


Abstract
We develop a statistical tool SNVer for calling common and rare variants in analysis of pooled or individual next-generation sequencing (NGS) data. We formulate variant calling as a hypothesis testing problem and employ a binomial–binomial model to test the significance of observed allele frequency against sequencing error. SNVer reports one single overall P-value for evaluating the significance of a candidate locus being a variant based on which multiplicity control can be obtained. This is particularly desirable because tens of thousands loci are simultaneously examined in typical NGS experiments. Each user can choose the false-positive error rate threshold he or she considers appropriate, instead of just the dichotomous decisions of whether to ‘accept or reject the candidates’ provided by most existing methods. We use both simulated data and real data to demonstrate the superior performance of our program in comparison with existing methods. SNVer runs very fast and can complete testing 300 K loci within an hour. This excellent scalability makes it feasible for analysis of whole-exome sequencing data, or even whole-genome sequencing data using high performance computing cluster. SNVer is freely available at http://snver.sourceforge.net/.
INTRODUCTION
The past few years have seen a dramatic development in sequencing technology, which has made the per-base cost of DNA sequencing plummet by ~100 000-fold over the past decade (1). Because of the affordable cost and high digital resolution, the new or ‘next-generation’ sequencing (NGS) technology is replacing the traditional hybridization-based microarray technology in many applications (2). For genetics studies, NGS holds the promise to revolutionize genome-wide association studies (GWAS). The recently completed phase of GWAS mainly addresses common SNPs with Minor allele frequency (MAF) >5%, based upon the common disease/common variant (CD/CV) hypothesis (3). However, the identified common variants explain only a small proportion of heritability (4). Rare variants therefore have been hypothesized to account for the missing heritability (5,6). To identify rare variants, a direct and more powerful approach is to sequence a large number of individuals (7). This line of thought also implicitly motivates the recent 1000 Genomes Project, which will sequence the genomes of 1200 individuals of various ethnicities by NGS (8). It is expected to extend the catalog of known human variants down to a frequency ~1%.
Although the cost of whole-genome or exome sequencing of all enrolled subjects is prohibitively high now, such studies will eventually be carried out in a manner similar to GWAS with very large sample sizes (9). While the cost is being brought down to as low as $1000 for sequencing a whole genome (10), in the interim, a cost-effective strategy has to be taken in order to take the full advantage of NGS. Such issues with cost and labor are not new as similar problems were confronted in the early expensive stage of GWAS and were circumvented by focusing on small candidate regions and the use of pooling of genomic DNA (11,12). Borrowing the same idea, many targeted re-sequencing applications utilizing pooling have been seen in the past few years (13–16).
The first-step analysis of NGS data for genetics study is often to identify genomic variants among sequenced samples. Quite a few SNP calling tools have been implemented to identify SNPs from sequencing of individual genomes. SNP calling is a relatively straightforward problem in analysis of sequencing data of individual genomes, because the frequency of a candidate allele can be only 0 (non-variant), 0.5 (heterozygous) or 1 (alternate homozygous) for a diploid genome. Despite (high) sequencing error of NGS, a reliable call can be easily made given a high depth of coverage, say 20× to 30×. Consequently, statistical models for SNP calling have been developed and integrated as one simple functional module in many NGS short reads analysis tools such as SAMtools (17), MAQ (18), GATK (19) and VarScan (20). SAMtools and MAQ use a Bayesian statistical model to compute the posterior probabilities of the three possible genotypes. Specifically, for the likelihood part, they employ a binomial distribution to characterize sampling of the two haplotypes, and the prior probability, like other Bayesian approaches, is pre-specified. SAMtools and MAQ empirically set the prior probability of observing a heterozygote to be 0.001 for the discovery of new SNPs, and 0.2 for inferring genotypes at known SNP sites. A similar Bayesian algorithm is used by GATK followed by sophisticated filtering. Such Bayesian approaches may not be ideal for multiplicity control because of the subjectivity of assigning the prior probability. VarScan implements a heuristic/statistical method. For each candidate site, it applies several heuristic filters such as having a minimum number of supporting reads and allele frequency reaching a minimum threshold. It also conducts a Fisher's exact test for testing the deviation of the read counts supporting variant alleles from being generated because of sequencing error. Those heuristic filters overlap with the Fisher's exact test in terms of reducing false positives. When not systematically considered, they may distort the statistics distribution under null and thus void the resultant P-values for multiplicity control. The variant call program we develop here is based on a frequentist approach, which will systematically consider all relevant factors and output P-values valid for multiplicity control.
