

Abstract
Cilia are mechanosensing organelles that communicate extracellular signals into intracellular responses. Altered functions of primary cilia play a key role in the development of various diseases including polycystic kidney disease. Here, we show that endothelial cells from the oak ridge polycystic kidney (Tg737orpk/orpk) mouse, with impaired cilia assembly, exhibit a reduction in the actin stress fibers and focal adhesions compared to wild type. In contrast, endothelial cells from polycystin-1 deficient mice (pkd1null/null), with impaired cilia function, display robust stress fibers and focal adhesion assembly. We found that the Tg737orpk/orpk cells exhibit impaired directional migration and endothelial cell monolayer permeability compared to the wild type and pkd1null/null cells. Finally, we found that the expression of heat shock protein 27 (hsp27) and the phosphorylation of FAK are down regulated in the Tg737orpk/orpk cells and overexpression of hsp27 restored both FAK phosphorylation and cell migration. Taken together, these results demonstrate that disruption of the primary cilia structure or function compromises the endothelium through the suppression of hsp27 dependent actin organization and focal adhesion formation, which may contribute to the vascular dysfunction in ciliopathies.
Keywords: primary cilia, hsp27, cardiovascular, endothelial
Introduction
Primary cilia are microtubule-based organelles that play a critical role in embryonic development and differentiation (Berbari et al., 2009). Their dysfunction has been associated with numerous disease states termed ciliopathies, which includes Polycystic Kidney Disease (PKD) (Kolb and Nauli, 2008; Nauli et al., 2003; Nauli et al., 2008). PKD itself is a unique ciliopathy as it can develop due to either change in the ciliary structure (deletion of intraflagellar transport proteins Tg737 or kif3) or in its signaling function (deletions of polycystin1 or 2), both of which can contribute to the disease development and result in the proliferation of renal epithelial cells and cyst formation in the kidney (Murcia et al., 1999). Physiologically, it has been shown that epithelial cells from Tg737orpk/orpk and pkd1null/null mouse models of PKD failed to induce calcium influx in response to flow mediated shear stress (Nauli et al., 2003; Nauli et al., 2006).
Although, dysfunction in the cilia have a major impact on kidney epithelial cells (Nauli et al., 2006; Siroky et al., 2006; Torres and Harris, 2006), PKD patients tend to present with cardiovascular complications such as hypertension approximately ten years prior to clinical manifestation of renal impairment (Ecder and Schrier, 2001; Kelleher et al., 2004). Moreover, these patients also exhibit endothelial dysfunctions not observed in normal patients suggesting that the cardiovascular complications seen in PKD (Kocaman et al., 2004; Turgut et al., 2007; Wang et al., 2000) may be mediated through alterations in endothelial cell function.
Primary cilia sense and respond to both chemical and mechanical signals. For example, they can coordinate platelet derived growth factor (PDGF)-induced cell migration in fibroblasts (Schneider et al., 2010). Cilia have also been implicated in transducing the mechanical signals induced by fluid shear stress in cholangiocytes (Masyuk et al., 2006), MDCK cells (Praetorius and Spring, 2001; Praetorius and Spring, 2003) and in pkd null kidney epithelial cells (Nauli et al., 2003; Nauli et al., 2006). Interestingly, primary cilia have the ability to sensitize the endothelial cells to fluid shear stress (Hierck et al., 2008). In fact, endothelial cilia were found in areas of disturbed flow and at the base of atherosclerotic lesions (Van der Heiden et al., 2008). Moreover, endothelial cells from Tg737orpk/orpk and pkd1null/null PKD mouse models failed to induce calcium influx in response to shear with a reduction in nitric oxide production (AbouAlaiwi et al., 2009; Nauli et al., 2008). These findings suggest that fluid shear induced bending of the primary cilia can transduce mechanical signals through the basal body and into the cell via interactions with the microtubules and actin cytoskeleton (Berbari et al., 2009). Although, this macro mechanosensing ability of cilia has been demonstrated for shear stress, research by Donnelly et.al. has also demonstrated that local micro mechanical cues from the extracellular matrix (ECM) can also influence ciliary function (Donnelly et al., 2010). The actin cytoskeleton is a critical mediator of mechanotransduction, since it can transfer contractile forces on to the ECM and also convey the mechanical forces applied to the ECM in to the cell (Ingber, 2006). Importantly, the mechanotransduction ability of the cilia to trigger calcium influx is governed and regulated by the actin cytoskeleton and intact adhesions to the ECM (Alenghat et al., 2004). However, there are currently no reports of alterations in primary cilia structure or function influencing the organization of the actin cytoskeleton or focal adhesion formation. In this study we addressed the role of primary cilia in actin cytoskeleton and focal adhesion assembly by using endothelial cells from the primary cilia deficient Tg737orpk/orpk and the primary cilia dysfunctional pkd1null/null mice (Murcia et al., 2000; Taulman et al., 2001). Our results present the primary cilia as a regulator of endothelial actin organization, focal adhesion formation, as well as directional migration and cell permeability in part through the modulation of hsp27 expression and signaling.
Experimental Procedures
Cell culture
Aortic endothelial cells isolated from Tg737orpk/orpk and pkd1null/null mice along with their wild type (WT) heterozygous controls were used in this study (Nauli et al., 2008). Cells were maintained in high glucose Dulbecco's Modification of Eagle’s Medium (DMEM) supplemented with 2% fetal bovine serum (FBS) and 5% Penicillin/Streptomycin (complete medium) as described previously (Nauli et al., 2008). These cells were previously characterized as bonafide endothelial cells by using different endothelial markers and functional signaling such as calcium and NO in response to stress (Nauli et al., 2008).
