

Abstract
Gene set analyses have become a standard approach for increasing the sensitivity of transcriptomic studies. However, analytical methods incorporating gene sets require the availability of pre-defined gene sets relevant to the underlying physiology being studied. For novel physiological problems, relevant gene sets may be unavailable or existing gene set databases may bias the results towards only the best-studied of the relevant biological processes. We describe a successful attempt to mine novel functional gene sets for translational projects where the underlying physiology is not necessarily well characterized in existing annotation databases. We choose targeted training data from public expression data repositories and define new criteria for selecting biclusters to serve as candidate gene sets. Many of the discovered gene sets show little or no enrichment for informative Gene Ontology terms or other functional annotation. However, we observe that such gene sets show coherent differential expression in new clinical test data sets, even if derived from different species, tissues, and disease states. We demonstrate the efficacy of this method on a human metabolic data set, where we discover novel, uncharacterized gene sets that are diagnostic of diabetes, and on additional data sets related to neuronal processes and human development. Our results suggest that our approach may be an efficient way to generate a collection of gene sets relevant to the analysis of data for novel clinical applications where existing functional annotation is relatively incomplete.
1. Introduction
Genome-wide expression studies are producing large quantities of experimental data characterizing a growing range of human diseases. Yet the biological interpretation of results obtained from these experiments is still a challenge, and clinical applications remain relatively elusive. Typically, microarray data are analyzed at the single gene level to identify transcripts with statistically significant differences between phenotypes, and a functional analysis is then performed on the gene list. Originally, such functional annotation was performed manually1,2, but soon many tools to automate the process were developed3–6.
More recently, analysis at the level of gene sets has emerged as a powerful alternative to individual-gene analyses to reflect the functional relationship between genes in a set. Mootha et al. initially demonstrated the power of using pre-defined gene sets in a case where no individual gene’s expression was significantly different between normal and diabetic patients7. Since then, many gene set analysis methods have been developed8–14. The goal of all gene set analysis methods is to identify functionally related genes that display coordinated expression changes. Typically, gene set analysis methods can be distinguished by their statistical criteria for differential expression, null hypotheses, and p-value calculations15.
However, all analytical methods incorporating gene sets depend on the knowledge of sets or pathways relevant to the underlying physiology. For fields such as diabetes and cancer, there has been considerable effort toward manual and computational curation of relevant gene function16. The Gene Ontology17 contains controlled descriptions of gene function that are frequently used to define gene sets. Pathway databases such as KEGG18, BioCyc19, and BioCarta (www.biocarta.com) can also be used to generate gene sets. However, for many complex physiological processes, there is still a need to identify relevant groups of functionally linked genes. Recent work studying gene expression in human development suggests that this area is one in which additional annotation is needed20.
Clustering approaches have long been used to find meaningful patterns in gene expression data and to identify functional gene sets from microarray data7,21–23. However, such methods do not necessarily generalize to inform the analysis of novel data sets since functionally related genes may be co-expressed only in a subset of conditions, and such gene sets would be missed by traditional clustering methods. Biclustering methods have emerged as an alternative to traditional clustering methods in such cases. Biclustering24 finds subgroups of genes that exhibit similar expression patterns over a subset of conditions. Many biclustering algorithms have been proposed25,26. More sophisticated biclustering algorithms search for coherent expression changes within subsets of conditions27–29. Coherence of a bicluster refers to coordinated changes of the genes’ expression patterns across a subset of conditions (as in Figure 1). Gene sets with coherent expression patterns in a data set may be functionally linked to the phenotype of interest.