Identifying SNPs from pooled NGS data is more challenging in that pooled DNA is sampled from a number of individuals, which consequently will give rise to variant allele frequencies other than simply 0, 0.5 or 1. Driven by the need for analysis of increasing amount of pooled NGS data, several programs/methods for the detection of variants from the pooled data have been developed. SNPSeeker employs the large deviation theory for SNP detection (21). It compares observed allele frequencies against the distribution of sequencing errors as measured by the Kullback Leibler (KL) distance (22). One limitation of this approach is that its error model has to be estimated from negative control data. SNPSeeker was recently extended to SPLINTER with two main improvements (23). First, it is capable of detecting rare short indels. Second, it provides a good cutoff after ranking all candidate variants to balance power and type I error rate, which, however, requires an additional positive control data. CRISP (24) models the number of reads of the reference and alternate alleles at a particular position across all pools as a contingency table, which is then tested by the Fisher's exact test. Its working hypothesis is that, due to rareness, presence of rare variants in all pools will be sporadic and then results in an excess of reads with the alternate allele as compared with the other pools, which is expected to be captured by the Fisher's exact test. CRISP then conducts a complementary test for the overabundance of alternate alleles within each pool against the sequencing error rate. Although it is shown that CRISP outperforms SNPSeeker, MAQ and VarScan (24), it has the following limitations. First, its working hypothesis does not hold well for common variants. When the MAF is large and/or the number of individuals in each pool is large, sporadic presence will disappear and result in no prominent excess of reads that can be captured by the Fisher's exact test. Second, their method is not applicable for single-pool data. Third, rareness and overabundance of alternate alleles are related but are captured separately using two different models, which may not be an efficient approach. In addition, these two separate tests make it hard to obtain an overall multiplicity control. Finally, its computational efficiency makes scalability an issue and may prevent its application in analysis of whole-exome or genome sequencing data. The main bottleneck comes from computing the P-value of a large number of contingency tables in the Fisher's exact test.
In addition to the above direct SNP calling programs, there are also other relevant studies for analysis of pooled NGS data, including estimating allele frequencies from pooled sequencing (25), evaluating the ability to detect rare SNPs (15) and investigating the power of variant detection in pooled DNA for NGS and the optimal pooling designs (26), among others. In this article, we develop a statistical tool SNVer (single nucleotide variant caller/seeker) for detecting variants in analysis of NGS data. SNVer is applicable to both pooled and individual data, and in particular it addresses the limitations that pre-existing methods have.
MATERIAL AND METHODS
Statistical models for single-pool data
For a genomic locus, let θ be its MAF in a population. If θ is larger than a threshold θ0 (θ > θ0), then we call it a single nucleotide polymorphism (SNP). Suppose that we sample N individuals (haploids) from this population for pooled sequencing. We assume that the number of individuals (n) carrying the minor allele follows a binomial distribution b(N, θ), namely,
with
Now we re-sequence this genomic region. Suppose that K short reads cover this locus, if no sequencing error, given n individuals carrying the minor allele, the number of minor alleles X we observe from the K short sequence reads follows also a binomial distribution b(K, n/N), namely:
with
Now we assume sequencing error rate to be ε, under which the minor allele will be flipped to one of the other three alternate alleles, and vice versa. So the observed X follows a binomial distribution 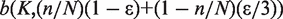 , namely,
, namely,
with
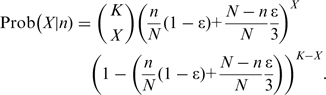 |
Since n is not observable, we sum it out and obtain the statistical model for X as
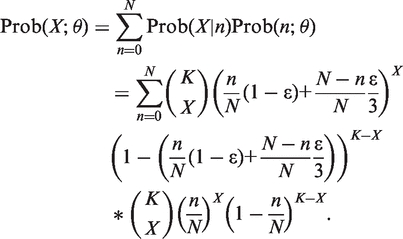 |
Now we consider the hypothesis test of whether this locus is a (rare) variant (θ > θ0)
Its significance P-value will be
Partial conjunction test for multiple-pool data
The above statistical model is for testing a locus in one single-pool data. For M pools, we propose to test it in each pool separately. We therefore obtain a set of M hypotheses for each candidate variant. The problem of making a variant call at one specific locus involves the simultaneous testing of hypotheses at the set level. Typical questions considered in the multiple-testing framework include: (i) Are all M hypotheses in the set true? (ii) Are all M hypotheses in the set false? (iii) Are at least u out of M hypotheses in the set false? These questions are referred to as conjunction test, disjunction test and partial conjunction test, respectively (27). Testing whether a locus is a variant based on multiple-pool data is equivalent to the partial conjunction test that at least u = 1 out of the M hypotheses for that locus is false. Let 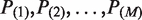 be the ordered P-values obtained from each single-pool test. Following (27), we employ the Simes method to calculate the pooled P-value for the partial conjunction test as
be the ordered P-values obtained from each single-pool test. Following (27), we employ the Simes method to calculate the pooled P-value for the partial conjunction test as
If the set of M null P-values at the tested locus are independent, Benjamini and Heller (27) show that p1/M is a valid P-value for testing the partial conjunction null. The Benjamini Hochberg (BH) procedure (28) and other multiple-test adjustments can then be applied to the pooled Simes’ P-values for multiplicity control when testing a large number of loci. It has been shown that this Simes–BH procedure controls the false discovery rate (FDR) at the pre-specified nominal level (27).
Data sets
Simulated data
We simulate synthetic data to investigate the numerical performances of our approach. For the single-pool scenario, a total of 10 000 data sets are generated under each combination of several conditions:
Sequencing coverage: low (10×) and high (30×).
