

Abstract
It has been only 5 years since the identification of TDP-43 as the major protein component of the ubiquitinated inclusions in FTLD-U. At that time, there were approximately a dozen papers about TDP-43; today, a “TDP-43” search reveals almost 600 papers. It is now clear that the majority of FTLD cases containing tau- and alpha-synuclein-negative, ubiquitin-positive inclusions (FTLD-U) are FTLD-TDP. The spectrum of TDP-43 proteinopathies includes FTLD-TDP with or without ALS, with or without mutations in GRN, VCP, or TARDBP, with or without chromosome 9p linkage, and sporadic and non-SOD1 familial ALS with or without FTLD-TDP. There are four sub-types of FTLD-TDP, and these correlate with specific clinical and genetic profiles. Sub-types are determined by the presence, predominance, and distribution of the various TDP-43 immunopositive insoluble aggregates—neuronal cytoplasmic inclusions, neuronal intranuclear inclusions, and dystrophic neurites. In this paper, FTLD-TDP pathologic sub-types will be described, and examples of each sub-type will be shown, and implications for future research will be discussed.
Keywords: FTLD-TDP; TAR-DNA binding protein-43; TDP-43; Progranulin; GRN; TARDBP, FTLD-U; FTLD-U; Frontotemporal dementia; FTD; Amyotrophic lateral sclerosis; ALS
Introduction
It has been only 5 years since transactive response (TAR) DNA-binding protein of 43 kDa molecular weight (TDP-43) was reported to be the major protein component of the abnormal aggregates found in frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U) (Neumann et al. 2006; Arai et al. 2006). Prior to this discovery, a literature search for TDP-43 revealed about a dozen papers, mostly about its role in HIVand cystic fibrosis. However, FTD researchers who had been eagerly anticipating the identification of the FTLD-U protein immediately began exploring the molecular and pathological implications that TDP-43 was a key protein in FTLD-U. This resulted in a profound expansion of the TDP-43 literature such that at the end of March 2011, a search for TDP-43 revealed almost 600 papers.
TDP-43 is an ubiquitously expressed, highly conserved RNA- and DNA-binding nuclear protein involved in exon skipping and translation regulation encoded by the TARDBP gene on chromosome 1 (Ou et al. 1995). In FTLD-U, TDP-43 is abnormally phosphorylated, ubiquitinated, and cleaved, resulting in the generation of toxic C-terminal fragments, and nuclear to cytoplasmic translocation (Fig. 1). Because nuclear TDP-43 is lost from neurons with cytoplasmic inclusions and because the C-terminal fragments are aggregated in the urea soluble fraction of human brain homogenates, it is likely that loss of function plays a role in the neurodegenerative process (Kwong et al. 2007). Importantly, impairing nuclear TDP-43 import with siRNAs results in cytoplasmic TDP-43 accumulation and FTLD (Nishimura et al. 2010). Finally, recent evidence has shown that TDP-43 resides in the dendritic processing body of somatodendrites, in the form of RNA granules colocalized with the postsynaptic protein PSD95, where it acts as a translational repressor and thus likely helps regulate neuronal plasticity (Wang et al. 2008).
Fig. 1.

Ubiquitin (a) and TDP-43 (b) immunohistochemistry of superficial frontal cortex of an FTLD-U case. Note neuronal cytoplasmic inclusions (NCIs) (open arrows), neuronal intra-nuclear inclusions (NIIs) (arrows), and dystrophic neurites (DNs) (asterisks). This TDP-43 antibody labels normal as well as abnormal TDP-43, so nuclei of neurons without abnormal TDP-43 aggregates are also labeled in b (arrowhead). Note that TDP-43 reveals slightly greater pathology than does ubiquitin. Dako polyclonal ubiquitin and ProteinTech polyclonal TDP-43 antibodies. Magnification, ×600
Abnormal protein aggregates in most cases of FTLD-U were ultimately shown to be composed primarily of abnormal phosphorylated TDP-43, and the recommended diagnostic terminology for such cases became FTLD-TDP (Neumann et al. 2006; Arai et al. 2006; Cairns et al. 2007a; Davidson et al. 2007; Mackenzie et al. 2010). A subset of FTLD-U cases were ultimately found to be negative for TDP-43, and these were called “atypical FTLD-U” (a-FTLD-U) (Cairns et al. 2007a; Mackenzie et al. 2008; Roeber et al. 2008). Of the 63 cases of FTLD-U in the Northwestern ADC Neuropathology Core Brain Bank, three (5%) are a-FTLD-U, conflicting with reports that a-FTLD-U cases comprise 7–20% of FTLD-U cases (Mackenzie et al. 2008; Roeber et al. 2008). Inclusions in most a-FTLD-U cases, including the three Northwestern a-FTLD-U cases, were subsequently found to be composed primarily of abnormal fused in sarcoma (FUS) protein (Neumann et al. 2009a). A few FTLD-U cases remain that are negative with both TDP-43 and FUS immunohistochemistry. These are predominantly FTD-3 families with CHMP2B mutations and may possibly include rare additional a-FTLD-U cases. The recommended diagnostic terminology in such cases is FTLD-UPS, as a nod to the fact that the ubiquitin proteasome system likely plays a role in these disorders, without (yet) apparent involvement of another major protein (Mackenzie et al. 2010). Interestingly, although the lower motor neuron (LMN) skein-like inclusions (SLIs) and Lewy-body-like inclusions (LBLIs) in amyotrophic lateral sclerosis (ALS) are also composed primarily of abnormal TDP-43 protein, the SLIs and LBLIs and other LMN inclusions in cases of familial ALS (FALS) with SOD1 mutations do not appear to contain TDP-43 (Mackenzie et al. 2007).
