

Abstract
Intracellular signaling events play fundamental roles in regulating physiological function. In neurons, these include inducing growth and differentiation, secretion, gene expression, and controlling processes associated with learning and memory. All of these processes have in common the vital dependence on changes in intracellular Ca2+ regulation- [Ca2+]i. Numerous toxicants, including metals, polychlorinated biphenyls, and biological neurotoxins can disrupt [Ca2+]i.
Understanding how toxicants disrupt Ca2+-dependent neuronal signaling, and thus induce neuronal death or dysfunction requires the ability to monitor [Ca2+]i at the level of individual cells. A series of fluorophores which can report on changes in [Ca2+]i has been pivotal in this process. This section describes how to use these fluorophores to study effects of neurotoxicants on two types of process in [Ca2+]i individual cells; and in mitochondrial [Ca2+] membrane potential. Similar techniques using distinct fluorophores can be applied to other physiological processes.
Keywords: fluorescence, calcium
Application of Single-Cell Microfluorimetry to Neurotoxicology Assays
Single-cell microfluorimetry allows for real-time measurement of changes in essential cellular functions which directly affect neuronal intracellular signaling and viability. These include maintenance of intracellular calcium concentration ([Ca2+]i), plasma membrane potential, pH and mitochondrial membrane potential. Such information is useful for elucidating the pathway(s) which contribute to neuronal damage and death. Moreover, they can give insight into possible points of intervention. These approaches are applicable to all cells, and similar methods and material would be used. Examples of similar approaches in other cells include ventricular myocytes (Ward and Moffat, 1992), renal tubule cells (Friedman and Gesek, 1995), endothelial cells (Neve et al., 1995) and T-Cells (Choi et al., 1995). However this chapter will deal specifically with their use in neurons. Protocols for measuring cellular viability are provided elsewhere in this series.
This unit includes basic protocols for monitoring two essential cellular parameters - intracellular Ca2+ concentration: [Ca2+]i (using fura-2), and mitochondrial membrane potential (ψM) (using tetramethylrhodamine ethyl ester-TMRE). Both protocols can be carried out using the same experimental setup consisting of a fluorescence microscope coupled to a fluorescence emission and detection system. However, the appropriate fluorescence filters must be used for each indicator. Numerous other fluorescent indicators have been developed. These can be used to monitor other cellular processes, including concentrations of other ions (Zn2+, Cl−, Na+, H+), pH, and cell membrane potential. A very useful primer on the types of fluorophores available for measuring various cellular processes can be found on the Molecular Probes, Invitrogen website (http://www.probes.com).
Ideally, the type of experiments described here are carried out using a perfusion system that allows continual, rapid exchange of fluid in the cell chamber at a constant rate, yet should not cause cell movement or sheer forces due to fluid flow (see Peng et al., 2005). Using continuous perfusion permits one to examine time course of effects of toxicants on cell function. All experiments use cells attached to glass coverslips to prevent artifactual changes in fluorescence associated with plastic dishes and coverslips. All sample figures in this chapter depict fluorescence changes in either primary cells (rat cerebellar granule neurons) or a transformed neuronal cell line (i.e. rat pheochromocytoma - PC12 cells) attached to glass coverslips which were coated with poly-L-lysine. Note that the same types of studies can be carried out with high precision using individual cells of different origin-hippocampal pyramidal cells, cortical cells, etc., non-neuronal primary or dissociated cells as well as transformed cell lines. Examples of such cultures include undifferentiated PC12 cells, neuroblastoma-glioma (NG108-15, N1E115, etc.) and similar homogeneous transformed cell lines, chromaffin cells, or homogenous primary cells (T cells, smooth muscle cells, certain types of glia). When cells are homogeneous unless spatial resolution of a change is needed--i.e. a localized change in a neuronal process or the cell-type is heterogenous in the culture (as is the case with virtually all primary neuronal cultures) one can make these measurements from cells in bulk culture using a spectrofluorimeter. In this case, an average fluorescence signal is detected for all cells in the population. However, spatial resolution is lost. Similarly, subcellular fractions such as synaptosomes can be used equally well for “bulk” fluorimetry measurements (see Denny et al., 1993). The specific techniques associated with these “bulk preparations” will not be considered here. In addition, these methods can also be applied to more complex biological systems, such as isolated brain slices (Yuan and Atchison, 2005). Additional methodological limitations are imposed by these experimental systems, and are beyond the scope of this chapter. However, many of the components of experimental design and methodologies are common to these various types of preparations.