Sequencing error: low (0.01) and high (0.05)
MAF: rare variants with θ ~ U(0.001, 0.01), less common variants with θ ~ U(0.01, 0.05) and very common variants θ ~ U(0.05, 0.5)
The number of sequenced individuals from low to high with N = 10, 20, 50, 100, 200, 500, 1000, 1500, 2000
For each MAF setting θ ~ U(θmin, θmax), we calculate the power of our approach for detecting variants by testing the null hypothesis H0: θ < θmin. Meanwhile, to demonstrate that type I error is controlled at the nominal level by our proposed test, we simulate θ ~ U(0, θmin), and evaluate how likely the same null hypothesis H0: θ < θmin will be rejected by mistake. For both power and type I error evaluations, we call a variant at the nominal level 0.05.
For the multiple-pool scenario, we follow the above single-pool simulation settings except that we simulate five pools with the same number of individuals in each pool and the total N = 10, 20, 50, 100, 200, 500, 1000, 1500, 2000.
Real data
We also assess the performance of our method in analysis of two pooled and one individual real NGS data sets as summarized in Table 1. The first one was an in-house Autism data set generated using ABI SOLiD platform from sequencing three genomic regions, denoted as Core, CDH9 and CDH10, of size 187, 158 and 158 kb, respectively, on chromosome 5 of the human genome. We made 24 pools with six individuals in each, totaling 144 samples. We have 12 pools for Autism case samples and the other half 12 pools for control samples. One case pool experiment failed and we therefore have 23 pools in total for analysis. We aligned short sequence reads by the Bioscope software from ABI SOLiD with default parameters. The mapped short sequence reads cover >96% of the three target regions with average 90× depth of coverage per individual. Meanwhile, we collected individual genotyping data for each sample, which were generated from Illumina HumanHap550v3 SNP arrays with approximately 550 000 markers. With individual genotyping data, we may calculate the concordance of identified variants between pooled sequencing data and individual genotyping data for evaluating variant call quality.
Table 1.
Summary of T1D and Autism pooled sequencing and ADHD individual sequencing data sets
| Disease | Platform | Total reads | Reads length | #Pool |
#Individual per pool | Region | Coverage per individual | |
|---|---|---|---|---|---|---|---|---|
| Case | Ctrl | |||||||
| Autism | SOLiD | ~402 M | 50 bp | 11 | 12 | 6 | ~503 kb | ~90× |
| T1D | 454 | ~9.4 M | ~250 bp | 10 | 10 | 48 | ~31 kb | ~80× |
| ADHD | Illumina | ~57 M | 76 bp × 2 | three individuals | ~38 Mb | ~20× | ||
The second data set was collected in a recent study of causative Type 1 Diabetes (T1D) variants (14). Exons and splice sites of 10 candidate genes were re-sequenced by the 454 sequencing system. Ten pooled samples each comprising equal amounts of DNA from 48 T1D patients and 10 pooled samples each comprising equal amounts of DNA from 48 healthy controls were made, totaling 480 T1D patients and 480 healthy controls from Great Britain. For each of the 20 pooled DNA samples, the numbers of produced short reads range from 281 270 to 579 102, with average length of 250 bases and 9 416 365 reads in total. We mapped these reads by BWA-SW (29) with default parameters and the average depth of coverage is 80× per individual.
The third one was an in-house individual sequencing data set. We performed paired end exome sequencing on three members affected with attention deficit/hyperactivity disorder (ADHD) in a pedigree, using the Illumina Genome Analyzer IIx platform with read lengths of 76 bp. It targets all human exonic regions totaling ~38 Mb. We aligned the short reads by BWA with default parameters and removed duplicates by picard (http://sourceforge.net/projects/picard/). These mapped and cleaned short reads were then re-aligned locally by the GATK IndelRealigner tool (30). The average depth of coverage is ~20× for each patient. Meanwhile, we also collected the genotyping data of these three patients, generated from the Illumina Human610-Quad version 1 SNP arrays with ~610 000 markers (including ~20 000 non-polymorphic markers).
For pooled sequencing data, CRISP has been shown to outperform other existing methods (24), so we focus on the comparison of our program with CRISP in performance evaluation. We also include SAMtools for comparison although it is not designed for pooled sequencing data. For the ADHD individual data, we compare SNVer with SAMtools and GATK. Variant positions were called and filtered by SAMtools with all default settings plus using awk ‘($3 = = “*” &$6> = 50) || ($3! =‘*’ &$6> = 20)’, as suggested by the SAMtools website. For the ADHD data, SAMtools with the suggested setting returned so many variants that we also report SAMtools results with an additional filtering −d20 to remove variant calls with sequencing coverage less than 20, for getting comparable numbers of variant calls as SNVer. We also called variants using the GATK UnifiedGenotyper, followed by further filtering based on the latest recommendations from the authors of GATK (see Supplementary Data for the detailed settings). SNVer utilizes SAMtools (17) to process and pile up mapped short reads. CRISP has its own pileup procedure integrated in its analysis pipeline. To make a fair comparison, following CRISP (24), we perform similar quality control and set the same processing parameters such as mapping quality and base quality filtering thresholds.