Except for FTLD-FUS, FTLD-UPS, and FALS (SOD1) cases described above, all FTLD-U cases with and without ALS are TDP-43 variants of FTLD (FTLD-TDP). In addition, many, or most, cases of hippocampal sclerosis (HS) have TDP-43-positive inclusions and may represent frontotemporal dementia (Hatanpaa et al. 2004; Amador-Ortiz et al. 2007; Probst et al. 2007), but will not be discussed in the current manuscript. The clinical and genetic associations are described below together because characteristic histological patterns are associated with specific gene mutations or linkages. Two different FTLD-TDP pathological subtype classification systems that rely on similar histological features, but number the subtypes differently, have been published by Sampathu et al. (2006 and Mackenzie et al. (2006). Mackenzie FTLD-TDP subtype 1 corresponds to Sampathu subtype 3, Mackenzie type 2 to Sampathu type 1, and Mackenzie type 3 to Sampathu type 2. FTLD-TDP type 4 is the same in both classification systems. The Mackenzie classification system will be used in the current paper. In this paper, the subtypes will first be described, followed by brief clinicopathological–genetic case reports, with illustrations, of each subtype.
Pathological, Clinical, and Genetic Associations
The inclusions in FTLD-TDP type 1 (Cairns et al. 2007a; Sampathu et al. 2006; Mackenzie et al. 2006; Josephs et al. 2009; Armstrong et al. 2010) include neuronal cytoplasmic inclusions (NCIs), neuronal intranuclear inclusions (NIIs), and dystrophic neurites (DNs), predominantly in upper cortical layers. The dentate gyrus generally has few NCIs and occasional NIIs and few to absent DNs. Hippocampal sclerosis (HS) is uncommon, and subcortical gray matter and brainstem TDP pathology can be mild to severe. The clinical presentation can be that of either behavioral variant frontotemporal dementia (bvFTD) or primary progressive aphasia (PPA), and those with aphasia have either primary non-fluent aphasia or, less commonly, semantic dementia (SD). Clinical and pathologic ALS is sometimes present. A positive family history is common, present in up to 50% of cases, and mutations in progranulin (GRN) are most often found in familial cases. Males are affected slightly more frequently than females at an approximate ratio of 3:2, and the average duration of disease is 8.4 years.
In FTLD-TDP type 2 (Cairns et al. 2007a; Sampathu et al. 2006; Mackenzie et al. 2006; Josephs et al. 2009; Armstrong et al. 2010), DNs, sometimes described as “long,” predominate over NCIs and may be predominantly located in upper cortical layers. NIIs are infrequent to absent. The dentate gyrus may have few to moderate NCIs, but NIIs are absent and DNs are few to absent. HS can be prominent. Subcortical gray matter TDP pathology is generally moderate, and brainstem TDP pathology is rare. The clinical presentation can be bvFTD but is more commonly PPA, most often SD. ALS is uncommon. Family history is positive in approximately 30% of cases, but there are as yet no known associated genetic mutations. Males and females are equally affected, and the average duration of disease is 8.1 years.
Type 3 FTLD-TDP (Cairns et al. 2007a; Sampathu et al. 2006; Mackenzie et al. 2006; Josephs et al. 2009; Armstrong et al. 2010; Brandmeir et al. 2008; Gitcho et al. 2009) pathology consists predominantly of NCIs, which may be present in lower as well as upper cortical layers. NIIs and DNs are rare. In many type 3 cases, NCIs are frequent in the dentate gyrus, and in some of these, the cortical DNs are few. In others, however, dentate gyrus NCIs are few. HS is often prominent, while subcortical gray and brainstem TDP pathology can be mild to severe. The clinical presentation is most often bvFTD and rarely PPA, and ALS is often present. About one-third of cases have a positive family history; some of these are due to mutations in TARDBP, and some of these are in families with linkage to chromosome 9p. Males are affected more often than females at a ratio of 3:1. The average duration of disease is shorter than in the other two groups at 5.4 years, likely because more of these cases have ALS.
FTLD-TDP Type 4 pathology (Cairns et al. 2007a; Mackenzie et al. 2010; Forman et al. 2006; Guinto et al. 2007) consists almost entirely of frequent NIIs and infrequent NCIs and DNs in upper layers with relative sparing of the dentate gyrus. HS is usually absent. Subcortical gray matter TDP pathology may be moderate, and brainstem TDP pathology is often mild. Patients are equally likely to present with bvFTD or PPA. ALS is not part of the disease, but inclusion body myositis may be present. The full clinical spectrum of the disease, not always present in each affected individual, includes inclusion body myositis, Paget’s disease of the bone, and frontotemporal dementia (IBM-PFD). Family history is positive in virtually all cases, as all are related to mutations in the gene for valosin-containing protein (VCP). Males and females are equally affected, and the average duration of disease is 13.3 years.