Figure 1. Heatmap of a representative bicluster that shows coherent change across samples.
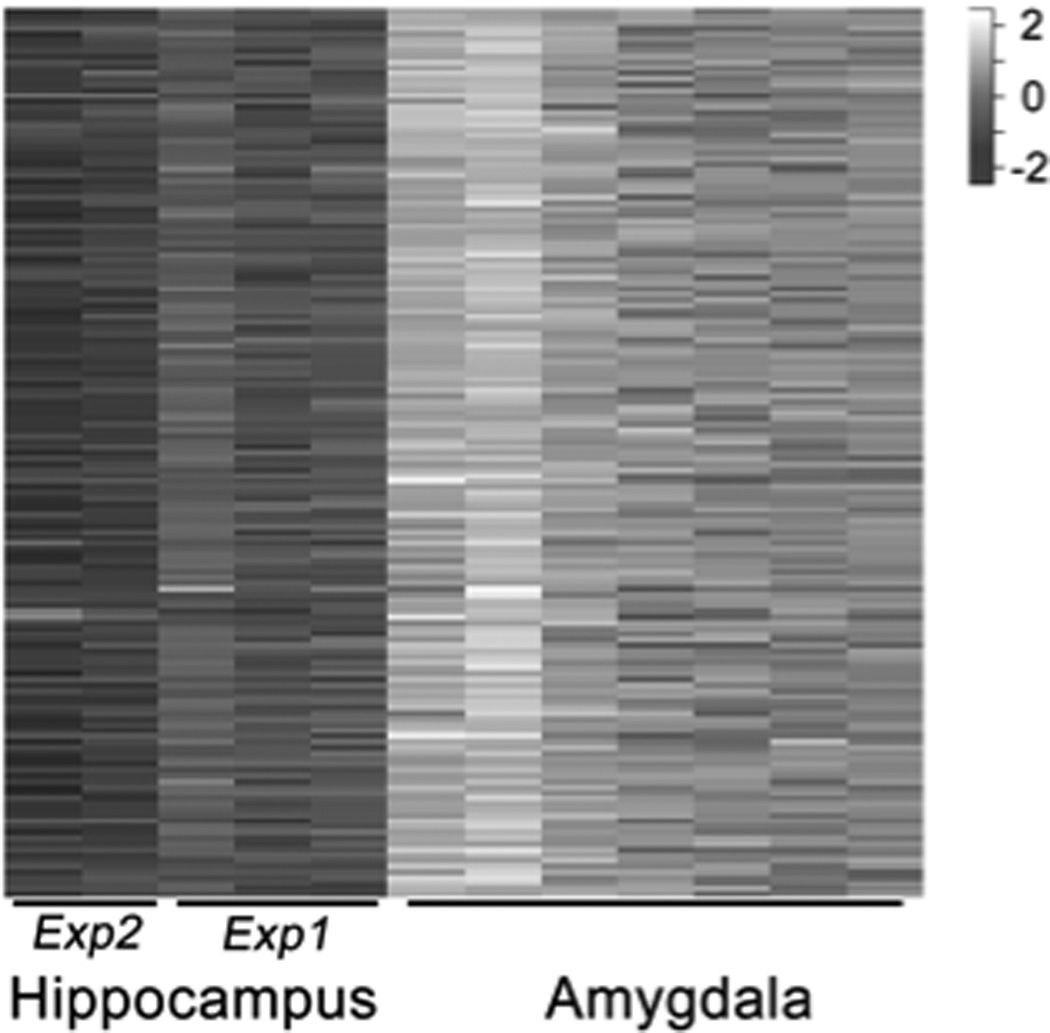
Samples from two studies on the hippocampus show lower gene expression when compared to samples from amygdala. Within each tissue type, coherent changes in expression are also apparent.
Here, we describe a novel approach to identifying candidate gene sets using new criteria for selecting coherent biclusters across multiple experiments somewhat related to the desired clinical application. Previous efforts have looked for coherent functional modules showing enrichment in a particular gene expression data set, often by incorporating network, pathway, or clinical information30–32. Our method differs from these approaches in that we identify gene sets showing coherent expression patterns across multiple related studies, and then assess the general relevance of our candidate sets by using them for gene set analysis of novel clinical data. In this sense, our work is closest to that of Liu et al.33, who find processes dysregulated across many related experiments. However, their work still requires pre-defined gene sets relevant to the phenotype being studied. The goal of our method is to systematically identify novel gene sets that generalize well for the analysis of new data in fields where molecular annotation is sparse, such as development or neuronal function. We use careful dataset selection, biclustering, and filtering to identify novel candidate gene sets, and we observe that several of these show coherent differential expression patterns in clinical test data sets from different yet related physiological processes. This method works even when the training data sets come from different tissues or species than the test data, allowing us to find clinically-applicable gene sets using existing data from model organisms. Several of the gene sets differentially expressed in the test data show enrichment for informative Gene Ontology terms, but many others have no significant overlap with previously known functional categories. Nonetheless, they can be useful as diagnostics and can help direct future translational research into gene-gene and gene-disease relationships, particularly in medical fields where the underlying molecular physiology is not yet well understood.
2. Methods
2.1. Algorithm overview
We start by integrating publicly available gene expression data from several studies that are related, but not too closely related, to each other and to the test data set we wish to analyze. We apply a biclustering algorithm that finds coherent changes within and across studies (Figure 1) to the combined training data. Subsequently, we filter out biclusters that do not meet certain quality criteria. We consider the remaining biclusters as candidate gene sets, which we use for the analysis of human clinical gene expression test data distinct from the data used for gene set discovery. Details of each of these steps in our method are discussed below.
2.2. Data acquisition and normalization
We downloaded single channel Affymetrix microarray data (as .CEL files) from the Gene Expression Omnibus (GEO) (Table 1). The Affymetrix CEL files for each medical area of interest were imported into the R statistical software (v2.8.1; http://www.R-project.org), and all training data for that area were normalized at once. Normalization was performed with the AffyPLM package in BioConductor (v2.4), using RMA background correction, quantile normalization, and the Tukey biweight summary method. After normalization, the variances of all probes were computed across all samples, and the 50% of the probes with the lowest variance were removed, eliminating probes that are not expressed in the relevant tissues or whose expression does not vary enough to be informative for our purposes.
Table 1.