RESULTS
Power and type I error evaluations
The single-pool results are shown in Figure 1. We can see that our method can control type I error rate at the nominal level 0.05 in all settings. The number of sampled individuals (sample size) and the depth of coverage are both shown to be helpful in improving power. The largest improvement of ~10% attributed to depth of coverage (from 10× to 30×) is observed in the rare variants and high sequencing error (up–right panel). The improvement contributed by larger sample size keeps increasing at a decreasing rate until saturated. These power improvement curves would be helpful for pooling experiment design and provide guidance as to how to balance sample size (cost) and desired power. As expected, rare variants are much harder to be detected than common variants. A large sample size is required for achieving high power to detect them. Finally, higher sequencing error (0.05 versus 0.01) puts a small dent to power.
Figure 1.

Power (PW) and Type I error rate (Err) of SNVer using single-pool data at low (10×) and high (30×) coverage.
Figure 2 shows similar results for the multiple-pool scenario. Again, type I error rate is controlled at the nominal level 0.05. We also observe that given the same number of sequenced individuals, single-pool design yields a bit higher power with lower type I error rate in comparison with multiple-pool design, for example, 1000 individuals using one single pool versus five pools with 200 individuals in each. CRISP selects candidate SNPs by the Fisher's exact test, which is then followed by additional filtering steps. In the multiple-pool scenario, we show that the rankings of candidates SNPs by our test is superior to those by the Fisher's exact test employed by CRISP. To compare the efficiencies of these two rankings, we divide the 10 000 positives with θ ~ U(θmin, θmax) and 10 000 negatives with θ ~ U(0, θmin) into 100 groups, each with 100 positives and 100 negatives. These 200 loci are then ranked by their significance levels of testing the null H0: θ < θmin using our statistical models. Rankings based the Fisher's exact test are also generated. The area under the curve (AUC) score averaged over 100 groups is used to evaluate these two rankings as shown in Figure 3 for the typical scenario of 30× coverage and 0.05 sequencing error. We can see that the Fisher's exact test is very inefficient for detecting common and less common variants. CRISP therefore has to rely on additional sequencing error models to complement the Fisher's exact test for detecting common variants. We apply the BH procedure to control FDR at the nominal level of 0.1 and 0.05. As shown in Supplementary Table S1, the FDR for the Fisher's exact test is inflated, particularly dramatically for common and less common variants; SNVer controls the FDR very well. The number of sequenced individuals is modeled in our test and is shown to be helpful. This information is not explicitly utilized by CRISP in its Fisher's exact test and therefore contributes very little for detecting common and less common variants, although CRISP models it at the later filtering step.
Figure 2.

Power (PW) and Type I error rate (Err) of SNVer using multiple-pool data at low (10×) and high (30×) coverage.
Figure 3.

Ranking efficiency of the binomial models employed by SNVer versus the Fisher's exact test employed by CRISP.
The accuracy of allele frequency estimation has an impact on variant call, and is more critical for establishing association in genetics studies. Therefore we also plot the estimated MAF against the actual MAF when ε = 0.01 in Figure 4. For a moderate sample size of 250, we observe good concordance with correlation coefficients r2 = 0.9828 and r2 = 0.9318 for the single-pool design and the multiple-pool design, respectively. When the sample size increases to 1000, the concordance improves to r2 = 0.9955 and r2 = 0.9769 for the single- and the multiple-pool design, respectively. The lower concordance of the multiple design may be attributed to its additional between-pool variance. It also explains why singe-pool design yields fewer false positives than the multiple-pool design for the same set of samples.
Figure 4.

Correlation between the minor allele frequencies and its estimates in pooled sequencing.
Real data application
Better performance
The user of SNVer only needs to set the sequencing error rate ε and the variant threshold θ0. SNVer will then report the significance P-values of the tested loci of how likely their MAF θ < θ0. We assume ε = 0.01 for all real data sets. CRISP calls both rare and common variants, so we set θ0 = 0 for SNVer to compare their performance in calling variants. CRISP will output the variants it calls, while SNVer will report overall significance P-values for each locus, based on which the user can choose a threshold he/she feels appropriate and make variant calls. To make a comparison, we rank loci by their P-values output by SNVer and take the significance threshold that gives the same number of variants called by CRISP. The loci identified as variants by these two programs are then annotated by SeattleSeq (http://gvs.gs.washington.edu/SeattleSeqAnnotation/), and we count how many of them have been confirmed as variants in dbSNP. Following (30), we evaluate variant call quality by examining dbSNP rate, transition/transversion (Ti/Tv) ratio and concordance of sequencing and individual genotyping calls. A higher Ti/Tv ratio generally indicates a higher accuracy; this metrics is particularly helpful for assessing novel single nucleotide variant calls (30). The variant call results are summarized in Table 2. For the Autism and T1D pooled sequencing data sets, SNVer has the higher dbSNP rates, the higher overall Ti/Tv ratios and the higher Ti/Tv ratios for new sites, in comparison with CRISP. It indicates the better quality of the call sets SNVer produced. In contrast, SAMtools made much fewer SNP calls which led to much lower sensitivities, despite its higher Ti/Tv ratios. Out of the 110 SNPs that have been genotyped by SNP arrays in the Autism data set, SAMtools identified only 16 SNPs with 100% genotyping concordance, while both SNVer and CRISP called about 100 SNPs with 100% genotyping concordance. This confirms that SAMtools may not be appropriate for pooled sequencing data. The correlation between alternate allele frequencies in individually genotyped DNA samples and frequency estimates in the sequenced DNA pools is plotted in Figure 5, with r2 = 0.92 and r2 = 0.94 for the Autism case and control, respectively. The achieved 100% genotype concordance with less perfect frequency estimates is not surprising because accurate estimate of allele frequency θ is only critical for rare variants when testing θ > 0.