Case Reports
Case 1, FTLD-TDP Type 1
This 53-year-old man died after a 3.5-year history of bvFTD with combined PPA. He had deficits in attention, comportment, judgment, insight, and language. He had no motor dysfunction. Neither parent was affected, but all six of his mother’s siblings had “dementia.” Two of these were autopsied, and later studies revealed similar pathology. Genetic analysis revealed no pathogenic GRN mutation. At autopsy, the brain weighed 1,250 g. There was moderate to severe frontal and temporal and mild parietal atrophy and no hippocampal atrophy. There was mild ventricular dilatation and caudate atrophy. Hematoxylin and eosin (H&E) stains revealed marked frontal and mild patchy temporal superficial microvacuolation and gliosis. There was mild gliosis of the caudate and putamen and mild depopulation and gliosis of the substantia nigra. Hippocampal sclerosis was not present. TDP-43 immunostains using both a polyclonal antibody (Protein Tech, Chicago, IL, USA) and a monoclonal antibody to phosphorylated TDP-43 (CosmoBio, Tokyo Japan) revealed NCIs, short DNs, and occasional NIIs that were moderate to frequent in superficial frontal and temporal cortex (Fig. 2) and sparse to moderate in deeper cortical layers (Fig. 3). NCIs and DNs were also frequent in superficial motor cortex (Fig. 4). In the dentate gyrus, there were sparse NCIs and NIIs (Fig. 5a), and in the striatum, NCIs and DNs were moderate and NIIs were sparse (Fig. 5b).
Fig. 2.

Case 1, FTLD-TDP type 1. Immunostains comparing labeling with polyclonal TDP-43 antibody (a) and monclonal antibody to phosphorylated TDP-43 (p-TDP-43) (b). Note frequent NCIs and DNs, with more DNs demonstrated with the phosphorylated TDP-43 antibody. NIIs indicated by arrows. Magnification, ×400
Fig. 3.

Case 1. Sparse to moderate NCIs, DNs, and NIIs in deep cortex with polyclonal TDP-43 (a) and monoclonal p-TDP-43 antibodies (b). NIIs indicated by arrows. Magnification, ×400
Fig. 4.
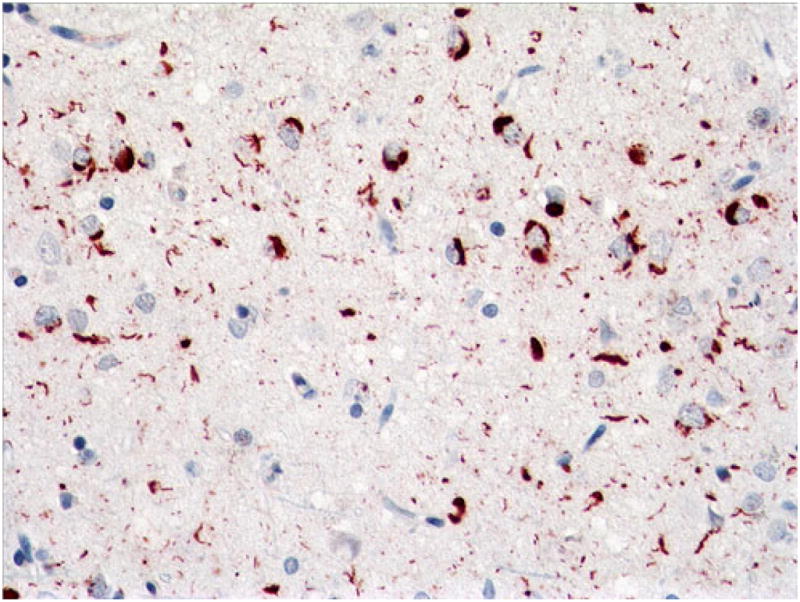
Case 1. Frequent NCIs and DNs in superficial motor cortex. p-TDP-43 antibody. Magnification, ×400
Fig. 5.
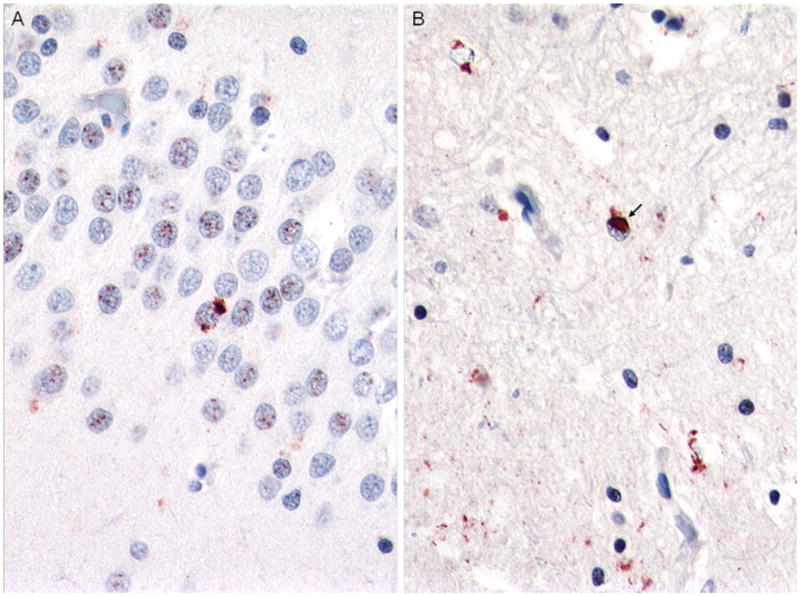
Case 1. Sparse dentate gyrus NCIs (a) and sparse to moderate NCIs, DNs, and NIIs in the striatum (b). p-TDP-43 antibody. Magnification, ×400
Case 2, FTLD-TDP Type 2
This 53-year-old woman presented with naming and word-finding difficulties and, within a few months, developed compulsive behaviors and social disinhibition. Later, she became mute. Her sweets intake increased dramatically, and executive function and judgment became impaired. Neurological examination revealed no motor dysfunction. She died at age 65, after a 12.5-year course of disease. Family history was negative. At autopsy, the brain weighed 900 g. There was severe frontal, temporal, and parietal atrophy, greatest in anterior temporal regions where it was “knife edge-like” (Fig. 6). There was also severe caudate and brainstem atrophy, mild occipital and hippocampal atrophy, and severe ventricular dilatation. Nigral pallor was mild. TDP-43 IHC revealed a predominance of long neurites in frontal (including motor), temporal, and parietal cortex, most numerous in superficial cortex but also present in lower layers (Fig. 7). The dentate gyrus had frequent dense and granular NCIs, and the striatum had frequent long and short DNs (Fig. 8).
