aPKC activity is known to be regulated via protein-protein crosstalk [38]. For instance, it has been demonstrated that aPKC signaling is negatively regulated by PAR4 (prostate-apoptosis-response 4) in various cell types, where PAR4 selectively binds to aPKCs and inhibits their enzymatic activity [39]. Consistently, PAR-4 knockout resulted in enhanced PKCζ-mediated IL-4/STAT6 activation and exacerbated concavalalin A-induced liver damage [33]. Based on these studies, we examined potential effect of γ-TE on PAR-4 expression and PAR4-aPKC (PKCζ or PKCι/λ) interactions. Immunoblotting data showed that γ-TE treatment caused an increase of PAR4 expression (Figure 5B). To study the effect on PAR4-PKC interaction, cell lysates from vehicle- or γ-TE-treated cells were immunoprecipitated with Par4 antibody, and the immunoprecipitants were resolved by SDS-PAGE and analyzed by immunoblotting with an antibody for aPKCs (PKCζ and PKCλ/ι). An immunoreactive band corresponding to aPKCs was clearly stronger in the immunoprecipitants from cells treated by γ-TE than those from DMSO-treated controls (Figure 5C). This result indicates a stronger interaction between PKCζ/PKCλ/ι and PAR4 in γ-TE treated cells than vehicle-treated controls, which has been shown to cause impairment of aPKC signaling [39].
DISCUSSION
In the present study, we demonstrated that vitamin E forms but not their metabolites dose-dependently inhibited IL-13-induced eotaxin-3 secretion, and γ-TE was the most effective among the tested vitamin E forms. γ-TE treatment led to inhibition of IL-13-stimulated STAT6 phosphorylation and STAT6 DNA binding acitivity, which are critical to activation of eotaxin expression. Further mechanistic studies revealed that the aPKCs rather than classic PKCs or other signaling is pivotal to IL-13-stimulated STAT6 activation in A549 cells. Interestingly, γ-TE treatment resulted in up-regulation of PAR4 expression, which has been shown to interact with atypical PKCs and consequently inhibits aPKC-mediated signaling [39]. Consistently, the immunoprecipitation results indicated that γ-TE treatment resulted in enhanced interaction between PAR4 and aPKC. Our study provides the first evidence that vitamin E forms suppressed IL-13/STAT6 activation by induction of PAR4 expression and enhancement of aPKC-PAR4 interaction that negatively regulates aPKCs (Figure 6) [39].
Figure 6. The mechanism underlying γ-TE-caused inhibition of eotaxin.
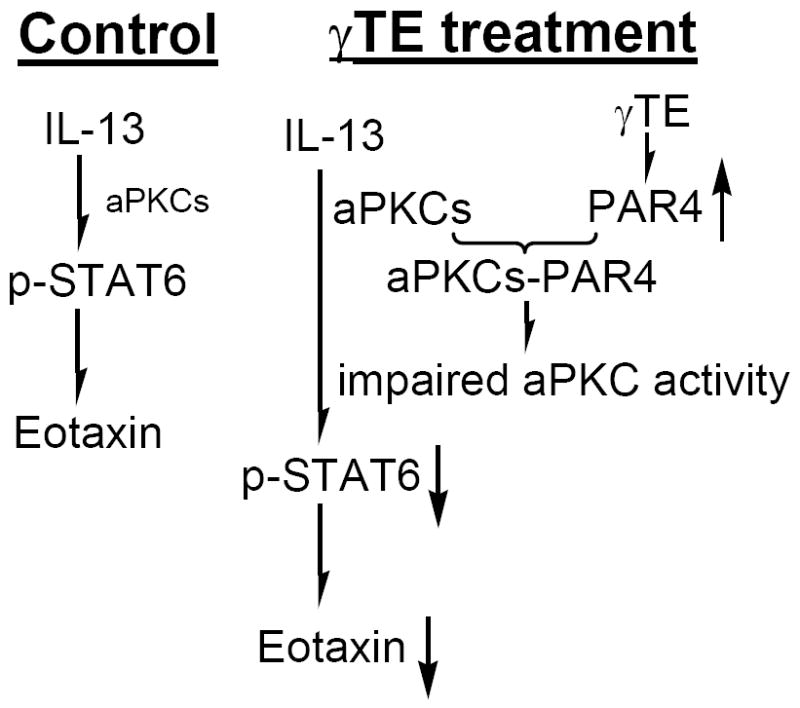
Our data indicate that aPKCs are critically important to STAT6 phosphorylation and eotaxin generation. γ-TE treatment enhanced PAR4 expression and aPKC-PAR4 complex formation, which negatively regulates aPKCs activity and therefore resulted in decreased eotaxin secretion.