Table 2.
Comparison of SNP calling by CRISP, SAMtools, GATK and SNVer
| Data | No. of SNP |
Ti/Tva |
Concordanceb | |||||
|---|---|---|---|---|---|---|---|---|
| All | Known | Novel | dbSNP% | All | Known | Novel | TP/P (%) | |
| Autism (pooled) | ||||||||
| Case | ||||||||
| CRISP | 2182 | 1791 | 391 | 82.1 | 1.68 | 1.79 | 1.26 | 101/101 (100) |
| SNVer | 2182 | 1795 | 387 | 82.3 | 1.71 | 1.81 | 1.35 | 102/102 (100) |
| SAMtools | 261 | 260 | 1 | 99.6 | 2.26 | 2.29 | 0/1 | 16/16 (100) |
| Control | ||||||||
| CRISP | 2063 | 1610 | 453 | 78.0 | 1.68 | 1.83 | 1.27 | 96/96 (100) |
| SNVer | 2063 | 1617 | 446 | 78.4 | 1.78 | 1.89 | 1.45 | 95/95 (100) |
| SAMtools | 239 | 238 | 1 | 99.6 | 2.06 | 2.05 | 1/0 | 16/16 (100) |
| T1D (pooled) | ||||||||
| Case | ||||||||
| CRISP | 306 | 93 | 213 | 30.3 | 0.95 | 2.58 | 0.63 | N/A |
| SNVer | 306 | 126 | 180 | 41.2 | 1.71 | 2.15 | 1.47 | |
| SAMtools | 14 | 9 | 5 | 64.3 | 10/4 | 8/1 | 2/3 | |
| Control | ||||||||
| CRISP | 167 | 110 | 57 | 65.9 | 1.49 | 2.93 | 0.46 | |
| SNVer | 167 | 120 | 47 | 71.9 | 2.34 | 3.00 | 1.35 | |
| SAMtools | 18 | 12 | 6 | 66.7 | 14/4 | 11/1 | 3/3 | |
| ADHD (Individual) | ||||||||
| 84 060 | ||||||||
| SNVer | 18 001 | 17 535 | 466 | 97.4 | 2.89 | 2.89 | 2.73 | 4158/4183 (99.4) |
| SAMtools | 48 988 | 47 513 | 1475 | 97.0 | 2.66 | 2.68 | 2.16 | 4437/8116 (54.7) |
| SAMtools20× | 15 038 | 14 538 | 500 | 96.7 | 2.70 | 2.72 | 2.11 | 2034/3158 (64.4) |
| GATK | 19 655 | 19 713 | 482 | 97.6 | 2.91 | 2.94 | 2.15 | 4649/4657 (99.8) |
| 84 615 | ||||||||
| SNVer | 17 436 | 16 914 | 522 | 97.0 | 2.85 | 2.87 | 2.22 | 4032/4063 (99.2) |
| SAMtools | 46 037 | 44 489 | 1548 | 96.6 | 2.64 | 2.67 | 1.94 | 4173/7643 (54.4) |
| SAMtools20× | 15 510 | 14 942 | 568 | 96.3 | 2.74 | 2.77 | 2.02 | 2062/3247 (63.5) |
| GATK | 18 892 | 18 419 | 473 | 97.5 | 2.89 | 2.92 | 2.03 | 4537/4566 (99.4) |
| 92 157 | ||||||||
| SNVer | 18 676 | 18 208 | 468 | 97.5 | 2.90 | 2.92 | 2.37 | 4192/4224 (99.2) |
| SAMtools | 49 729 | 47 693 | 2036 | 95.9 | 2.69 | 2.73 | 2.03 | 4251/7996 (53.2) |
| SAMtools20× | 15 881 | 15 370 | 511 | 96.8 | 2.80 | 2.83 | 1.99 | 2028/3259 (62.2) |
| GATK | 20 100 | 19 631 | 469 | 97.7 | 2.98 | 3.00 | 2.35 | 4700/4710 (99.8) |
aTransition and transversion ratio for the identified variants. When the number of variants is small we just report the numbers but not calculate the ratio, e.g. 10/4 for all variants in T1D case by SAMtools means 10 transitions and 4 transversions.
bGenotype concordance. P represents the number of variants called by each program and also genotyped. TP represents the number of variant calls concordant between sequencing data and individual genotyping data.
20×: Additional filtering of sequencing depth ≥20 is applied.
Figure 5.