For measurements in single cells or bulk preparations, chemicals, drugs or toxins are added directly to the perfusing solution. Real time changes in cell fluorescence can then be measured continually during and following exposure to the test agent.
Basic protocol 1: Measuring Changes in [Ca2+]i
Change in [Ca2+]i underlie numerous cellular signaling processes. These changes are generally transitory and localized. The transient and local nature of the signals are crucial to the cell to prevent metabolic rundown, frank cytotoxicity and to allow accurate signaling. Maintenance of [Ca2+]i homeostasis is critical for maintaining normal cellular function and viability. By measuring changes in [Ca2+]i, one can both examine individual signaling events, as well as dissect the events leading up to cell death. The procedure is similar to that for other fluorescence assays: cells are attached to glass coverslip, loaded with fluorescent indicator, and then monitored in a fluorescence system to determine the effect of exposure to pharmacological agents and/or toxicants. Fura-2 remains the most commonly used fluorophore for measurement of [Ca2+]I although a number of other indicators have been developed, including the ratiometric UV indicator Indo, as well as several non-ratiometric indicators useful for visible wavelengths (Fluo-3, Calcium Green). These latter are typically chosen for measurements made using confocal microscopy. Furthermore, more specialized Ca-sensitive probes have also been developed with specific purposes in mind - i.e. low Ca2+ affinity -Fura FF, Mag-Fura 2). The distinctions, advantages and uses of these agents can again be found at: www.probes.com.
Fura-2 has a number of advantages to its use, the most important being that it is a so-called “ratiometric indicator”. This property is demonstrated in Figure 1, which depicts properties described from the original paper on this topic (Grynkiewicz et al., 1985). Using a cell free system, the fluorescence spectrum of fura-2 recording at 505nm emission is elicited across wavelengths from 300 to 400 nm. The response to increasing [Ca2+] is shown as a peak signal at ~340nm and increases as [Ca2+] increases. Conversely, a minimum (though not absolute) is attained at 380nm. Thus using the peak 340 signal/peak 380 signal allows an assessment of [Ca2+]i. The theoretical aspects of this, along with this initial description of the fluorescent properties of Fura-2 are also found in Grynkiewicz et al. (1985).
Figure 1.

Fluorescence excitation spectra of fura-2 (free acid) in solutions containing 0–39.8 μM free Ca2+. Excitation spectrum was scanned between 300–400 nm, and emitted fluorescence was detected at 505 nm. When bound to Ca2+, Fura 2 the fluorescence spectrum shifts with an increase in the maximum and minimum fluorescence. These occur at ~ 340 and ~380 nm respectively. Note that the emitted fluorescence does not change upon altering free Ca for excitation at 360 nm. This “Ca-insensitive” wavelength is known as the “isobestic point”.
Fura-2 like many other agents used to study changes in fluorescence which occur intracellularly is coupled to an acetoxy methyl ester (AM, i.e. fura-2AM). This allows fluorophore to be loaded into cell, where endogenons esterases will cleave the AM thereby trapping the fluorophere. Differences in loading efficiency and differences in fluorescence are obviated by use of ratiometric imagery. In some cases, its possible to correlate the change in fluorescence directly to an absolute value, i.e. [Ca2+]i, pH, membrane potential. However, in other cases fluorescence changes can only be measured in a relative fashion because of interactions of other ions with the fluorophore.
Newer agents in which the fluorophore is conjugated to a dextran have also been developed. While these derivatives overcome some problems inherent to use of AM-forms of fluorophore, they are not as readily loaded into cells due to their size. Thus more elaborate methods are needed to load these forms of fluorophore. Details of the use of these agents can also be found at www.probes.com.