It is intriguing that previous studies have indicated that the atypical PKCs including PKCζ and PKCλ/ι play key roles in modulation of airway inflammation and may therefore be therapeutic targets for asthma [34, 35]. For instance, the expression of PKCζ increases during Th2 but not Th1 differentiation of CD4+ T cells. The loss of PKCζ blocks IL-4/STAT6 activation and decreases Th2 cytokine secretion. As a result, PKCζ-/- mice displayed profound inhibition of ovalbumin-induced allergic airway disease [34]. Recently, it is reported that PKCλ/ι knockout mice have impaired activation of NFκB in Th2 cells and dampened airway inflammation [35]. In the current study, we showed that both aPKC isoforms play important roles in IL-13-stimulated STAT6 activation and downregulation of PKCλ/ι appears to have profound effects on STAT6 protein status in human lung epithelial cells.
The atypical PKCs belong to a subgroup of the protein kinase C family. Unlike classic PKC isoforms, the aPKCs cannot be regulated by calcium, phorbol esters or diacylglycerol [40, 41]. Instead, the activity of aPKCs is shown to be controlled via their interaction with regulatory proteins. PAR4 appears to be an endogenous inhibitor for PKC activity by interaction with the zinc finger domain of PKCζ and PKCλ/ι [39]. Although PAR4 was initially discovered in prostate cells undergoing apoptosis [42], recent studies have revealed that PAR4 is generally expressed in many tissues including the lung [43]. Because of its regulation of aPKC-mediated signaling, PAR4 is believed to be able to modulate inflammatory responses [38, 44]. Thus, the loss of PAR4 is found to enhance PKCζ-mediated activation of IL-4/STAT6, and as a result, PAR4 knockout exacerbates hepatitis induced by Concanavalin A [33]. Our present study demonstrated that aPKCs are critical to IL-13-stimulated STAT6 phosphorylation in lung epithelial A549 cells, which mimics the regulatory role of PKCζ in IL-4/STAT6 activation in Th2 cells [34]. γ-TE treatment led to up-regulation of PAR4 and increased PAR4/aPKC crosstalk, which consistently correlates with impaired activation of IL-13/STAT6.
γ-TE has previously been shown to suppress NFκB activation via inhibition of IκBα phosphorylation in various cell types [45]. Because aPKC and PAR4 have been implicated in regulation of NFκB [38, 46], we investigated whether γ-TE modulates TNF-α-stimulated NFκB activation in A549 cells. However, γ-TE at 20 or 50 μM did not show any effects on TNFα-induced IκBα phosphorylation in confluent cells (Y Wang, unpublished observations). This is consistent with our previous observation that γ-TE does not have any effect on NFκB-upregulated cyclooxygenase-2 expression induced by IL-1β in A549 cells [12]. In addition, Regala et al [37] have demonstrated that knock down aPKC has no impact on TNF-α induced NFκB activation in A549 cells. Therefore, aPKC-regulated NFκB activation and γ-TE’s inhibition of NFκB are likely cell-type dependent.