Correlation between alternate allele frequencies in individually genotyped DNA samples and its estimates in the sequenced DNA pools for the Autism data set. Different symbols represent different depth of coverage ranges as shown in the legend.
As shown in Table 2, for the ADHD individual sequencing data, under family-wise error rate 0.05 level, SNVer also obtained the variant call sets with good quality. This is evidenced by the ~97% dbSNP rates, the approximately 2.9 overall Ti/Tv ratios, the 2.22–2.73 Ti/Tv ratios for novel sites, and the 99% genotype concordance. SAMtools with suggested parameters/filters made 2+ times more variant calls than SNVer (e.g. ~49 K versus ~18 K). The lower Ti/Tv ratios and genotype concordance suggest poorer quality for these larger call sets made by SAMtools. When applied with an additional filtering of sequencing depth ≥20×, SAMtools identified fewer SNPs than SNVer. But it still has lower quality as indicated by the lower Ti/Tv ratios and genotype concordance. Compared with GATK, SNVer has similar performance, while with the higher Ti/Tv ratios for novel variants in all three individuals.
We note that the Ti/Tv ratios for novel variants in the pooled sequencing data are low for both programs. It suggests that they may not perform well for novel variants if we estimate the false-positive rates based on the Ti/Tv ratios following (30). It confirms that variant calling is more challenging for pooled sequencing. Meanwhile, estimating false-positive rates using this summary statistic should be cautious for pooled sequencing. First, Ti/Tv estimate for pooled samples is not as accurate as for individual samples. Second, targeted resequencing regions are usually small, e.g. 31 kb for the T1D data and 503 kb for the Autism data, and therefore may exhibit higher genomic and statistical variances. For example, the ADHD individual 840 60 has an exome-wide Ti/Tv ratio of 2.89 for all variants; if we calculate Ti/Tv ratios based on only 500-kb regions, then the smallest Ti/Tv ratio we obtain is 1.31, and the largest 7.00 with SD = 1.53 (we consider only 500-kb regions with at least 30 variants for having stable Ti/Tv ratio estimates).
Better scalability
SNVer and SAMtools exhibit similar efficiency in terms of running time. The running time of SNVer and CRISP in analysis of the T1D and Autism data sets is given in Figure 6. The main bottleneck of CRISP comes from computing the P-value of a large number of contingency tables in the Fisher's exact test. Therefore, in additional to the number of tests, its time efficiency is also largely dependent on the number of pools and the depth of coverage. In contrast, these two factors have little impact on SNVer and its running time is roughly linear with the region size (the number of tests). For example, SNVer spends 0.1 h on 31 kb and 1.5 h on 503 kb for the two data sets, respectively. SNVer is much faster than CRISP. Taking the T1D case for example, SNVer is ~500-fold faster than CRISP and achieves 300 kb/h. Such efficiency makes feasible the application of SNVer to analysis of whole-exome sequencing data, or even whole-genome sequencing data using high performance computing cluster, both of which, however, will take prohibitively longer time for CRISP.
Figure 6.

(a and b) Comparison of running time of SNVer and CRISP for testing testing (a) the T1D 31 kb region and (b) the Autism 503 kb region. Running time of SNVer is mainly determined by the region size (the number of tests), while larger pool numbers and sequencing depth will take additional time for CRISP.
Informative ranking and multiplicity control
SNVer reports one single overall significance P-value for each locus, based on which the rankings of all tested loci can be produced. Such rankings are more informative and accurate than the dichotomous decision of whether to ‘accept or reject the candidate as a variant’ provided by CRISP and most other existing methods. For example, four rare variants have been found to be associated with T1D based on the T1D data set by comparing the estimated MAF in cases and controls (14). We use SNVer to call these four variants by testing the null hypothesis θ ≤ θ0 = 0.01. We give the rankings of them by SNVer in Table 3, as well as the dichotomous decisions made by CRISP. For SNVer, we observe very significant ranking changes of these four SNPs, which are consistent with their MAFs (relative to the threshold 0.01) and the MAF differences. CRISP identifies three of them, rs35337543, ss107794688 and ss107794687, as variants in both cases and controls, exhibiting no informative differential changes. It should be noted that the ranking difference may only reflect frequency difference. Large frequency difference between case and control of those variants may suggest their potential association with the phenotype, but their functional importance to the phenotype is yet to be assessed by further experiments.
Table 3.
Informative rankings of four rare variants with the null hypothesis θ ≤ θ0 = 0.01
| SNP | T1D case |
T1D control |
||||
|---|---|---|---|---|---|---|
| Estimated MAF (%) | SNVer ranking | CRISP CALL | Estimated MAF (%) | SNVer ranking | CRISP CALL | |
| rs35337543 | 0.36 | 17 557 | Y | 2.51 | 45 | Y |
| rs35667974 | 0.72 | 17 557 | N | 2.42 | 59 | Y |
| ss107794688 | 0.50 | 17 557 | Y | 1.79 | 56 | Y |
| ss107794687 | 1.07 | 145 | Y | 2.45 | 51 | Y |
In addition to ranking, valid P-values given by SNVer also make multiplicity control possible. Tens of thousands or millions loci are usually simultaneously examined in typical NGS experiments. It is particularly desirable to have multiplicity control, which gives the user an idea of the chance of making any errors and/or the proportion of false positives among the variant calls they make. Each user can choose the type I error rate threshold he or she considers appropriate, instead of just the dichotomous decisions of whether to ‘accept or reject the candidates’ provided by most existing methods.