Materials
(HBS) HEPES- Buffered Saline Solution (see recipe)
Fura-2 acetoxymethylester (fura-2 AM) (see recipe)
40 mM K+ buffer (see recipe)
EGTA- HBS (see recipe)
Ethanol
Cotton swab (such as a Q-tip®)
Kimwipes®
Inverted fluorescence microscope system coupled to excitation light source (such as a xenon lamp in a covered housing) with fluorescence filters for 340 and 380 nm excitation (preferably mounted on a rotating filter wheel for rapid switching between excitation wavelengths), and a 510 nm emission barrier
Perfusion system (consisting of perfusion pump, tubing to lead from bottle of buffer to microscope and out to waste container)
-
Temperature control system integrated into microscope stage (optional)
At the beginning of each experiment, it is important to verify cell viability and excitability, as well as the ability of the cell to buffer changes in [Ca2+]i. One approach that can be used with excitable cells such as neurons or muscle cells, is to apply a brief pulse 40 mM K+ solution for 1 − 1 ½min. This will depolarize the plasma membrane, which causes a brief, rapidly reversed influx of extracellular Ca2+ (Ca2+e) into the cells (Figure 2). A viable cell will respond to the K challenge with an increase in fluorescence, and recover baseline [Ca2+]i following removal of the K+. In some cases, if voltage-gated Ca2+ channels are not found in large numbers on the cell, it may not respond to the KCl-stimulation. If this is the case, an alternative for neuronal and glial cells is to use a glutamate analog such as kainic acid to depolarize.While it is tempting simply to record the ratio of fluorescence induced by excitation at the two wavelengths, this can be problematic. Because fura-2 interacts with a number of polyvalent cations (albeit with variable affinity) changes in other endogenous cations - Mg2+, Zn2+, Cu2+, Mn2+ can also affect fura fluorescence (Grynkiewicz et al., 1985) (see Figure 3). Depending upon the specific effect of a particular cation, it can increase or decrease the fluorescence at either or both 340 and 380 nm excitability wavelengths. This, in turn, can affect the ratio of fluorescence recorded, yet have little if anything, to do with changes in [Ca2+]i. These effects can be spotted by examining the nonratioed “raw” data obtained at 340 and 380 nm. Alternatively, the fluorescence generated at 360 nm excitation can be monitored. Recall from Figure 1, that this wavelength is known as the isobestic point, or Ca-insensitive wavelength. Altered fluorescence at 360 nm indicates a contribution of some non-Ca2+ divalent cation. If need be, the contribution of this “contaminating divalent” can be removed by measuring effects of either the cell-permeant heavy metal chelator TPEN or the cell-impermeant heavy metal chelator DTPA on elevations in 340/380 nm ratio.When appropriate, [Ca2+]i is calculated from the equation of Grynkiewicz, et al. (1985): [Ca2+]i=Kd[(R-Rmin)/(Rmax-R)]×(Sf2/Sb2): where Rmax is the 340/380 nm ratio during Ca2+ saturation, and Rmin is the 340/380 nm ratio during Ca2+-free conditions. Sf2 and Sb2 are the emission intensities at 380 nm excitation during Ca2+-free and Ca2+-saturating conditions, respectively. Rmax is determined by lysing the cells with 0.1% (v/v) TritonX-100 in 2 mM Ca2+. Rmin is determined on the same sample by adding 50 mM EGTA (pH remained at 7.4). Alternatively Ca2+ entry can be stimulated with either 4-Br-A23187 (3 μM) or ionomycin (2 μM) in Ca2+-containing HBS in order to determine Rmax followed by ionophore and 10 mM EGTA to determine Rmin.
FIG. 2.
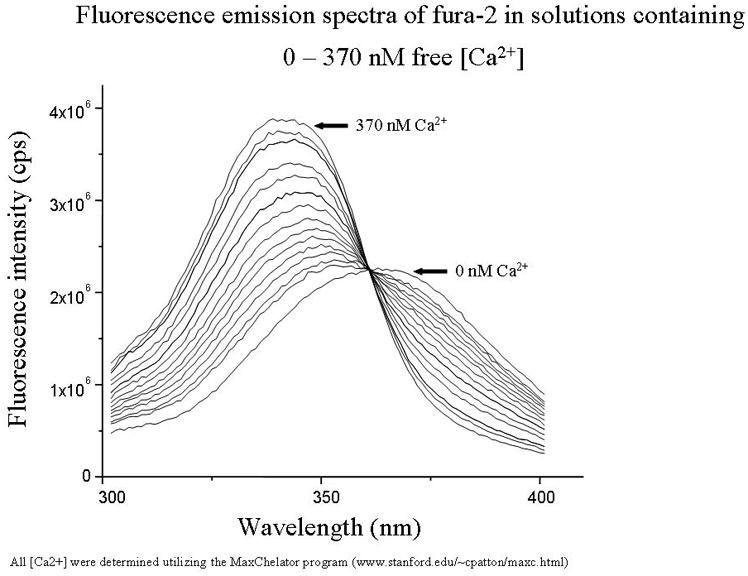
(A) Representative tracing of the fura-2 fluorescence from a cerebellar granule cell exposed to 40 mM K1 for 1 min (as a viability test), followed by 10 min 0.08% ethanol (as a vehicle control for 5 mM CsA) and 45 min 0.5 mM MeHg. The ratio of fluorescence emitted at excitation wavelengths of 340 and 380 nm gives the approximate level of [Ca21]i, which increases in a biphasic manner.. (From Limke and Atchison, 2001, TAAP with permission)
Figure 3.