The present study was initially inspired by our recent observations that γ-T supplementation attenuated ovalbumin-stimulated eosinophilic infiltration in bronchoalveolar lavage fluid in Brown Norway rats [17, 18]. Because eotaxin is secreted by lung epithelium in response to IL-13 or IL-4 stimulation and its only known function is to recruit eosinophils to the airway, our current observation that vitamin E forms dose-dependently inhibited IL-13-activated eotaxin secretion provides a molecular basis for the observed in vivo anti-asthmatic activity of γ-T [17, 18]. It is noteworthy that the IC50s of γ-T, δ-T and γ-TE for inhibition of IL-13-induced eotaxin secretion are pharmacologically achievable, as these concentrations have been reported in rodents and humans shortly after supplementation with these vitamin E forms [47-49]. Since compared with γT, δ-T and γ-TE are similarly or more effective in inhibition of eotaxin secretion (this study) and suppression of cyclooxygenase- and 5-lipoxygenase-mediated proinflammatory eicosanoids [12, 13], δ-T and γ-TE may have comparable or even stronger anti-asthmatic activity than γ-T, which warrants further investigation. In addition, future studies are necessary to elucidate the signaling that is responsible for PAR4 induction, and the role of vitamin E forms in modulating that signaling pathway(s).
Acknowledgments
GRANTS This work was supported by a NIH-NCCAM grant NIH P01AT002620.
ABBREVIATION
- α-T, β-T, γ-T, or δ-T
α, β, γ, or δ-tocopherol
- α-, β-, γ-and δ-TE
α-, β-, γ-and δ-tocotrienol
- CEHC
7,8-dimethyl 2-(β-carboxyethyl)-6- hydroxychroman
- aPKC
the atypical protein kinase C
- cPKC
classic protein kinase C
- IL-13
interleukin-13
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
BIBLIOGRAPHY
- 1.Lim Y, Vasu VT, Valacchi G, Leonard S, Aung HH, Schock BC, et al. Severe vitamin E deficiency modulates airway allergic inflammatory responses in the murine asthma model. Free radical research. 2008;42:387–96. doi: 10.1080/10715760801976600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Montano Velazquez BB, Jauregui-Renaud K, Banuelos Arias Adel C, Ayala JC, Martinez MD, Campillo Navarrete R, et al. Vitamin E effects on nasal symptoms and serum specific IgE levels in patients with perennial allergic rhinitis. Ann Allergy Asthma Immunol. 2006;96:45–50. doi: 10.1016/s1081-1206(10)61039-3. [DOI] [PubMed] [Google Scholar]
- 3.Pearson PJ, Lewis SA, Britton J, Fogarty A. Vitamin E supplements in asthma: a parallel group randomised placebo controlled trial. Thorax. 2004;59:652–6. doi: 10.1136/thx.2004.022616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reiter E, Jiang Q, Christen S. Anti-inflammatory properties of alpha- and gamma-tocopherol. Molecular aspects of medicine. 2007;28:668–91. doi: 10.1016/j.mam.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shahar E, Hassoun G, Pollack S. Effect of vitamin E supplementation on the regular treatment of seasonal allergic rhinitis. Ann Allergy Asthma Immunol. 2004;92:654–8. doi: 10.1016/S1081-1206(10)61432-9. [DOI] [PubMed] [Google Scholar]
- 6.Zheng K, Adjei AA, Shinjo M, Shinjo S, Todoriki H, Ariizumi M. Effect of dietary vitamin E supplementation on murine nasal allergy. The American journal of the medical sciences. 1999;318:49–54. doi: 10.1097/00000441-199907000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Dietrich M, Traber MG, Jacques PF, Cross CE, Hu Y, Block G. Does gamma-tocopherol play a role in the primary prevention of heart disease and cancer? A review Journal of the American College of Nutrition. 2006;25:292–9. doi: 10.1080/07315724.2006.10719538. [DOI] [PubMed] [Google Scholar]