Selected gene expression data sets for gene set discovery.
| Data Set | GEO Accession # |
Title | Tissue | Samples |
|---|---|---|---|---|
| Metabolic (Human) | GSE5090 | Polycystic ovary syndrome patients vs control subjects | Adipose | PCOS patients, controls |
| GSE9105 | Effect of acute physiologic hyperinsulinemia | Vastus lateralis | 240 mins of insulin infusion | |
| GSE474 | Obesity and fatty acid oxidation | Rectus abdominus and vastus lateralis | Lean, obese | |
| Developmental (Mouse) | GSE6882 | Embryonic ovary development | Ovary | Embryonic |
| GSE8065 | Early postnatal development of the small intestine | Intestine | Postnatal | |
| GSE12769 | Testis developmental time course | Testis | Postnatal | |
| GSE13103 | Early mouse embryo eye development | Optic fissure | Embryonic | |
| Neuronal (Mouse) | GSE9803 | Striatal gene expression data | Striatum | wild-type |
| GSE4040 | Gene expression in murine hippocampus | Hippocampus | wild-type | |
| GSE4034 | Gene expression in amygdala and hippocampus | Amygdala, Hippocampus | wild-type |
2.3. Biclustering
Next, we biclustered the normalized, filtered gene expression data using the Iterative Signature Algorithm (ISA)27,34. We have found that ISA identifies more coherent and potentially biologically relevant biclusters than several other biclustering methods35,36. Briefly, ISA starts with a random initial set of genes. All samples are scored for coherence with respect to this gene set and samples are chosen for which the score exceeds a predefined condition threshold (tC). Next, all genes are scored across the selected samples and a new set of genes is selected based on a predefined gene threshold (tG). The entire procedure is repeated until it converges. We used the BiCAT implementation35 of the ISA algorithm with tG = 2 and tC =1, parameters recommended for the identification of coherent patterns in a prior study37.
2.4. Selecting biclusters as candidate gene sets
Although we chose the ISA biclustering approach because the algorithm is able to find coherent biclusters that include samples from multiple experiments, there is no guarantee that the resulting biclusters have the generalizable-coherence property that we want for our candidate gene sets. In addition, ISA often identifies multiple overlapping biclusters. While some degree of overlap between gene sets might accurately represent genes involved in more than one cellular process, a high degree of overlap of both genes and samples likely occurs when different random starting points of the iterative algorithm converge to similar solutions. Additionally, some of the resulting biclusters can be noisy and their genes’ expression patterns only poorly correlated with each other. Therefore, we subject the biclusters to several quality measures before selecting certain ones as candidate gene sets.
First, we remove any biclusters that do not show coherent expression changes across samples from two or more experiments. That is, if the samples selected for a bicluster do not come from at least two different source data sets, we discard the gene set as being less likely to generalize to new conditions and tissues. Our experience suggests that this criterion, given an appropriate choice of training data, is most responsible for the applicability of these discovered gene sets in new contexts (data not shown).
We next assess the overlap between the gene sets defined by the biclusters. If any pair of gene sets G and H overlap such that at least 80% of the genes in G are in H and at least 80% of the genes in H are in G, we select only the bicluster with fewer genes. We reason that the smaller bicluster contains a core group of genes with a stronger functional association with the phenotype.
To enforce expression homogeneity within the biclusters, we use a recently proposed measure of bicluster quality, the average correlation value (ACV)38, to score biclusters for homogeneity. The ACV measures the average pairwise expression correlation between all pairs of genes in a cluster. The maximum ACV score of 1.0 denotes a highly correlated bicluster. ACV has been shown to be more robust than the widely-used mean squared residue score25. We discard biclusters with ACV < 0.5 (though results are quite robust to varying this threshold). Biclusters that remain after all of these filtering steps are considered as candidate gene sets.
Finally, we note that normalization in meta-analyses is an important challenge, since many experiment-specific factors may persist even after normalization, and over-normalization may suppress real signal. In order to assess normalization bias in our resulting biclusters, we calculate a score called the chip correlation value (CCV). The CCV is measured by calculating the correlation between sample averages for genes in a given bicluster with the sample averages over the entire gene expression matrix. Although biclusters are not discarded based on their CCV scores, it should be noted that extreme correlations might reflect insufficient normalization.
2.5. Applying candidate gene sets to analyze test data
If our novel gene sets show coherent expression changes in a new setting, we can assume that their genes have some functional relationship, even if the exact nature of that relationship is unknown. Any gene-set data analysis method can be applied to assess coherent expression changes in test data; here, we choose Gene Set Enrichment Analysis (GSEA)16. GSEA is a statistical framework that determines if members of a given gene set show collective expression changes linked to sample phenotypes by calculating a Kolmogorov-Smirnov running sum called the enrichment score (ES). We report the normalized enrichment score (NES) because this measure accounts for the gene set size, thus allowing for comparison between different experiments. The magnitude of the NES reflects the degree of enrichment for a given gene set. We accept a gene set as differentially expressed using an FDR q-value cut-off of 25%, as suggested by the GSEA authors16. For time series data (the developmental data sets), we used the Pearson metric for ranking genes. For the maternal blood data set20 (see Results), we used the GSEA-preranked option on genes ranked by the closer-to-zero (i.e., approximately the less-significant) of two t-scores, one comparing paired antepartum and postpartum maternal blood samples, and the other comparing paired neonatal cord blood and postpartum maternal blood samples.
Subsequently, in order to gain biological insight into the biclusters, we used the Database for Annotation, Visualization and Integrated Discovery (DAVID)39,40 (the April, 2008 release) to identify functional annotation terms significantly over-represented in the gene sets. A functional term is considered to be significantly enriched if its Benjamini-Hochberg-adjusted p-value, as reported by DAVID, is less than 0.05.