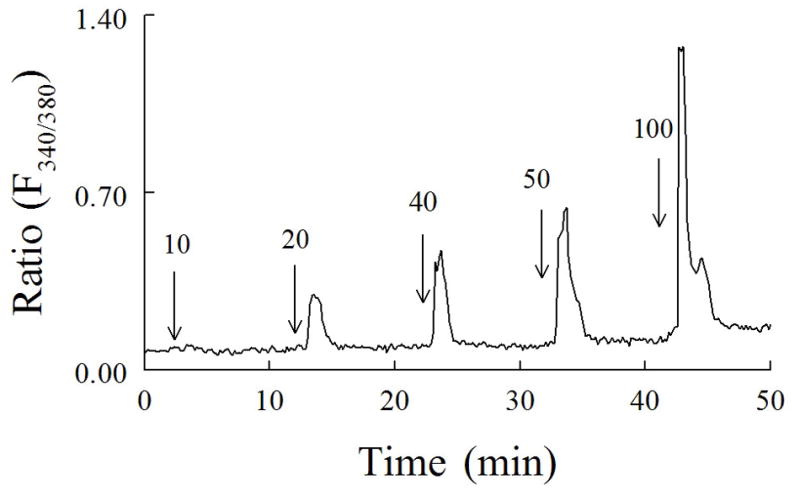
representative example of changes in fura-2 fluorescence [ratio (F340/380)] in a single cerebellar granule neuron exposed to increasing concentrations of K_(10–100 mM). Each K_ solution was applied for 1 min starting at the arrow, with the [K_] (millimolar).. indicated by the number above the arrow. (From Limke et al., 2002, JPET with permission).
Loading cells with Fura-2
Warm HBS in water bath (37°C).
In a glass or plastic vial, mix 3.0 ml warm HBS with 9–12 μl fura-2. Briefly vortex to mix.
-
Remove cells from incubator, and transfer to a sterile culture hood. Gently wash 2–3 times with warm HBS, then overlay with recently mixed fura-2 in HBS. Place cells back in incubator for 15–60 min.
Initial experiments need to be performed to determine optimal loading time for specific cells (and whether temperature affects the loading). The final concentration of fura-2 and duration of fluorophone loading will vary by cell type, but generally fall within the range of 0.1–5 μM. For rat cerebellar granule cells, a final concentration of 3–4 μM is ideal. Similarly, in some cell types Pluronic F-127 is used in loading fura. This has been reported to affect the fura-2-mediated response to KCl-induced depolarization (Sutachan et al., 2006). This is a case where “more” is not “better”. Investigators should strive to attain shorter loading periods and be the lowest amount of feasible fura. One common mistake with fura-2 is over-loading of cells with fluorophore. When cells retain excess dye, it is difficult to measure small changes in [Ca2+]i. When using a system that gives a color scale to measure relative fluorescence intensity, a baseline color of blue-green in the non-excited state is ideal (red or white fluorescence intensity in unstimulated cells suggests over-loading with indicator). Ideally, cells should stably retain dye, without significant loss of fluorescence, for at least an hour during control experiments. Remove cells from incubator. Wash 2–3 times with HBS to remove extracellular fura-2. Using sharp forceps, gently lift the coverslip from the culture dish and transfer it to a secure chamber (such as an O-ring ring chamber that securely holds the coverslip in place). Dip the cotton swab in ethanol, and gently wipe across the non-cell side of the coverslip to clean. Gently dry the bottom of the coverslip using a Kimwipe and test for chamber leakage. Invert the chamber, and add sufficient HBS to cover the cells without flooding the chamber. Place the cells in a dark box to transport to the microscope room.
Measuring changes in [Ca2+]i
-
Gently insert the chamber onto the microscope stage. Add perfusion tubes to opposite sides of the chamber to create flow across the top of the coverslip, and, if part of your system, a probe to maintain the system at a constant temperature. Rinse with HBS for 15–30 min to allow leakage of excess dye from cells. During the rinse, locate cells to monitor and set up the experimental parameters such as measurement location, frequency of data acquisition, etc. If using a non-confocal microscope system, be sure to focus the microscope on the sharp edges of the cells (the cells will seem to disappear under regular light but will have sharp edges when viewed using fluorescence).