- 8.Jiang Q, Christen S, Shigenaga MK, Ames BN. gamma-tocopherol, the major form of vitamin E in the US diet, deserves more attention. Am J Clin Nutr. 2001;74:714–22. doi: 10.1093/ajcn/74.6.714. [DOI] [PubMed] [Google Scholar]
- 9.Jiang Q, Ames BN. Gamma-tocopherol, but not alpha-tocopherol, decreases proinflammatory eicosanoids and inflammation damage in rats. Faseb J. 2003;17:816–22. doi: 10.1096/fj.02-0877com. [DOI] [PubMed] [Google Scholar]
- 10.Jiang Q, Elson-Schwab I, Courtemanche C, Ames BN. gamma-tocopherol and its major metabolite, in contrast to alpha-tocopherol, inhibit cyclooxygenase activity in macrophages and epithelial cells. Proc Natl Acad Sci U S A. 2000;97:11494–9. doi: 10.1073/pnas.200357097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Q, Lykkesfeldt J, Shigenaga MK, Shigeno ET, Christen S, Ames BN. Gamma-tocopherol supplementation inhibits protein nitration and ascorbate oxidation in rats with inflammation. Free Radic Biol Med. 2002;33:1534–42. doi: 10.1016/s0891-5849(02)01091-2. [DOI] [PubMed] [Google Scholar]
- 12.Jiang Q, Yin X, Lill MA, Danielson ML, Freiser H, Huang J. Long-chain carboxychromanols, metabolites of vitamin E, are potent inhibitors of cyclooxygenases. Proc Natl Acad Sci U S A. 2008;105:20464–9. doi: 10.1073/pnas.0810962106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang Z, Yin X, Jiang Q. Natural forms of vitamin E and 13′-carboxychromanol, a long-chain vitamin E metabolite, inhibit leukotriene generation from stimulated neutrophils by blocking calcium influx and suppressing 5-lipoxygenase activity, respectively. J Immunology. 2011;186:1173–9. doi: 10.4049/jimmunol.1002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christen S, Woodall AA, Shigenaga MK, Southwell-Keely PT, Duncan MW, Ames BN. gamma-tocopherol traps mutagenic electrophiles such as NO(X) and complements alpha-tocopherol: physiological implications. Proc Natl Acad Sci U S A. 1997;94:3217–22. doi: 10.1073/pnas.94.7.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooney RV, Franke AA, Harwood PJ, Hatch-Pigott V, Custer LJ, Mordan LJ. γ-Tocopherol detoxification of nitrogen dioxide: Superiority to α-tocopherol. Proc Natl Acad Sci USA. 1993;90:1771–5. doi: 10.1073/pnas.90.5.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamahata A, Enkhbaatar P, Kraft ER, Lange M, Leonard SW, Traber MG, et al. gamma-Tocopherol nebulization by a lipid aerosolization device improves pulmonary function in sheep with burn and smoke inhalation injury. Free Radic Biol Med. 2008;45:425–33. doi: 10.1016/j.freeradbiomed.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner JG, Jiang Q, Harkema JR, Ames BN, Illek B, Roubey RA, et al. gamma-Tocopherol prevents airway eosinophilia and mucous cell hyperplasia in experimentally induced allergic rhinitis and asthma. Clin Exp Allergy. 2007 doi: 10.1111/j.1365-2222.2007.02855.x. [DOI] [PubMed] [Google Scholar]
- 18.Wagner JG, Jiang Q, Harkema JR, Illek B, Patel DD, Ames BN, et al. Ozone enhancement of lower airway allergic inflammation is prevented by gamma-tocopherol. Free radical biology & medicine. 2007;43:1176–88. doi: 10.1016/j.freeradbiomed.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wynn TA. IL-13 effector functions. Annual review of immunology. 2003;21:425–56. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 20.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annual review of immunology. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 21.Holgate ST, Lackie P, Wilson S, Roche W, Davies D. Bronchial epithelium as a key regulator of airway allergen sensitization and remodeling in asthma. American journal of respiratory and critical care medicine. 2000;162:S113–7. doi: 10.1164/ajrccm.162.supplement_2.ras-12. [DOI] [PubMed] [Google Scholar]