2.6. Orthology
In some cases, we derived biclusters based on gene expression data in model organisms and evaluated their utility for interpreting human gene expression data from clinical samples. In these cases, mouse-derived biclusters were mapped to their human gene symbols using DAVID’s Gene ID Conversion Tool. Further, probe sets from human Affymetrix Chips are collapsed to their gene symbols using GSEA. In such cases, the gene symbols are used instead of their Affymetrix probe set identifiers.
3. Results
We applied this approach to three different functional areas to highlight its utility for functional interpretation of clinical data. We start by applying our method to the well-studied metabolic field and follow with two other areas where annotation is relatively sparse: neuronal function and development. Table 2 summarizes the characteristics of the resulting biclusters from each field.
Table 2.
Characterization of resulting biclusters.
| Study | # of genes | # of conditions | ACV | CCV | ||||
|---|---|---|---|---|---|---|---|---|
| min | mean | max | min | mean | max | mean ± stdev | mean ± stdev | |
| Metabolic | 12 | 63.8 | 154 | 5 | 10.1 | 17 | 0.74 ± 0.12 | −0.23 ± 0.30 |
| Neuronal | 7 | 122.6 | 436 | 3 | 9.0 | 19 | 0.95 ± 0.03 | 0.12 ± 0.49 |
| Developmental | 4 | 528.8 | 893 | 6 | 9.1 | 12 | 0.94 ± 0.03 | 0.07 ± 0.37 |
3.1. Metabolic data set
Metabolic disorders include a broad array of medical conditions such as diabetes, obesity, hypertension, and insulin resistance. We compiled gene expression data from publicly available metabolic studies involving human tissue samples hybridized to Affymetrix GeneChip HG-U133A arrays. The initial experiments include adipose tissue samples from polycystic ovary syndrome (PCOS) patients compared with control subjects (GSE5090), vastus lateralis muscle samples during acute physiologic hyperinsulinemia (GSE9105), and vastus lateralis muscle samples from obese and lean subjects. PCOS is a common endocrine disorder that is associated with metabolic abnormalities including insulin resistance, increased risk for diabetes mellitus, obesity and hyperlipidemia41.
The entire metabolic data set consisting of 53 samples and 11,141 genes was used as input for biclustering. Overall, ISA identified 15 biclusters for the metabolic data. Filtering resulted in 11 biclusters selected as candidate metabolic gene sets. One bicluster was discarded based on low ACV; three biclusters were filtered because of high degree (>80%) of overlap (Figure 2). In such cases, the biclusters with fewer genes were selected because they were likely to be more specific. On average, the selected biclusters contain 64 genes and 10 conditions with more than 73% correlation between genes. Further, average CCV is relatively low (−0.23 ± 0.3) suggesting that the clusters are not due to normalization artifacts (Table 2).
Figure 2. Metabolic bicluster overlap before filtering.
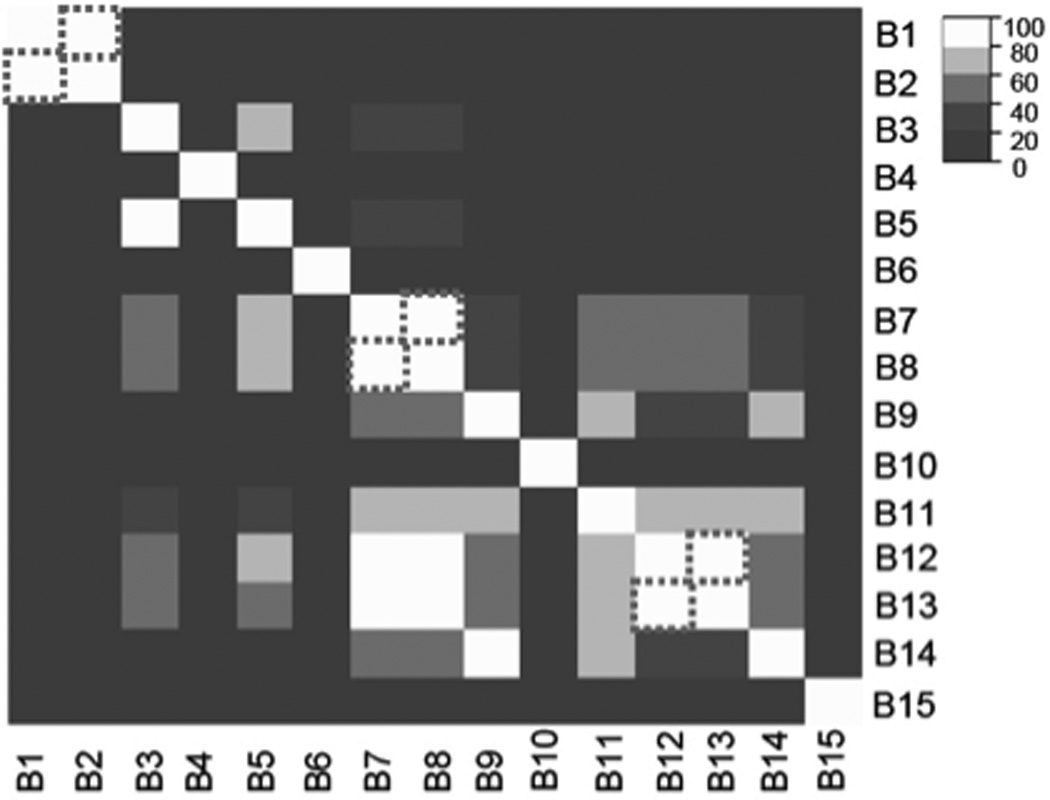
A heatmap of overlap between biclusters from the metabolic study is shown. Biclusters with >80% overlap with each other are outlined in dashed boxes. In such cases, the bicluster with fewer genes is chosen as a candidate gene set. Note that biclusters 7 and 13 are both retained because the high overlap is in one direction only. In such cases, it is possible that both gene sets represent interesting biological functions.