Optimal rinse time also needs to be determined for each cell type prior to beginning real experiments. After loading, it is normal to see a decrease in fluorescence which stabilizes with time; at this point the experiment can begin. Fluorescence can decrease if the cells become bleached by exposure to too much light, so try to minimize the excitation exposure times as much as possible. To measure changes in fura-2 fluorescence, alternately excite at 340 and 380 nm, and measure the fluorescence above 380 nm (this is achieved by selecting for an appropriate light filter in your microscope). The ratio of the emission caused by excitation at 340/380, measured above 510 nm, gives the relative [Ca2+]i. In some cases, a ratio of 360/380 is used in lieu of 340/380. This is typically done in instances in which there is concern about “contamination” of the “Ca2+ signal” with that of other divalent cations. This technique makes use of the property of fura-2 that its’ isobestic point (Ca2+-insensitive wavelength) is ~360nm (See Fig 1). Because the normal response of fura to the Ca2+ bound form is a reduction in fluorescence at 380nm (See Fig 1), this technique involves a reduction in the denominator (380nm response) with no change in the numerator (360 response), and thus still reports an increase in ratio, and hence an increase in [Ca2+]i.
Basic Protocol 2: Measuring Changes in Mitochondrial Membrane Potential
Mitochondria control a number of cellular processes, in particular energy levels through ATP production. They are also a critical regulator of apoptosis. Similarly to the plasma membrane, mitochondria maintain a negative membrane potential that regulates opening of channels within the inner mitochondrial membranes; when depolarized, these channels open, causing release of pro-apoptotic factors, such as cytochrome c, and hindering production of ATP. The following protocol allows measurement of the inner mitochondrial membrane potential in groups of mitochondria within a cell in real time. Unlike fura-2, which is trapped inside the cell by the AM group (which prevents the dye from leaking back across the plasma membrane), Tetramethylrhodamine ethyl ester (TMRE) can easily pass through the cell membrane at any time, necessitating its inclusion in all experimental solutions to maintain dye availability.
Materials
HBS buffer (see recipe)
Tetramethylrhodamine ethyl ester (TMRE) (see recipe)
40 mM K+ buffer (see recipe)
Ethanol
Cotton swab (such as a Q-tip)
Kimwipes
Inverted fluorescence microscope system coupled to excitation light source (such as a xenon lamp in a covered housing) with fluorescent filter for 540 nm excitation (preferably mounted on a rotating filter wheel for rapid periods of excitation), and 590 nm emission barrier
Perfusion system (consisting of perfusion pump, tubing to lead from bottle of buffer to microscope and out to waste container)
Temperature control system integrated into microscope stage (optional)
Loading cells with TMRE
Warm HBS in water bath (37°C). Make up 100 nM TMRE in HBS from stock solution (see recipe for details), and warm in water bath (37°C).
-
Remove cells from incubator, and transfer to a sterile culture hood. Gently wash 2–3 times with warm HBS, then overlay with 100 nM TMRE. Place cells back in incubator for 20–60 min.
Initial experiments need to be performed to determine optimal loading time for specific cells. For rat cerebellar granule neurons, 30 min in 100 nM TMRE is ideal. Remove cells from incubator. Wash 2–3 times with HBS to remove excess dye. Using sharp forceps, gently lift the coverslip from the culture dish and transfer to secure chamber (such as an O-ring chamber that securely holds the coverslip in place). Dip the cotton swab in ethanol, and gently wipe across the non-cell side of the coverslip to clean. Use a kimwipe to dry the bottom of the coverslip and test for chamber leakage. Flip the chamber over, and add sufficient 10 nM TMRE (diluted in HBS) to the top of the chamber to cover cells without flooding chamber. Place cells in dark box to transport to microscope room.
Measuring changes in mitochondrial membrane potential
-
Gently place chamber onto microscope stage. Add perfusion tubes to opposite sides of chamber to create flow across the top of the coverslip, and, if part of your system, a probe to maintain constant temperature. Rinse with 10 nM TMRE in HBS for 15–30 min to allow leakage of excess dye from cells. During the rinse, locate cells to monitor and set up experimental parameters such as measurements location, frequency of data acquisition, etc. If using a non-confocal microscope system, be sure to focus the microscope on the sharp edges of the cells (the cells will seem to disappear under regular light but will have sharp edges when viewed using fluorescence).