- 22.Berkman N, Ohnona S, Chung FK, Breuer R. Eotaxin-3 but not eotaxin gene expression is upregulated in asthmatics 24 hours after allergen challenge. American journal of respiratory cell and molecular biology. 2001;24:682–7. doi: 10.1165/ajrcmb.24.6.4301. [DOI] [PubMed] [Google Scholar]
- 23.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 24.Blanchard C, Durual S, Estienne M, Emami S, Vasseur S, Cuber JC. Eotaxin-3/CCL26 gene expression in intestinal epithelial cells is up-regulated by interleukin-4 and interleukin-13 via the signal transducer and activator of transcription 6. The international journal of biochemistry & cell biology. 2005;37:2559–73. doi: 10.1016/j.biocel.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Jedrzkiewicz S, Nakamura H, Silverman ES, Luster AD, Mansharamani N, In KH, et al. IL-1beta induces eotaxin gene transcription in A549 airway epithelial cells through NF-kappaB. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1058–65. doi: 10.1152/ajplung.2000.279.6.L1058. [DOI] [PubMed] [Google Scholar]
- 26.Jiang Q, Freiser H, Wood KV, Yin X. Identification and quantitation of novel vitamin E metabolites, sulfated long-chain carboxychromanols, in human A549 cells and in rats. J Lipid Res. 2007;48:1221–30. doi: 10.1194/jlr.D700001-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang Q, Wong J, Fyrst H, Saba JD, Ames BN. {gamma}-Tocopherol or combinations of vitamin E forms induce cell death in human prostate cancer cells by interrupting sphingolipid synthesis. Proc Natl Acad Sci U S A. 2004;101:17825–30. doi: 10.1073/pnas.0408340102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kagami S, Saeki H, Komine M, Kakinuma T, Tsunemi Y, Nakamura K, et al. Interleukin-4 and interleukin-13 enhance CCL26 production in a human keratinocyte cell line, HaCaT cells. Clinical and experimental immunology. 2005;141:459–66. doi: 10.1111/j.1365-2249.2005.02875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siemasko K, Chong AS, Jack HM, Gong H, Williams JW, Finnegan A. Inhibition of JAK3 and STAT6 tyrosine phosphorylation by the immunosuppressive drug leflunomide leads to a block in IgG1 production. J Immunol. 1998;160:1581–8. [PubMed] [Google Scholar]
- 30.Zhang D, Qiu L, Jin X, Guo Z, Guo C. Nuclear factor-kappaB inhibition by parthenolide potentiates the efficacy of Taxol in non-small cell lung cancer in vitro and in vivo. Mol Cancer Res. 2009;7:1139–49. doi: 10.1158/1541-7786.MCR-08-0410. [DOI] [PubMed] [Google Scholar]
- 31.Moynihan B, Tolloczko B, Michoud MC, Tamaoka M, Ferraro P, Martin JG. MAP kinases mediate interleukin-13 effects on calcium signaling in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2008;295:L171–7. doi: 10.1152/ajplung.00457.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pesu M, Aittomaki S, Takaluoma K, Lagerstedt A, Silvennoinen O. p38 Mitogen-activated protein kinase regulates interleukin-4-induced gene expression by stimulating STAT6-mediated transcription. J Biol Chem. 2002;277:38254–61. doi: 10.1074/jbc.M201427200. [DOI] [PubMed] [Google Scholar]
- 33.Duran A, Rodriguez A, Martin P, Serrano M, Flores JM, Leitges M, et al. Crosstalk between PKCzeta and the IL4/Stat6 pathway during T-cell-mediated hepatitis. The EMBO journal. 2004;23:4595–605. doi: 10.1038/sj.emboj.7600468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin P, Villares R, Rodriguez-Mascarenhas S, Zaballos A, Leitges M, Kovac J, et al. Control of T helper 2 cell function and allergic airway inflammation by PKCzeta. Proc Natl Acad Sci U S A. 2005;102:9866–71. doi: 10.1073/pnas.0501202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang JQ, Leitges M, Duran A, Diaz-Meco MT, Moscat J. Loss of PKC lambda/iota impairs Th2 establishment and allergic airway inflammation in vivo. Proc Natl Acad Sci U S A. 2009;106:1099–104. doi: 10.1073/pnas.0805907106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eichholtz T, de Bont DB, de Widt J, Liskamp RM, Ploegh HL. A myristoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. J Biol Chem. 1993;268:1982–6. [PubMed] [Google Scholar]