We then applied these candidate metabolic gene sets in a GSEA analysis of data from Mootha, et al. comparing smooth muscle gene expression in diabetic patients and healthy controls7. Recall that this is the data set that was first used to demonstrate the GSEA approach; there are no individually differentially expressed genes, and gene sets related to oxidative phosphorylation were shown to be downregulated in diabetics in this data. However, no gene sets were shown to be significantly upregulated in diabetes7. In our experiments on the same data, out of our eleven candidate biclusters, three were significantly upregulated (FDR q-value < 0.25) in smooth muscle from diabetic patients: bicluster9, bicluster11 and bicluster14. The GSEA results for differential expression of these gene sets are summarized in Table 3A, and full functional enrichment results are listed in supplementary table S1.
Table 3.
Differential expression of candidate gene sets in test data.
| A) Metabolic biclusters | |||||||
| Species | Tissue | Bicluster # | # of genes | ES | NES | NOM p- val |
FDR q-val |
| Homo Sapiens | Smooth Muscle |
Bicluster14 | 31 | 0.57 | 1.71 | 0.01 | 0.08 |
| Bicluster9 | 39 | 0.54 | 1.64 | 0.03 | 0.07 | ||
| Bicluster11 | 32 | 0.50 | 1.60 | 0.04 | 0.08 | ||
| B) Neuronal biclusters | |||||||
| Species | Tissue | Bicluster # | # of genes | ES | NES | NOM p- val |
FDR q-val |
| Homo Sapiens | Dorsolateral prefrontal cortex |
Bicluster4 | 128 | 0.65 | 1.52 | 0.00 | 0.21 |
| Bicluster12 | 65 | 0.53 | 1.39 | 0.05 | 0.22 | ||
| Bicluster1 | 197 | 0.58 | 1.38 | 0.13 | 0.19 | ||
| Bicluster3 | 219 | 0.51 | 1.37 | 0.10 | 0.17 | ||
| C) Developmental biclusters | |||||||
| Species | Tissue | Bicluster # | # of genes | ES | NES | NOM p- val |
FDR q-val |
| Homo Sapiens | Blood | Bicluster4 | 239 | 0.31 | 1.54 | 0.000 | 0.005 |
In an attempt to interpret the functional role of these gene sets, we evaluated the enriched biclusters using functional annotation tools in DAVID. However, these differentially expressed biclusters either showed no statistically significant overlap with current ontology classes (bicluster11) or overlapped only with broad GO terms such as developmental process (bicluster14) or multicellular organismal process and biological regulation (bicluster9).
We had originally expected that any gene sets we discovered in our metabolic data would overlap heavily with existing functional annotation, reflecting the wealth of research about the molecular mechanisms of diabetes and obesity. However, we instead discovered new gene sets that exhibited coherent changes across diverse experiments and that also showed significant coordinated upregulation in diabetics. While the exploratory q-value cutoff suggested for GSEA analysis16 allows for a one-in-four false-positive rate, all three of the gene sets identified in this analysis had much lower q-values. Thus, although any of these findings might be a false-positive, it is unlikely (probability ≤ 0.0005) that all three of them are. We believe these results suggest that there may be previously unrecognized functional links among the members of each of these gene sets, warranting further study. In clinical applications where diagnosis is difficult or early diagnosis is critical, such gene sets might also be useful as diagnostic tools even before their functional roles are understood.
3.2. Neuronal data set
Motivated by an interest in the impact of loss of nicotinic activity on cochlear synapse formation42, we collected gene expression data from substructures of the mouse central nervous system: striatum (GSE9803), hippocampus (GSE4040) and amygdala (GSE4034). Gene expression data from only wild-type mice were considered and all studies utilized Affymetrix Mouse430.2 GeneChips. This neuronal data set included 32 samples and 22,550 genes. ISA initially identified 33 biclusters for the neuronal data42; filtering resulted in 25 candidate neuronal gene sets, whose characteristics are summarized in Table 2.
We applied the neuronal candidate gene sets to analyze human gene expression data from postmortem brains (specifically, dorsolateral prefrontal cortex) of adults with Down syndrome (DS) and healthy control subjects (GSE5390). In this data set bicluster4, bicluster12, bicluster1 and bicluster3 were upregulated in DS patients (Table 3B).