Unlike AM dyes, which trap the dye inside the cell membrane, TMRE is a highly lipophilic cationic derivative of rhodamine 123 which is attracted to the highly negative transmembrane potential of the inner mitochondrial membrane. Because TMRE can easily pass through membranes, it is critical to maintain a small, constant concentration of TMRE in all solutions, as loss of dye availability will rapidly result in loss of fluorescence. -
To measure changes in mitochondrial membrane potential, excite the cells at 540 nm and measure the fluorescence above 590 nm (this is achieved by selecting for an appropriate light filter in your microscope).
Because TMRE fluorescence intensity will always be relative to the number of mitochondria in a cell, data are presented as relative to the starting fluorescence of 100%. Irreversible loss of TMRE fluorescence indicates widespread mitochondrial depolarization, a strong indicator of impending cell death.
Reagents and Solutions
HBS
Stock solutions of inorganic ions can be maintained at room temperature for several months. Once HBS is made up, can be stored in 4°C for up to 1 wk.,
| Stock solution (made in diH2O) or powder | Molarity in Final Solution (mM) | For 1.0 L HBS |
| NaCl (1.45 M) | 150 | 103.0 mL |
| KCl (500 mM) | 5.4 | 10.7 mL |
| CaCl2 (200 mM) | 1.8 | 9.0 mL |
| MgSO4 (100 mM) | 0.8 | 8.0 mL |
| Dextrose | 20 | 3.604 g |
| HEPES | 20 | 4.766 g |
| diH2O | 869.3 mL |
Mix until dissolved and clear. Adjust pH to 7.30 with Tris; filter with sterile suction filter (such as a Whatman #1 filter).
EGTA-HBS
Stock solutions can be maintained at room temperature for several months. Once EGTA-HBS is made up, can be stored in 4°C for up to 1 wk.
| Stock solution (made in diH2O) or powder | Molarity in Final Solution (mM) | For 1.0 L EGTA-HBS |
| NaCl (1.45 M) | 150 | 103.0 mL |
| KCl (500 mM) | 5.4 | 10.7 mL |
| MgSO4 (100 mM) | 0.8 | 8.0 mL |
| EGTA | 0.2 (20 μM) | 0.0076 g |
| Dextrose | 20 | 3.604 g |
| HEPES | 20 | 4.766 g |
| diH2O | 869.3 mL |
Mix until dissolved and clear. Adjust pH to 7.30 with Tris; filter with sterile suction filter (such as a Whatman #1 filter).
40 mM K+
Stock solutions can be maintained at room temperature for several months. Once 40 mM K+/HBS is made up, can be stored in 4°C for up to 1 week.
| Stock solution (made in diH2O) or powder | Molarity in Final Solution (mM) | For 100 mL 40 mM K+ |
| NaCl (1.45 M) | 105 | 7.27 mL |
| KCl (500 mM) | 40 | 8.0 mL |
| MgSO4 (100 mM) | 0.8 | 0.8 mL |
| CaCl2 (200 mM) | 1.8 | 0.9 mL |
| Dextrose | 20 | 0.360 g |
| HEPES | 20 | 0.477 g |
| diH2O | 86.93 mL |
Mix until dissolved and clear. Adjust pH to 7.30 with Tris; filter with sterile suction filter (such as a Whatman #1 filter).
Fura-2
Add 50 μl anhydrous DMSO to 50 μg vial of fura-2 (creates a solution of approximately 1 mM). Cover vial with aluminum foil. Store at ≤−20°C until use; thaw at room temperature prior to use.
TMRE
Mix 25 mg of powdered TMRE with 48.5 mL ethanol to produce 1 mM stock; store at −20°C in a dark container (such as a glass bottle covered with aluminum foil). Dilute to 100 nM in HBS for loading cells, and 10 nM in all experimental solutions to maintain dye availability.
Commentary
Ca2+ signaling is critical for a number of cellular processes, including cell division, axonal outgrowth, and cell death in a regulated fashion (apoptosis). Cells maintain tight regulation of Ca2+ signaling through a number of mechanisms, including plasma membrane transporters (e.g. the Na+/Ca2+ exchanger), intracellular Ca2+ binding proteins (such as calmodulin), and intracellular organelles that uptake and release Ca2+ in a regulated manner (primarily the mitochondria and smooth endoplasmic reticulum). Unregulated, irreversible increases in intracellular Ca2+ (Ca2+i) usually signal cell death, either through necrosis or apoptosis. However, brief bursts of increased [Ca2+]i are typically involved in non-toxic cellular signaling.