- 37.Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005;280:31109–15. doi: 10.1074/jbc.M505402200. [DOI] [PubMed] [Google Scholar]
- 38.Moscat J, Rennert P, Diaz-Meco MT. PKCzeta at the crossroad of NF-kappaB and Jak1/Stat6 signaling pathways. Cell death and differentiation. 2006;13:702–11. doi: 10.1038/sj.cdd.4401823. [DOI] [PubMed] [Google Scholar]
- 39.Diaz-Meco MT, Municio MM, Frutos S, Sanchez P, Lozano J, Sanz L, et al. The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell. 1996;86:777–86. doi: 10.1016/s0092-8674(00)80152-x. [DOI] [PubMed] [Google Scholar]
- 40.Akimoto K, Mizuno K, Osada S, Hirai S, Tanuma S, Suzuki K, et al. A new member of the third class in the protein kinase C family, PKC lambda, expressed dominantly in an undifferentiated mouse embryonal carcinoma cell line and also in many tissues and cells. J Biol Chem. 1994;269:12677–83. [PubMed] [Google Scholar]
- 41.Mellor H, Parker PJ. The extended protein kinase C superfamily. The Biochemical journal. 1998;332(Pt 2):281–92. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sells SF, Wood DP, Jr, Joshi-Barve SS, Muthukumar S, Jacob RJ, Crist SA, et al. Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell Growth Differ. 1994;5:457–66. [PubMed] [Google Scholar]
- 43.Boghaert ER, Sells SF, Walid AJ, Malone P, Williams NM, Weinstein MH, et al. Immunohistochemical analysis of the proapoptotic protein Par-4 in normal rat tissues. Cell Growth Differ. 1997;8:881–90. [PubMed] [Google Scholar]
- 44.Goswami A, Ranganathan P, Rangnekar VM. The phosphoinositide 3-kinase/Akt1/Par-4 axis: a cancer-selective therapeutic target. Cancer research. 2006;66:2889–92. doi: 10.1158/0008-5472.CAN-05-4458. [DOI] [PubMed] [Google Scholar]
- 45.Ahn KS, Sethi G, Krishnan K, Aggarwal BB. Gamma-tocotrienol inhibits nuclear factor-kappaB signaling pathway through inhibition of receptor-interacting protein and TAK1 leading to suppression of antiapoptotic gene products and potentiation of apoptosis. J Biol Chem. 2007;282:809–20. doi: 10.1074/jbc.M610028200. [DOI] [PubMed] [Google Scholar]
- 46.Diaz-Meco MT, Lallena MJ, Monjas A, Frutos S, Moscat J. Inactivation of the inhibitory kappaB protein kinase/nuclear factor kappaB pathway by Par-4 expression potentiates tumor necrosis factor alpha-induced apoptosis. J Biol Chem. 1999;274:19606–12. doi: 10.1074/jbc.274.28.19606. [DOI] [PubMed] [Google Scholar]
- 47.Freiser H, Jiang Q. Gamma-tocotrienol and gamma-tocopherol are primarily metabolized to conjugated 2-(beta-carboxyethyl)-6-hydroxy-2,7,8-trimethylchroman and sulfated long-chain carboxychromanols in rats. J Nutr. 2009;139:884–9. doi: 10.3945/jn.108.103309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Husain K, Francois RA, Hutchinson SZ, Neuger AM, Lush R, Coppola D, et al. Vitamin E delta-tocotrienol levels in tumor and pancreatic tissue of mice after oral administration. Pharmacology. 2009;83:157–63. doi: 10.1159/000190792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiser J, Alexis NE, Jiang Q, Wu W, Robinette C, Roubey R, et al. In vivo gamma-tocopherol supplementation decreases systemic oxidative stress and cytokine responses of human monocytes in normal and asthmatic subjects. Free Radic Biol Med. 2008;45:40–9. doi: 10.1016/j.freeradbiomed.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]