Bicluster4 showed statistically significant enrichment for the GO biological process term, lipid metabolic process, and several PANTHER terms including lipid, fatty acid and steroid metabolism; mRNA transcription regulation; voltage-gated K channel; and transferase. Bicluster1 is enriched for several GO categories including nervous system development, myelination, and regulation of action potential. Enriched GO terms for bicluster 3 include developmental process, localization, cell adhesion and death. Enriched PANTHER categories for this bicluster include neuronal activities, receptor mediated endocytosis, cytoskeletal protein, cell junction protein, and cadherin. On the other hand, bicluster12 did not exhibit statistically significant overlap with any functional annotation terms.
Cadherins are proteins involved in calcium-ion-mediated cell adhesion. Abnormalities in myelination, cell adhesion, and lipid classes have been implicated in DS43–45. In addition, these results are consistent with our recent observation of increased oxidative stress, and apparent downstream disruption of ion signaling and cell structural integrity, in the DS fetus46. The functional roles of genes in these novel gene sets mined from diverse neuronal tissues in healthy mice may therefore help inform ongoing translational efforts to develop novel therapies for Down syndrome.
3.3. Developmental data set
We collected gene expression data representing mouse developmental time courses in various tissues, all hybridized to Affymetrix Mouse430.2 GeneChips. We only considered data from wild-type animals; treated samples and mutant strains were excluded. The data were derived from ovary (GSE6882) and optic fissure (GSE13103) during embryonic development, and intestine (GSE8065) and testis (GSE12769) during postnatal development. Overall, this data set contained 24 samples and 22,550 genes.
Initially, ISA identified 25 biclusters on this data set. Filtering resulted in 10 biclusters to be considered as candidate developmental gene sets, which are characterized in Table 2. We then applied these developmental biclusters to re-analyze expression data from our previous study of maternal and fetal gene expression20. This study confirmed the detection of fetal mRNA in maternal whole blood by SNP analysis after identifying candidate fetal transcripts that were upregulated in both antepartum maternal blood (at 37–40 weeks’ gestation) and umbilical cord blood compared to postpartum maternal blood. We used the GSEA “preranked” feature so that we could rank the genes based on their less significant performance in these two different comparisons (antepartum to postpartum, and antepartum to neonatal; see Methods).
In this analysis we found that developmental bicluster4 (Table 3C) was significantly upregulated (FDR q-value < 0.005) in both the antepartum mothers and the babies’ cord blood compared to the postpartum mothers, and therefore would be considered likely to include fetal transcripts in maternal circulation. Bicluster4 showed statistically significant overrepresentation of several GO terms, including digestion, lipid transport, and lipid binding. SP_PIR (Protein Information Resource) terms such as intestine, glycoprotein, neuropeptide, and inflammatory response were also overrepresented. Given that myelin membrane synthesis relies upon lipid and sterol metabolism47, expression of these genes may reflect the maturing neurological system of the near term fetus, necessary for coordinating the complex sequence of actions needed for feeding and breathing; or it may simply reflect direct preparation for digestion. In our previous analysis of this data20, we saw evidence of putative fetal expression of genes related to several functional processes likely to be needed at birth: immunity, sensory perception, lung maturation, and neurological function. However, no functional over-representation of digestive or metabolic proteins was detected as a set. Indeed, a painstaking manual annotation effort revealed hints that such proteins were among the likely fetal transcripts, but their significance was unclear. In contrast, the present work likely suggests that the healthy term fetus is preparing to feed.
The fact that such transcripts are detectable in maternal circulation helps support the proposal to use transcriptional analysis of maternal blood as a non-invasive approach to monitor fetal development. Translational applications of this work might include detecting potential feeding disorders before birth by identifying dysregulation of this gene set in individual fetuses.
4. Discussion
4.1 Implications
Our understanding of functional relationships among sets of genes is still in its infancy. Discovery of coherent gene sets that work together in different biological processes or disease states may help further annotate genomes by assigning function to unknown genes or discovering previously unsuspected relationships. Our method allows us to identify gene sets likely to have a common functional role in a given tissue or disease state. We found that many candidate gene sets selected in this way show statistically significant differential expression in new test data sets, suggesting that such gene sets may generalize well across tissues and relevant disease states.
Many gene set discovery methods rely upon annotation tools that utilize ontology or pathway databases. A potential issue with such functional enrichments is the dependency of p-values on bicluster sizes48. Smaller yet functionally-relevant biclusters may go unnoticed due to their insignificant enrichment p-values. Our approach of searching for coherent biclusters spanning conditions from multiple experiments allows us to extract biological phenotype features that generalize well across different tissues and species, even in the absence of enrichment for known functional pathways. Thus, this approach may be a way to generate a collection of gene sets relevant to the analysis of data from novel areas, where existing functional annotation is relatively incomplete.