Fig. 6.
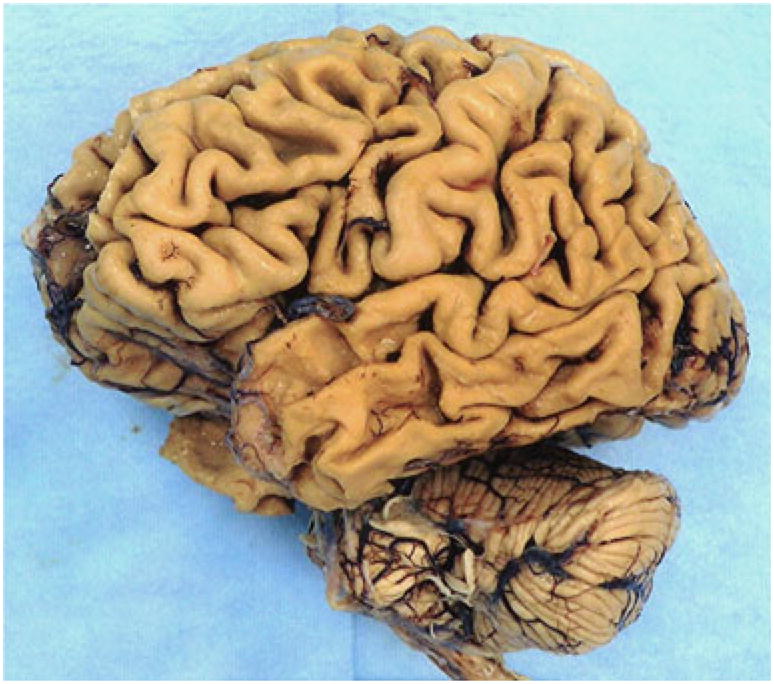
Case 2. Left aspect of gross brain, showing severe frontal, temporal, and parietal atrophy, greatest in temporal regions, where it is “knife edge-like,” with relative sparing of the posterior superior temporal gyrus
Fig. 7.

Case 2. Long neurites predominate in cortex and are present in superficial as well as deep layers. Superficial cortex (a), ×200 magnification. Low power (b) showing full-thickness cortex (b), ×40 magnification. p-TDP-43 antibody
Fig. 8.
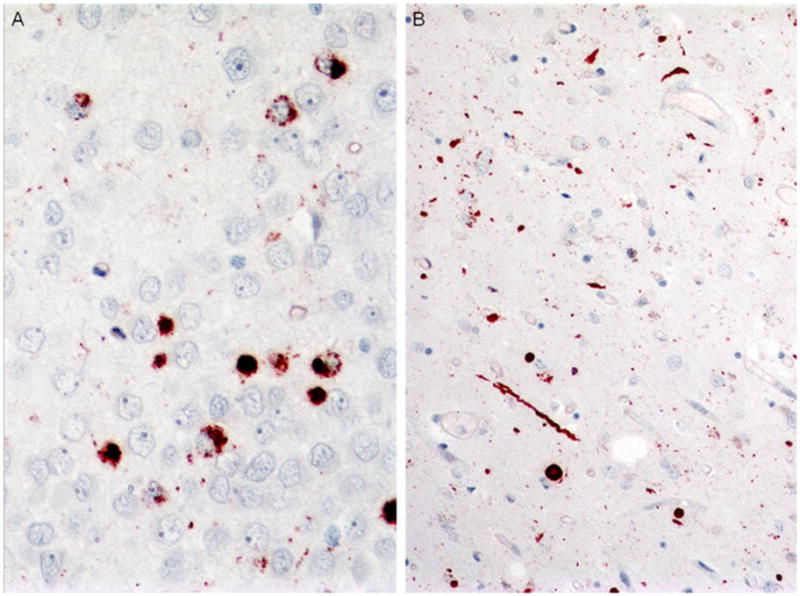
Case 3A. Frequent dense and granular dentate gyrus inclusions (a) and frequent long and short neurites in the caudate nucleus (b). p-TDP-43 antibody, a ×400, b ×200 magnification
Case 3A, FTLD-TDP Type 3
This 61-year-old man had onset of bvFTD approximately 3 years prior to death and onset of sporadic ALS approximately 6 months later. At autopsy, the brain weighed 1,400 g, and there was mild atrophy of frontal cortex only and moderate pallor of the substantia nigra and locus coeruleus. Histological H&E sections revealed mild to moderate superficial microvacuolation in frontal (including motor) and temporal cortex. p-TDP-43 immunostains highlighted frequent granular and dense NCIs in superficial cortex (Fig. 9) and sparse filamentous and dense, some Pick-like, NCIs in deep cortex (Fig. 10). Dentate gyrus NCIs were moderate (Fig. 11).
Fig. 9.