DISCUSSION
We have developed a novel statistical tool SNVer for calling SNPs in analysis of pooled or individual NGS data. Different from the previous models employed by CRISP, it analyzes common and rare variants in one integrated model, which considers and models all relevant factors including variant distribution and sequencing errors simultaneously. As a result, the user does not need to specify several filter cutoffs as required by CRISP. Some variant calling methods simply discard loci with low depth of coverage to achieve reliable variant calls. Our statistical model does not discriminate against poorly covered loci. Loci with any (low) coverage can be tested and depth of coverage will be quantitatively factored into the final significance calculation. SNVer reports one single overall significance P-value for evaluating the significance of a candidate being a variant. An advantage of reporting results on a more continuous scale, instead of just the dichotomous decision of whether to ‘accept or reject the candidate as a variant’ as most existing methods do, is that the user can choose the alpha threshold he or she considers appropriate. We have used both simulated data and real data to demonstrate the superior performance of our program in comparison with pre-existing methods. Although SNVer is motivated by the need for analysis of pooled NGS data, it can also be applied to individual NGS data as a special case (N = 2 for diploid species), as shown in the ADHD data set.
Sampling bias is a non-trivial problem in pooled sequencing, and in particular, rare variants are prone to sampling issues. Properly considering it may further improve the power. In this article, to make inference of the MAF θ of each site, we model the number of observed alleles conditional on the coverage from a frequentist standpoint. The power of detecting variants may be further improved if sampling bias is modeled properly so that we have more informative inference of the coverage rather than conditional on it. Since we have only one observation for each site, to model sampling bias or make any site-specific inference, e.g. base quality/error, we have to pool information across sites. Bayesian models may be a better, if not the only, way to this end. For example, the distribution of coverage of all sites can be approximated by the Gamma distribution for Illumina's short read alignments (31). Shen and colleagues (32) propose to estimate the posterior error rates for each substitution through a Bayesian formula, in which error models are learned from training data sets. Our frequentist approach does not model sampling bias; however, it has its own merits. First, the sampling bias issue may be very application specific. Different target enrichment kits may have different coverage uniformities. More variant sampling bias is expected for targeted re-sequencing, the current main pooling application, due to region-specific GC content. Mapping algorithms will also critically impact coverage. As a result, any approaches with sampling bias modeled may have to check carefully whether the sampling bias model/distribution fits well for every application. Second, our frequentist approach does not pool information across sites, which consequently has minimal requirement for input and wider applications. For example, when only one or few sites are tested, and without any help from external training data, sampling bias could not be modeled (well), but our frequentist approach still can be applied.
So, sampling bias is not considered in our frequentist approach, which consequently makes few assumptions, requires minimal input, and thus has wider applications. On the other hand, sampling issues may be addressed by more careful pooled re-sequencing designs (33). Companies such as NimbleGen and Agilent are also competing to improve their target enrichment kits to obtain coverage uniformity. With these upstream efforts, sampling bias may have a minimized impact on downstream variant call algorithms.
Our current program can be improved and extended in several ways. First, small indels are not supported. Indels impose a great challenge for NGS including DNA amplification and reads mapping which are under fast development. When those techniques become mature in handling indels, we may investigate their distribution and work out a proper calling strategy. Second, sequencing quality scores can be utilized to estimate site-specific sequencing error. Third, the majority loci of sequenced segments are known to carry no variants. The density of SNP is estimated to be around 1 out of 1000 bases. Such prior percentage of non-nulls information may help obtain more precise multiplicity control. Fourth, the dependency among tests will also be informative in increasing testing efficiency. We have shown that the LD dependency information is very informative in increasing the efficiency of conducting genome-wide association tests in analysis of GWAS data (34). We also found recently that dependency information is helpful for increasing the efficiency of testing hypotheses at the set level (35). For NGS data, one non-null (variant) is expected from every 1000 consecutive genomic bases. Such dependency patterns, if appropriately modeled, may help further improve testing efficiency. Lastly, our current program focuses on calling variants, namely, testing whether θ is larger than a threshold. Under the same framework, our models can be naturally extended for case-control association studies by testing whether θcase = θcontrol. We are currently working on these extensions.
In summary, we have developed a statistical tool SNVer for calling common and rare variants in analysis of both pooled and individual NGS data. As more and more NGS data become available, we expect more applications of our program.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Institute Development Fund to the Center for Applied Genomics from The Children's Hospital of Philadelphia (partial). The open access publication charge for this paper has been waived by Oxford University Press – NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Juvenile Diabetes Research Foundation and Wellcome Trust for providing the T1D NGS data used in the study. The authors thank Dan Koboldt for clarifying the usage of the Fisher's exact test in VarScan and Dr Vikas Bansal for helpful discussion. The authors also thank all four referees for their constructive comments, which have greatly helped improve the presentation of the article. The authors declare Juvenile Diabetes Research Foundation and Wellcome Trust bear no responsibility for interpreting the T1D results generated in the study.