Along with basic viability assays to distinguish between necrosis and apoptosis, monitoring changes in [Ca2+]i can provide important information regarding the mode and signaling pathways involved in cell death.
Critical parameters to consider before testing
One important control that is often overlooked is testing whether the test chemical of interest interferes with the fluorophore itself, either by quenching or increasing fluorescence. To test this, the full emission spectrum of the fluorophore is tested in a cell-free system in the presence and absence of the test article, to determine whether the emission pattern shifts in the presence of the test chemical.
For AM dyes such as fura-2, it is critical to note that the ability of the dye to convert from the lipophilic AM form to the free, membrane impermeant form is dependent on esterase activity to cleave the AM component from the dye. Esterase activity varies from cell to cell, which can result in cells with very low basal fluorescence in some types of cells. Esterase activity is promoted by loading the cells at 37°C; however, inherent low esterase activity cannot be overcome by exposing the cells to a large excess of dye. Cells can also be “leaky” when it comes to retaining dye within the cell membrane. Some cells actively pump out dye through anionic transporters; for example, AM esters are substrates for P-glycoprotein (PGP) transporters, which can extrude dye from the cell prior to being broken down into the AM-free, cell-trapped form. While this is generally not an issue for neurons in primary culture, it can become problematic when dealing with cells that express PGP, such as astrocytes.
Anticipated results
Trouble-shooting
One of the most common mistakes during fluorescence measurements is confusing artefact for real shifts in fluorescence. For example, if cells shift during the experiment (either within the visual field due to bumping of the microscope stage, or focally due to loss of focus on the sharp membrane edge of the cells), fluorescence changes will occur which have nothing to do with cell signaling. Certain drugs and metals can bind to fluorophores directly, which will interfere with fluorescence independently of any actual cellular affects. For example, fura-2 binds to other, non-Ca2+ divalent cations, including Zn2+ and Mg2+. For this reason, it is important to monitor carefully experiments both at the beginning and end to ensure constant parameters (focal integrity, flow rate of the perfusion system, location of cells within field, etc.), and perform sufficient control experiments in cell-free systems to verify that any changes in fluorescence are truly significant.
Additionally, it is very important to avoid over-loading cells with dye, especially when using dyes in an AM form. When esterases cleave the AM group from the fluorescent indicator, there is the generation of potentially toxic by-products, including formaldehyde and acetic acid. Excess hydrophilic dye also has the potential to become incorporated into the plasma membrane, which can interfere with fluorescence measurements. Finally, non-hydrolyzed fura-2 AM dye is fluorescent but does not bind Ca2+; thus, the presence of excess fura-2 AM can lead to underestimation of [Ca2+]i.
Time considerations
After dye loading into cells, fluorescent measurements are generally limited to 1 – 1½ hrs, depending on the initial fluorescence intensity and the rate of data acquisition. In the case of dyes trapped within the cell membrane (such as fura-2), repeated excitation of the fluorophore will gradually bleach the dye, resulting in loss of fluorescence. Overall experiment time will vary depending on the optimal time needed to load and rinse cells, which varies by the dye used and the type of cells utilized.
Figure 4.