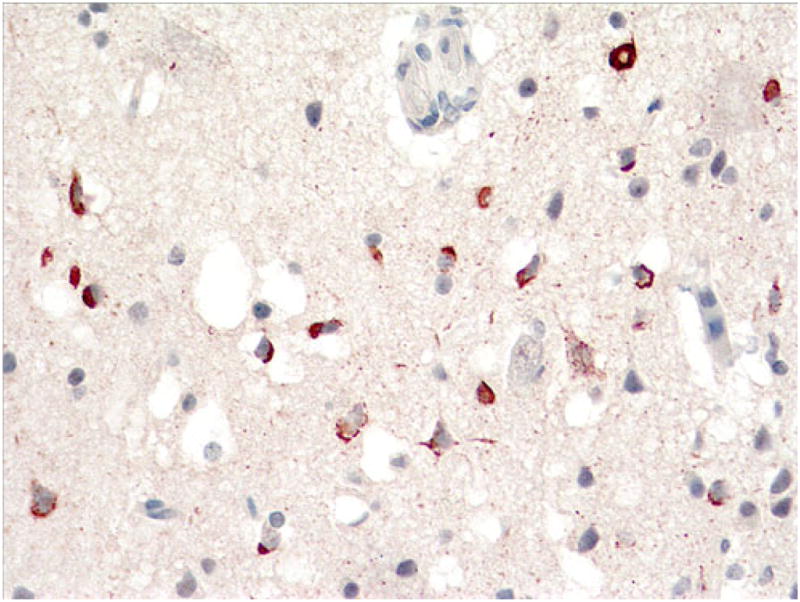
Case 3A. Frequent filamentous, dense, and granular NCIs and sparse DNs in superficial cortex. p-TDP-43 antibody. Magnification, ×400
Fig. 10.

Case 3A. Sparse dense NCIs and DNs, deep cortex. Arrow points to Pick-like body. p-TDP-43 antibody. Magnification, ×400
Fig. 11.
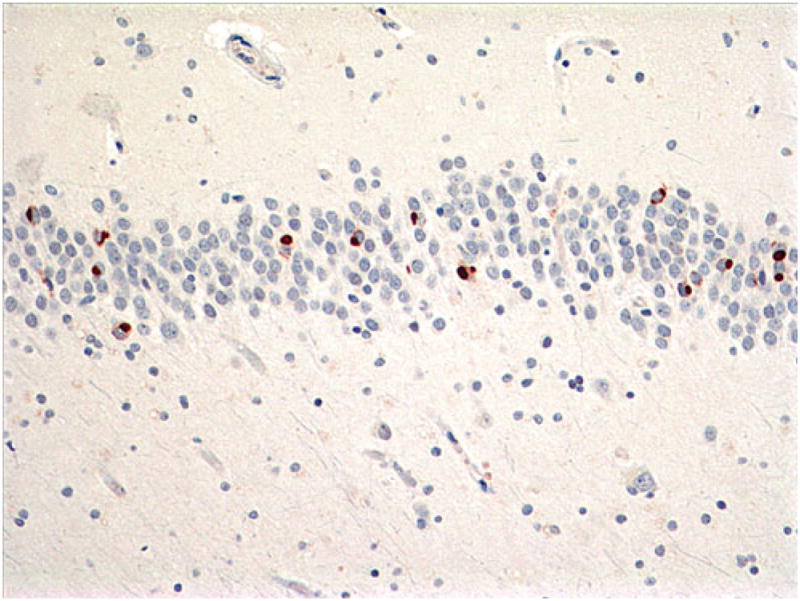
Case 3A. Moderate dense NCIs in dentate gyrus. Some are large, round, and Pick-like. p-TDP-43 antibody. Magnification, ×200
Case 3B, FTLD-TDP Type 3
This 58-year-old man died after a 3-year history of FALS and bvFTD. His father had died of ALS. Genetic analysis revealed no mutations in SOD1, FUS, or TARDBP. The brain weighed 1,400 g. There was mild atrophy of the frontal poles and caudate nuclei, moderate ventricular dilatation, and moderate pallor of the substantia nigra. TDP-43 immunostains showed moderate granular and sparse dense NCIs in all layers of cortex and sparse DNs in superficial cortex (Fig. 12). There were frequent granular and sparse dense NCIs in the dentate gyrus and sparse NCIs and moderate DNs in the caudate nucleus (not shown).
Fig. 12.

Case 3B. Moderate granular and sparse dense NCIs and sparse DNs in superficial cortex (a) and moderate granular inclusions in deep cortex (b). p-TDP-43 antibody. Magnification for both, ×400
Case 3C, FTLD-TDP Type 3, Chromosome 9p-Linked FTD-ALS
This 54-year-old man had a 10-year history of a psychiatric disorder, described as bipolar disorder versus cyclothymia, and a 3-year history of ALS. Family history was significant for six members who died of ALS and two others who had cognitive impairment, his mother who died at 67 of “Alzheimer disease,” and a cousin who died of “dementia.” The fresh brain weighed 1,600 g. There was no cortical, hippocampal, or caudate atrophy, and there was no ventricular dilatation. There was mild pallor of the substantia nigra. TDP-43 immunostains revealed frequent NCIs, predominantly granular, but also filamentous and dense, in superficial motor cortex (Fig. 13), and sparse to moderate NCIs, predominantly filamentous, but also “Pick-like,” in deep layers of motor cortex (Fig. 14). The frontal lobe was affected similarly but to a lesser degree. There were sparse dentate gyrus NCIs and sparse to moderate NCIs in the striatum (not shown).
Fig. 13.

Case 3C. Frequent NCIs in superficial motor cortex (upper left) but none in sensory cortex (lower right). Inset shows superficial NCIs at higher power. p-TDP-43 antibody, ×40 magnification; inset, ×400 magnification
Fig. 14.