The question of whether the enriched biclusters exhibit known functional coherence is itself of interest. The rationale behind using metabolic disease samples in our first experiment was to determine whether our method would capture meaningful functional annotation in a field where such annotation is relatively plentiful. Although one metabolic bicluster (Bicluster4) was enriched for expected metabolic terms such as UDP-glycosyltransferase activity and carbohydrate metabolism (Supplemental Table S1), we found several metabolic gene sets that were not statistically enriched for any informative pathway terms. This lack of enrichment may be due to the relatively small size of the metabolic biclusters. Importantly, despite the lack of enrichment, several of these biclusters were significantly differentially expressed in the test data. Furthermore, inspection of these biclusters revealed several genes with previously assigned roles in metabolic disorders. For example, consider bicluster9, which we found to be significantly upregulated in smooth muscles of diabetic individuals. The Phenopedia49 component of the Human Genome Epidemiology database (HuGE Navigator)50 suggests that several of the genes in this bicluster, including ADRA1A, ADRB1, APOC3, CACNA1A, MTHFR and TH, are disease susceptibility genes associated with cardiovascular diseases and obesity. However, no previous relationship between most of these genes was detected in the literature. These results suggest that our approach may help capture novel links among genes and between genes and phenotypes.
Equally important, several of our test data sets were from a different species than that of the original data used for biclustering. This is particularly important for biological processes such as development that rely on mammalian model systems. For example, for the developmental data set, candidate gene sets were acquired from several murine tissues: ovary, intestine, testis and optic fissure. Yet, orthologous gene sets were found to be upregulated during human development. Similarly, neuronal biclusters derived from mouse brain tissues provided information about expression in the dorsolateral prefrontal cortex of Down syndrome patients.
4.2 Future work
Future work will include obtaining a wider range of gene sets based on larger collections of training data, and exploring the impact of varying training set size or other parameters. Biclusters identified with ISA depend on the initially chosen set of genes and the threshold parameters tG and tC. By varying the threshold parameters and running ISA with different initial conditions, it is possible to generate a representative set of biclusters and to determine the method’s sensitivity to these changes. Additionally, it is preferable to identify smaller biclusters that consist of tightly linked genes. This goal can be realized by either refining our smaller discovered biclusters or by clustering the larger ones into smaller subsets. The impact of using different biclustering methods should also be explored further. To expand the training data sets, integration of data from different microarray platforms and multiple species, though non-trivial, is feasible51,52 and desirable. Furthermore, it is important to determine how best to select training data to facilitate discovering new gene sets for the analysis of particular test data sets. Future work might explore the effectiveness of this approach as a function of, for example, distances between MeSH terms describing the training and test data. Finally, future experiments are needed to identify and validate new functional relationships between genes that are suggested by our results.
Supplementary Material
Acknowledgments
This work was supported by R01HD058880 of the National Institutes of Health to DKS.
Contributor Information
Sevin Turcan, Department of Biomedical Engineering, Tufts University, 4 Colby St., Medford, MA, 02155, USA.
Douglas E. Vetter, Department of Neuroscience, Tufts School of Medicine, Boston, MA 02111, USA
Jill L. Maron, Department of Pediatrics, Tufts Medical Center, 800 Washington St., Boston, MA 02111, USA
Xintao Wei, Department of Computer Science, Tufts University, 161 College Ave., Medford, MA, 02155, USA.
Donna K. Slonim, Department of Computer Science, Tufts University, 161 College Ave., Medford, MA, 02155, USA
References
- 1.Iyer VR, et al. Science. 1999 Jan 1;283:83. [Google Scholar]
- 2.Wen X, et al. Proc Natl Acad Sci U S A. 1998 Jan 6;95:334. doi: 10.1073/pnas.95.1.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeeberg BR, et al. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hosack DA, Dennis G, Jr, Sherman BT, Lane HC, Lempicki RA. Genome Biol. 2003;4:R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khatri P, Draghici S, Ostermeier GC, Krawetz SA. Genomics. 2002 Feb;79:266. doi: 10.1006/geno.2002.6698. [DOI] [PubMed] [Google Scholar]
- 6.Khatri P, Draghici S. Bioinformatics. 2005 Sep 15;21:3587. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mootha VK, et al. Nat Genet. 2003 Jul;34:267. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 8.Hung JH, et al. Genome Biol. 2010;11:R23. doi: 10.1186/gb-2010-11-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barry WT, Nobel AB, Wright FA. Bioinformatics. 2005 May 1;21:1943. doi: 10.1093/bioinformatics/bti260. [DOI] [PubMed] [Google Scholar]