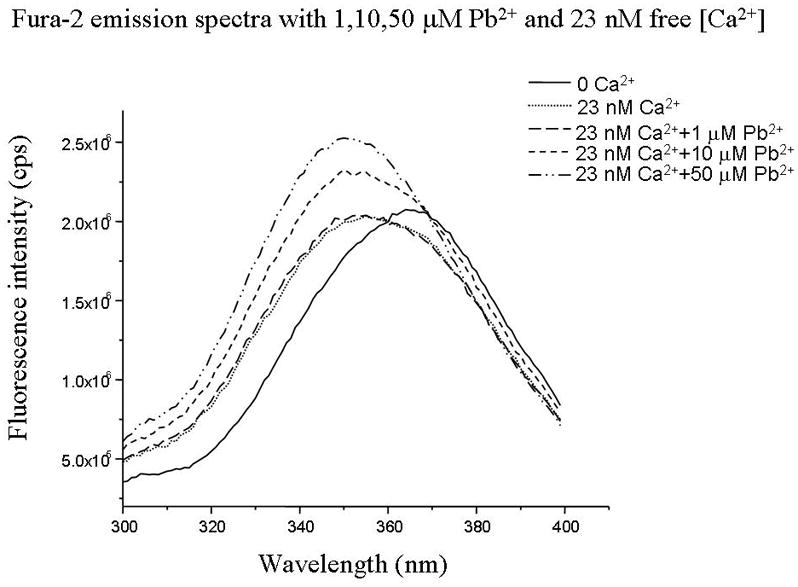
Fluorescence excitation spectra of fura-2 (free acid) in solutions containing 0–39.8 μM free Pb2+ in the absence and presence of Ca2+. Excitation spectrum was scanned between 300–400 nm, and emitted fluorescence was detected at 505 nm. When bound to Ca2+, Fura 2 the fluorescence spectrum shifts with an increase in the maximum and minimum fluorescence. These occur at ~ 340 and ~380 nm respectively. Note that the emitted fluorescence does not change upon altering free Ca for excitation at 360 nm. This “Ca-insensitive” wavelength is known as the “isobestic point”.
Acknowledgments
The authors gratefully acknowledge the contribution of Dr. Joshua Stuart Edwards for the fura-2 recordings in cell-free systems. The word processing assistance of Erin E. Koglin and Tara S. Oeschger is especially appreciated. This project was supported by NIEHS grants R01-ES03299, and R01-ES11662.
Literature cited
- Choi J, Sawant SG, Couch DB, Ho IK, Farley JM. Continuous measurement of changes in intracellular calcium concentration in mouse splenic T Cells attached to a glass substrate. J Biomed Sci. 1995;2(4):379–383. doi: 10.1007/BF02255225. [DOI] [PubMed] [Google Scholar]
- Denny MF, Atchison WD. Methylmercury-induced elevations in intrasynaptosomal zinc concentrations: an 19F-NMR study. J Neurochem. 1994;63(1):383–386. doi: 10.1046/j.1471-4159.1994.63010383.x. [DOI] [PubMed] [Google Scholar]
- Denny MF, Atchison WD. Mercurial-induced alterations in neuronal divalent cation homeostasis. Neurotoxicol. 1996;17(1):47–61. [PubMed] [Google Scholar]
- Edwards JR, Marty MS, Atchison WD. Comparative sensitivity of rat cerebellar neurons to dysregulation of divalent cation homeostasis and cytotoxicity caused by methylmercury. Toxicol Appl Pharmacol. 2005;208(3):222–232. doi: 10.1016/j.taap.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Friedman PA, Gesek FA. Stimulation of calcium transport by amiloride in mouse distal convoluted tubule cells. Kidney Int. 1995;48(5):1427–1434. doi: 10.1038/ki.1995.432. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260(6):3440–3450. [PubMed] [Google Scholar]
- Hare MF, McGinnis KM, Atchison WD. Methylmercury increases intracellular concentrations of Ca2+ and heavy metals in NG108-15 cells. J Pharmacol Exp Ther. 1993;266(3):1626–1635. [PubMed] [Google Scholar]
- Limke TL, Otero-Montanez JK, Atchison WD. Evidence for interactions between intracellular calcium stores during methylmercury-induced intracellular calcium dysregulation in rat cerebellar granule neurons. J Pharmacol Exp Ther. 2003;304(3):949–958. doi: 10.1124/jpet.102.042457. [DOI] [PubMed] [Google Scholar]
- Marty MS, Atchison WD. Pathways mediating Ca2+ entry in rat cerebellar granule cells following in vitro exposure to methyl mercury. Toxicol Appl Pharmacol. 1997;147(2):319–330. doi: 10.1006/taap.1997.8262. [DOI] [PubMed] [Google Scholar]
- Neve EP, Boyer CS, Moldeus P. N-ethyl maleimide stimulates arachidonic acid release through activation of the signal-responsive phospholipase A2 in endothelial cells. Biochem Pharmacol. 1995;49(1):57–63. doi: 10.1016/0006-2952(94)00308-9. [DOI] [PubMed] [Google Scholar]
- Ward CA, Moffat MP. Positive and negative inotropic effects of phorbol 12-myristate 13-acetate: relationship to PKC-dependence and changes in [Ca2+]i. J Mol Cell Cardiol. 1992;24(9):937–948. doi: 10.1016/0022-2828(92)91861-x. [DOI] [PubMed] [Google Scholar]