Case 3C. Sparse to moderate filamentous and dense NCIs and sparse DNs in deep layers of motor cortex (a). Pick-like dense NCI shown in (b). p-TDP-43 antibody. Magnifications, a ×200 and b ×400
Case 3D, FTLD-TDP Type 3 with TARDBP 3′UTR Variant
The decedent was a 70-year-old woman with a 4-year history of bvFTD characterized by obsessive–compulsive tendencies, deficits in attention, apathy, and socially inappropriate behavior. She had no clinical signs of ALS. Family history was significant for a brother who died of ALS but had no cognitive impairment. At autopsy, the brain weighed 1,150 g and showed moderate frontal and temporal and mild parietal atrophy. There was moderate atrophy of the hippocampus and mild caudate atrophy. There was mild pallor of the substantia nigra and locus coeruleus. The hippocampus had severe neuronal loss and gliosis in the CA1 region and subiculum consistent with hippocampal sclerosis. p-TDP-43 immunostains highlighted frequent granular NCIs and sparse long DNs in superficial frontal cortex (Fig. 15) and sparse filamentous and dense NCIs, some Pick-like, in deeper layers (Fig. 16). There were frequent granular NCIs in the dentate gyrus (Fig. 17a). Additionally, although the patient had no motor weakness or other signs of ALS, rare SLIs were seen in the hypoglossal nucleus (Fig. 17c). The brother with ALS and without bvFTD, on the other hand, had sparse dentate gyrus NCIs (Fig. 17b) and frequent LBLIs in the hypoglossal nucleus (Fig. 17c). Both siblings had a novel guanine to adenine variant in the 3′ segment of the untranslated region (3′-UTR) adjacent to exon 6 (NM_007375.3, position 2076G>A) of TARDBP (Gitcho et al. 2009).
Fig. 15.

Case 3D. Frequent granular NCIs and sparse long DNs (arrow), superficial frontal cortex. p-TDP-43 antibody. Magnification, ×400
Fig. 16.

Case 3D. Sparse dense and filamentous NCIs, deep frontal cortex. p-TDP-43 antibody. Magnification, ×400
Fig. 17.
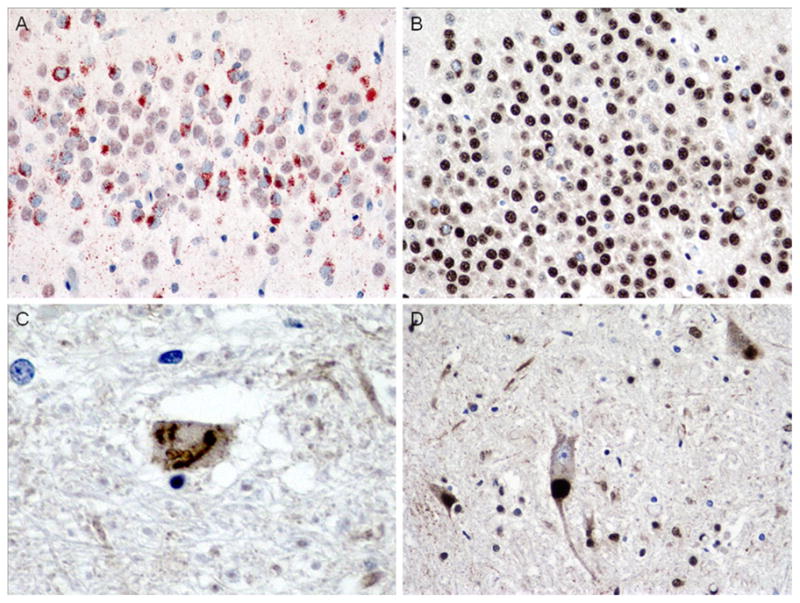
Case 3D. Comparison of NCIs in dentate gyrus (a, b) and SLIs and LBLIs in hypoglossal nucleus (c, d) in case 3C (a, c) compared to brother (b, d). Note frequent granular NCIs in case 3 with bvFTD (a) but only sparse NCIs in brother with clinical ALS (b). Additionally, hypoglossal SLIs were rare in case 3 (c), but LBLIs were frequent (and SLIs sparse, not shown) in brother (d). p-TDP-43 antibody (a), ×400 magnification, TDP-43 antibody (b, d), ×400 magnification and c ×600 magnification
Case 4, FTLD-TDP Type 4
This was a 60-year-old woman who had a 25-year history of Paget’s disease and an 8-year history of progressive cognitive impairment. She had no evidence during life, but did have histological evidence at autopsy, of IBM. There was a strong family history of either cognitive impairment or “psychiatric” diagnoses, “muscular dystrophy,” and Paget’s disease, and mutation analysis revealed the presence of the R155H VCP mutation in this family (Forman et al. 2006). At autopsy, there was severe diffuse atrophy (brain weight, 890 g) and severe atrophy of the hippocampus and amygdala, although there were only sparse cortical neuritic plaques and no neurofibrillary tangles. TDP-43 immunostains revealed rare NCIs and moderate to frequent DNs and NIIs in all neocortical sections (Fig. 18). DNs and NIIs were rare to sparse in deep gray nuclei and entorhinal cortex (not shown). There were no inclusions in the hippocampal pyramidal neurons or dentate gyrus.
Fig. 18.