- 10.Tian L, et al. Proc Natl Acad Sci U S A. 2005 Sep 20;102:13544. doi: 10.1073/pnas.0506577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goeman JJ, van de Geer SA, de Kort F, van Houwelingen HC. Bioinformatics. 2004 Jan 1;20:93. doi: 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- 12.Kong SW, Pu WT, Park PJ. Bioinformatics. 2006 Oct 1;22:2373. doi: 10.1093/bioinformatics/btl401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mansmann U, Meister R. Methods Inf Med. 2005;44:449. [PubMed] [Google Scholar]
- 14.Dinu I, et al. BMC Bioinformatics. 2007;8:242. doi: 10.1186/1471-2105-8-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goeman JJ, Buhlmann P. Bioinformatics. 2007 Apr 15;23:980. doi: 10.1093/bioinformatics/btm051. [DOI] [PubMed] [Google Scholar]
- 16.Subramanian A, et al. Proc Natl Acad Sci U S A. 2005 Oct 25;102:15545. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashburner M, et al. Nat Genet. 2000 May;25:25. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa M, Goto S. Nucleic Acids Res. 2000 Jan 1;28:27. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karp PD, et al. Nucleic Acids Res. 2005;33:6083. doi: 10.1093/nar/gki892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maron JL, et al. J Clin Invest. 2007 Oct;117:3007. doi: 10.1172/JCI29959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisen MB, Spellman PT, Brown PO, Botstein D. Proc Natl Acad Sci U S A. 1998 Dec 8;95:14863. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamayo P, et al. Proc Natl Acad Sci U S A. 1999 Mar 16;96:2907. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kankainen M, Brader G, Toronen P, Palva ET, Holm L. Nucleic Acids Res. 2006;34:e124. doi: 10.1093/nar/gkl694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartigan J. J. Am. Stat. Assoc. 1972;67:123. [Google Scholar]
- 25.Cheng Y, Church GM. Proc Int Conf Intell Syst Mol Biol. 2000;8:93. [PubMed] [Google Scholar]
- 26.Madeira SC, Oliveira AL. IEEE/ACM Trans Comput Biol Bioinform. 2004 Jan-Mar;1:24. doi: 10.1109/TCBB.2004.2. [DOI] [PubMed] [Google Scholar]
- 27.Ihmels J, Bergmann S, Barkai N. Bioinformatics. 2004 Sep 1;20:1993. doi: 10.1093/bioinformatics/bth166. [DOI] [PubMed] [Google Scholar]
- 28.Tanay A, Sharan R, Kupiec M, Shamir R. Proc Natl Acad Sci U S A. 2004 Mar 2;101:2981. doi: 10.1073/pnas.0308661100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gan X, Liew AW, Yan H. BMC Bioinformatics. 2008;9:209. doi: 10.1186/1471-2105-9-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dittrich MT, Klau GW, Rosenwald A, Dandekar T, Muller T. Bioinformatics. 2008 Jul 1;24:i223. doi: 10.1093/bioinformatics/btn161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keller A, et al. Bioinformatics. 2009 Nov 1;25:2787. doi: 10.1093/bioinformatics/btp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ulitsky I, Shamir R. Comput Syst Bioinformatics Conf. 2008;7:249. [PubMed] [Google Scholar]
- 33.Liu M, et al. PLoS Genet. 2007 Jun;3:e96. doi: 10.1371/journal.pgen.0030096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ihmels J, et al. Nat Genet. 2002 Aug;31:370. doi: 10.1038/ng941. [DOI] [PubMed] [Google Scholar]
- 35.Barkow S, Bleuler S, Prelic A, Zimmermann P, Zitzler E. Bioinformatics. 2006 May 15;22:1282. doi: 10.1093/bioinformatics/btl099. [DOI] [PubMed] [Google Scholar]
- 36.Wei X. PhD Dissertation. Computer Science, Tufts University; 2010. [Google Scholar]
- 37.Cheng KO, Law NF, Siu WC, Liew AW. BMC Bioinformatics. 2008;9:210. doi: 10.1186/1471-2105-9-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teng L, Chan L. Journal of Signal Processing Systems. 2007;50:267. [Google Scholar]
- 39.Dennis G, Jr, et al. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 40.Huang da W, Sherman BT, Lempicki RA. Nat Protoc. 2009;4:44. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 41.Urbanek M, Sam S, Legro RS, Dunaif A. J Clin Endocrinol Metab. 2007 Nov;92:4191. doi: 10.1210/jc.2007-0761. [DOI] [PubMed] [Google Scholar]
- 42.Turcan S, Slonim DK, Vetter DE. PLoS One. 2010;5:e9058. doi: 10.1371/journal.pone.0009058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wisniewski KE, Schmidt-Sidor B. Clin Neuropathol. 1989 Mar-Apr;8:55. [PubMed] [Google Scholar]
- 44.Lubec G, et al. J Neural Transm Suppl. 1999;57:161. doi: 10.1007/978-3-7091-6380-1_10. [DOI] [PubMed] [Google Scholar]
- 45.Brooksbank BW, Martinez M. Mol Chem Neuropathol. 1989 Dec;11:157. doi: 10.1007/BF03160049. [DOI] [PubMed] [Google Scholar]
- 46.Slonim DK, et al. Proc Natl Acad Sci U S A. 2009 Jun 9;106:9425. doi: 10.1073/pnas.0903909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verheijen MH, et al. Proc Natl Acad Sci U S A. 2009 Dec 15;106:21383. doi: 10.1073/pnas.0905633106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li G, Ma Q, Tang H, Paterson AH, Xu Y. Nucleic Acids Res. 2009 Aug;37:e101. doi: 10.1093/nar/gkp491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu W, Clyne M, Khoury MJ, Gwinn M. Bioinformatics. 2009 Oct 30; doi: 10.1093/bioinformatics/btp618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu W, Gwinn M, Clyne M, Yesupriya A, Khoury MJ. Nat Genet. 2008 Feb;40:124. doi: 10.1038/ng0208-124. [DOI] [PubMed] [Google Scholar]
- 51.Shi L, et al. Nat Biotechnol. 2006 Sep;24:1151. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsai J, et al. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-11-software0002. SOFTWARE0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.