REFERENCES
- 1.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 2.Mardis ER. A decade's perspective on DNA sequencing technology. Nature. 2011;470:198–203. doi: 10.1038/nature09796. [DOI] [PubMed] [Google Scholar]
- 3.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 2009;10:241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- 7.Li B, Leal SM. Discovery of rare variants via sequencing: implications for the design of complex trait association studies. PLoS Genet. 2009;5:e1000481. doi: 10.1371/journal.pgen.1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayden EC. International genome project launched. Nature. 2008;451:378–379. doi: 10.1038/451378b. [DOI] [PubMed] [Google Scholar]
- 9.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 10.Service RF. Gene sequencing. The race for the $1000 genome. Science. 2006;311:1544–1546. doi: 10.1126/science.311.5767.1544. [DOI] [PubMed] [Google Scholar]
- 11.Norton N, Williams NM, O'Donovan MC, Owen MJ. DNA pooling as a tool for large-scale association studies in complex traits. Ann. Med. 2004;36:146–152. doi: 10.1080/07853890310021724. [DOI] [PubMed] [Google Scholar]
- 12.Sham P, Bader JS, Craig I, O'Donovan M, Owen M. DNA Pooling: a tool for large-scale association studies. Nat. Rev. Genet. 2002;3:862–871. doi: 10.1038/nrg930. [DOI] [PubMed] [Google Scholar]
- 13.Momozawa Y, Mni M, Nakamura K, Coppieters W, Almer S, Amininejad L, Cleynen I, Colombel JF, de Rijk P, Dewit O, et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat. Genet. 2011;43:43–47. doi: 10.1038/ng.733. [DOI] [PubMed] [Google Scholar]
- 14.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Out AA, van Minderhout IJ, Goeman JJ, Ariyurek Y, Ossowski S, Schneeberger K, Weigel D, van Galen M, Taschner PE, Tops CM, et al. Deep sequencing to reveal new variants in pooled DNA samples. Hum. Mutat. 2009;30:1703–1712. doi: 10.1002/humu.21122. [DOI] [PubMed] [Google Scholar]
- 16.Calvo SE, Tucker EJ, Compton AG, Kirby DM, Crawford G, Burtt NP, Rivas M, Guiducci C, Bruno DL, Goldberger OA, et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet. 2010;42:851–858. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Druley TE, Vallania FL, Wegner DJ, Varley KE, Knowles OL, Bonds JA, Robison SW, Doniger SW, Hamvas A, Cole FS, et al. Quantification of rare allelic variants from pooled genomic DNA. Nat. Methods. 2009;6:263–265. doi: 10.1038/nmeth.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kullback S, Leibler RA. On information and sufficiency. Ann. Math. Stat. 1951;22:79–86. [Google Scholar]
- 23.Vallania FL, Druley TE, Ramos E, Wang J, Borecki I, Province M, Mitra RD. High-throughput discovery of rare insertions and deletions in large cohorts. Genome Res. 2010;20:1711–1718. doi: 10.1101/gr.109157.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bansal V. A statistical method for the detection of variants from next-generation resequencing of DNA pools. Bioinformatics. 2010;26:i318–i324. doi: 10.1093/bioinformatics/btq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ingman M, Gyllensten U. SNP frequency estimation using massively parallel sequencing of pooled DNA. Eur. J. Hum. Genet. 2009;17:383–386. doi: 10.1038/ejhg.2008.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JS, Choi M, Yan X, Lifton RP, Zhao H. On optimal pooling designs to identify rare variants through massive resequencing. Genet. Epidemiol. 2011;35:139–147. doi: 10.1002/gepi.20561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamini Y, Heller R. Screening for partial conjunction hypotheses. Biometrics. 2008;64:1215–1222. doi: 10.1111/j.1541-0420.2007.00984.x. [DOI] [PubMed] [Google Scholar]
- 28.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 29.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Depristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, Del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarin S, Prabhu S, O'Meara MM, Pe'er I, Hobert O. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat. Methods. 2008;5:865–867. doi: 10.1038/nmeth.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen Y, Wan Z, Coarfa C, Drabek R, Chen L, Ostrowski EA, Liu Y, Weinstock GM, Wheeler DA, Gibbs RA, et al. A SNP discovery method to assess variant allele probability from next-generation resequencing data. Genome Res. 2010;20:273–280. doi: 10.1101/gr.096388.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prabhu S, Pe'er I. Overlapping pools for high-throughput targeted resequencing. Genome Res. 2009;19:1254–1261. doi: 10.1101/gr.088559.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei Z, Sun W, Wang K, Hakonarson H. Multiple testing in genome-wide association studies via hidden Markov models. Bioinformatics. 2009;25:2802–2808. doi: 10.1093/bioinformatics/btp476. [DOI] [PubMed] [Google Scholar]
- 35.Sun W, Wei Z. Multiple testing for pattern identification, with applications to microarray time course experiments. J. Am. Stat. Assoc. 2011;106:73–78. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.