Case 4. Frequent NIIs (arrows) and DNs in cortex. Rare NCIs not shown. p-TDP-43 antibody. Magnification, ×400
FTLD-TDP Subtypes, Summary Statements
FTLD-TDP type 1, when familial, is likely to be associated with a mutation in GRN (Baker et al. 2006; Cruts et al. 2006). Type 1 is more likely to have neocortical NIIs and is less likely to have either HS or dentate gyrus NCIs (Davion et al. 2007; Hatanpaa et al. 2008). FTLD-TDP types 2 and 3 overlap clinically and pathologically and may form a continuum. Although the inclusions are predominantly DNs in type 2 and predominantly NCIs in type 3, they are both found in upper and lower cortical layers. HS may be prominent in both. Those with ALS have more dentate gyrus NCIs (Kovari et al. 2004). Familial cases may be linked to chromosome 9p or have TARDBP mutations (Cairns et al. 2007a, b; Mackenzie et al. 2010; Gitcho et al. 2009; Luty et al. 2008; LeBer et al. 2009; Boxer et al. 2010; Borroni et al. 2009; Borroni et al. 2010). FTLD-TDP type 4 is distinctive for cases with VCP mutations and NIIs predominate (Cairns et al. 2007a, b; Mackenzie et al. 2010; Forman et al. 2006; Guinto et al. 2007).
Conclusion
Interestingly, the original studies that led to the identification of TDP-43 as the major protein component of the inclusions in FTLD-U/FTLD-TDP were based on the development of a variety of specific monoclonal antibodies that labeled inclusions in FTLD-TDP types 2 and 3, but did not label inclusions in FTLD-TDP type 1 (Mackenzie classification) (Neumann et al. 2006; Sampathu et al. 2006; Mackenzie et al. 2006). However, through experiments with these specific monoclonal antibodies, TDP-43 was identified, and antibodies to TDP-43 labeled inclusions in all FTLD-TDP subtypes, and in all sporadic and familial ALS cases except those FALS cases due to SOD1 mutations (Neumann et al. 2006; Arai et al. 2006; Kwong et al. 2007; Cairns et al. 2007a; Davidson et al. 2007; Roeber et al. 2008; Mackenzie et al. 2007). Like tau in AD and alpha-synuclein in Lewy body dementia, TDP-43 in FTLD-TDP undergoes truncation, phosphorylation, and ubiquitination (Neumann et al. 2006, 2009b; Kwong et al. 2007; Sampathu et al. 2006; Hasegawa et al. 2008; Igaz et al. 2008). Abnormal TDP-43, normally a nuclear protein, becomes mislocalized, and aggregates in cytoplasmic inclusions and in extracellular dystrophic neurites, and, in FTLD-TDP type 3, also aggregates in neuronal intranuclear inclusions. The predominant type or types of aggregates vary according to FTLD-TDP sub-type. In this paper, the various sub-types have been described, and the case studies and associated figures have highlighted the clinical, genetic, and pathological differences and similarities. The advantages and disadvantages of “lumping” versus “splitting” entities can always be argued, but it is apparent that in FTLD-TDP, “splitting” has revealed specific correlations between the pathology and clinical presentations and genetic associations. These correlations have opened avenues of investigation for basic and clinical scientists alike and paved the way for continuing discoveries in future studies of FTLD-TDP.
Acknowledgments
We would like to gratefully acknowledge the NU CNADC directed by Dr. M-Marcel Mesulam, the NU Lois Insolia ALS Center directed by Dr. Teepu Siddique, Manjari Mishra, and Katherine Gasho for histological and immunohistochemical expertise, the UT Southwestern ADC for the FTLD-TDP type 4 case, and, most importantly, the generous patients and families without whom these studies would not be possible. This study is supported in part by NIH grant AG13854
Footnotes
This work was presented at the 7th International FTD Conference in Indianapolis, IN, USA on October 6, 2010.
References
- Amador-Ortiz C, Lin W-L, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Armstrong RA, Ellis W, Hamilton RL, Mackenzie IRA, Hedreen J, Gearing M, Montine T, Vonsattel J-P, Head E, Lieberman AP, Cairns NJ. Neuropathological heterogeneity in frontotemporal lobar degeneration with TDP-43 proteinopathy: a quantitative study of 94 cases using principal components analysis. J Neural Transm. 2010;117:227–239. doi: 10.1007/s00702-009-0350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick D, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Borroni B, Bonvicini C, Alberici A, Buratti E, Agosti C, Archetti S, Papetti A, Stuani C, Di Luca M, Gennarelli M, Padovani A. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Human Mutat. 2009;30:E974–E983. doi: 10.1002/humu.21100. [DOI] [PubMed] [Google Scholar]
- Borroni B, Archetti S, Del Bo R, Papetti A, Buratti E, Bonvicini C, Agosti C, Cosseddu M, Turla M, Di Lorenzo D, Pietro Comi G, Gennarelli M, Padovani A. TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuvenation Res. 2010;13:509–517. doi: 10.1089/rej.2010.1017. [DOI] [PubMed] [Google Scholar]
- Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R, Feldman H, Hsiung G-YR, Rutherford N, Laluz V, Whitwell J, Foti D, McDade E, Molano J, Karydas A, Wojtas A, Goldman J, Mirsky J, Sengdy P, DeArmond S, Miller BL, Rademakers R. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry. 2010;82:196–203. doi: 10.1136/jnnp.2009.204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandmeir NJ, Geser F, Kwong LK, Zimmerman E, Qian J, Lee VM-Y, Trojanowski JQ. Severe subcortical TDP-43 pathology in sporadic frontotemporal lobar degeneration with motor neuron disease. Acta Neuropathol. 2008;115:123–131. doi: 10.1007/s00401-007-0315-5. [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL, III, Schneider JA, Kretzschmar H, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Lee VMY, Trojanowski JQ, Mackenzie IR. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007a;171:227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Bigio EH, Mackenzie IRA, Neumann M, Lee VM-Y, Hatanpaa KJ, White CL, III, Schneider JA, Grinberg LT, Halliday G, Cuyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DMA. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007b;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin J-J, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du PD, Neary D, Snowden JS, Mann DM. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 2007;113:521–533. doi: 10.1007/s00401-006-0189-y. [DOI] [PubMed] [Google Scholar]
- Davion S, Johnson N, Weintraub S, Mesulam M-M, Engberg A, Mishra M, Baker M, Adamson J, Hutton M, Rademakers R, Bigio EH. Clinicopathologic correlations in PGRN mutations. Neurology. 2007;69:1113–1121. doi: 10.1212/01.wnl.0000267701.58488.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW, Tu P-H, Watts GDJ, Markesbery WR, Smith CH, Kimonis VE. Novel ubiquitin neuropathology in frontotemporal dementia with Valosin-Containing Protein gene mutations. J Neuropathol Exp Neurol. 2006;65:571–581. doi: 10.1097/00005072-200606000-00005. [DOI] [PubMed] [Google Scholar]
- Gitcho MA, Bigio EH, Mishra M, Johnson N, Weintraub S, Mesulam M, Rademakers R, Chakraverty S, Cruchaga C, Morris JC, Goate AM, Cairns NJ. TARDBP 3′ UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol. 2009;118:633–645. doi: 10.1007/s00401-009-0571-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinto JB, Ritson GP, Taylor JP, Forman MS. Valosin-containing protein and the pathogenesis of frontotemporal dementia associated with inclusion body myopathy. Acta Neuropathol. 2007;114:55–61. doi: 10.1007/s00401-007-0224-7. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, Nakano I, Oda T, Tsuchiya K, Akiyama H. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatanpaa KJ, Blass DM, Pletnikova O, Crain BJ, Bigio EH, Hedreen JC, White CL, III, Troncoso JC. Most cases of dementia with hippocampal sclerosis may represent fronto-temporal dementia. Neurology. 2004;63:538–542. doi: 10.1212/01.wnl.0000129543.46734.c0. [DOI] [PubMed] [Google Scholar]
- Hatanpaa KJ, Bigio EH, Cairns NJ, Womack KB, Weintraub S, Morris JC, Foong C, Xiao G, Hladik C, Mantanona TY, White CL., III TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a midwest-southwest consortium for FTLD study. J Neuropathol Exp Neurol. 2008;67:271–279. doi: 10.1097/NEN.0b013e31816a12a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, McCluskey LF, Trojanowski JQ, Lee VM-Y. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration anad amyotrophic lateral sclerosis. Am M Pathol. 2008;173:182–194. doi: 10.2353/ajpath.2008.080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Stroh A, Dugger B, Dickson DW. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol. 2009;118:349–358. doi: 10.1007/s00401-009-0547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovari E, Gold G, Giannakopolous P, Bouras D. Cortical ubiquitin-positive inclusions in frontotemporal dementia without motor neuron disease: a quantitative immunohistochemical study. Acta Neuropathol. 2004;108:207–212. doi: 10.1007/s00401-004-0881-8. [DOI] [PubMed] [Google Scholar]
- Kwong LK, Neumann M, Sampathu DM, Lee VM-Y, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 2007;114:63–70. doi: 10.1007/s00401-007-0226-5. [DOI] [PubMed] [Google Scholar]
- LeBer I, Camuzat A, Berger E, Hannequin D, Laquerriere A, Golfier V, Seilhean D, Viennet G, Couratier P, Verpillat P, Heath S, Camu W, Martinaud O, Lacomblez I, Vercelletto M, Salachas F, Sellal F, Didic M, Thomas-Anterion C, Puel M, Michel B-F, Besse C, Duyckaerts C, Meininger V, Campion D, Dubois B, Brice A. Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology. 2009;72:1669–1676. doi: 10.1212/WNL.0b013e3181a55f1c. [DOI] [PubMed] [Google Scholar]
- Luty AA, Kwok JBJ, Thompson EM, Blumbergs P, Brooks WS, Loy CT, Dobson-Stone C, Panegyres PK, Hecker J, Nicholson GA, Halliday GM, Schofield PR. Pedigree with frontotemporal lobar degeneration-motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol. 2008;8:1–11. doi: 10.1186/1471-2377-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Baborie A, Pickering-Brown S, Plessis DD, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–549. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- Mackenzie IRA, Foti D, Woulfe J, Hurwitz TA. Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain. 2008;131:1282–1293. doi: 10.1093/brain/awn061. [DOI] [PubMed] [Google Scholar]
- Mackenzie IRA, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJM, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DMA. Nomenclature and nosology for neuropathologic subtypes of fronto-temporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IRA. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009a;132:2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, Forman MS, Troost D, Kretzschmar HA, Trojanowski JQ, Lee VM-Y. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009b;117:137–149. doi: 10.1007/s00401-008-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, Gallo J-M, Hortobagyi T, Shaw CE, Rogelj B. Nuclear import impairment causes cytoplasmic transactivation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain. 2010;133:1763–1771. doi: 10.1093/brain/awq111. [DOI] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst A, Taylor KI, Tolnay M. Hippocampal sclerosis dementia: a reappraisal. Acta Neuropathol. 2007;114:335–345. doi: 10.1007/s00401-007-0262-1. [DOI] [PubMed] [Google Scholar]
- Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M. TDP-43-negative FTLD-U is a significant new clinicopathological subtype of FTLD. Acta Neuropathol. 2008;116:147–157. doi: 10.1007/s00401-008-0395-x. [DOI] [PubMed] [Google Scholar]
- Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VM. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–1352. doi: 10.2353/ajpath.2006.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang I-F, Wu L-S, Chang H-Y, Shen C-KJ. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]