Transfection
Tg737orpk/orpk cells were transfected with a plasmid containing mouse hsp-27- GFP (Origene) or GFP alone using Effectene transfection reagent (Qiagen).
Immunofluorescence staining and microscopy
Cells were grown on glass coverslips (#1, Fisherbrand) or MatTek dishes, rinsed with phosphate-buffered saline (PBS) and fixed for 20 min at room temperature in PBS containing 4% paraformaldehyde. Following fixation, cells were rinsed and permeabilized with PBS containing 0.25% Triton-X100. Permeabilized cells were again rinsed and blocked with normal serum for 20 min and then incubated at room temperature for 1 hour with the primary antibodies. Focal adhesions were stained with vinculin monoclonal antibody (Sigma) and cilia were stained with acetylated alpha tubulin (Sigma). Focal adhesion kinase phosphorylation was assessed by using a phoshospecific antibody against FAK-Tyr 397 (Cell Signaling). The cells were then washed with PBS and incubated with Alexa Fluor-conjugated secondary antibodies and with Alexa Fluor-594 conjugated phalloidin (Invitrogen) to stain actin stress fibers. After the incubation, cells were washed with PBS and mounted on glass slides using fluoromount. Images were obtained using a Olympus Confocal microscope using 60 X objective. Images were processed using Image J (NIH) software.
Cell migration/Scratch-wound assay
Cells were plated onto 6 well cell culture plates and cultured to a confluent monolayer. The monolayer was wounded using a 200uL pipette tip making scratch in the middle of the well. The cells were then washed with growth media and random areas were marked with pen and images were taken before and after 20–24 h of wounding using an Olympus microscope equipped with a CCD camera. Qcapture Pro software was used to measure the area of the wound.
SDS-PAGE and Western blot analysis
Cells were lysed in RIPA buffer (50mM Tris-HCl at pH 7.4, 150mM sodium chloride, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS) with protease and phosphates inhibitor cocktail (Boston Bioproducts). Proteins were resolved by electrophoresis on 4–20% gradient SDS- polyacrylamide gels (Laemmli, 1970) followed by transfer to Immobilon ® polyvinylidene difluoride membrane. Immunoblotting was performed with primary antibodies as follows: Anti-hsp27 (Stressgen), Anti-hsp90 (Stressgen) and Anti-α-tubulin (Abcam). The ECL method was used with anti-rabbit or mouse IgG-conjugated horseradish peroxidase (Pierce West Pico) 1:20,000 and developed with Kodak XAR film. Results were quantified using ImageJ software (Rasband, 1997–2007).
Cell permeability measurements
To determine endothelial permeability, cells were seeded (40000 cells/well) in Falcon HTS FluoroBlokTM 1.0 mm inserts (Becton Dickinson, Bedford, MA), and cultured to a confluent monolayer. After 24 h starvation, the upper chamber was replaced with 0.2 ml of Phenol Red-free medium containing the fluorescent Alexafluor-488-dextran (3 kDa, Invitrogen/Molecular Probes, Eugene, OR)(8ug/ul) with or without thrombin (1U/ml; Sigma). The lower chamber was filled with 0.7 ml of Phenol Red-free medium without Alexafluor-488-dextran. The fluorescence intensity in the lower chamber was read every min. using a Synergy 4 plate reader (BioTek) (Ex 485 nm and Em 528).
Data analysis
All the data shown is mean ± SEM from at least three independent experiments. Data was analyzed using student t-test and significance was set at p < 0.05.
Results
Primary Cilia deficient Tg737orpk/orpk endothelial cells exhibit reduced stress fibers and focal adhesions
Given that the actin cytoskeleton is required for mechanotransdcution by cilia (Alenghat et al., 2004), and deficits in cilia structure or function interfere with mechanotransduction (Nauli et al., 2008; Nauli and Zhou, 2004), we investigated if impaired cilia structure or function can alter the actin organization and focal adhesions. This was achieved using endothelial cells from the primary cilia deficient Tg737orpk/orpk and the primary cilia dysfunctional pkd1null/null mice (Murcia et al., 2000; Taulman et al., 2001). Initially, we confirmed an endothelial cell phenotype by immunostaining for VE-Cadherin and found that all the three cell lines used in this study express VE Cadherin at the plasma membrane (Fig.S1). Next, primary cilia expression was assessed by immunostaining with acetylated-alpha-tubulin antibody. Primary cilia were present in the wild type and pkd1null/null endothelial cells and were absent in Tg737orpk/orpk cells (Fig.S2). We then evaluated the assembly of actin stress fiber and focal adhesions using Alexa Fluor-594 phalloidin and vinculin staining, respectively. As shown in Fig.1, wild type cells formed stress fibers with distinct focal adhesions found throughout the cytoplasm and at the periphery. In contrast, Tg737orpk/orpk cells formed a limited stress fiber network with much smaller peripheral focal adhesions. However, the cilia dysfunctional pkd1null/null cells demonstrated a robust stress fiber formation with clear focal adhesions, mostly at the periphery of the cell. Quantitative analysis revealed that more than 90% of the Tg737orpk/orpk cells had weaker stress fiber formation and smaller peripheral focal adhesions compared to wild type and pkd1null/null cells (Fig.S3 and Fig.1). Overall, we found that the number and size of assembled focal adhesions was increased in the pkd1null/null cells compared to wild type and Tg737orpk/orpk cells (Fig.S3).
Figure 1. Impairement in primary cilia structure and function alters stress fiber formation and focal adhesion assembly in endothelial cells.
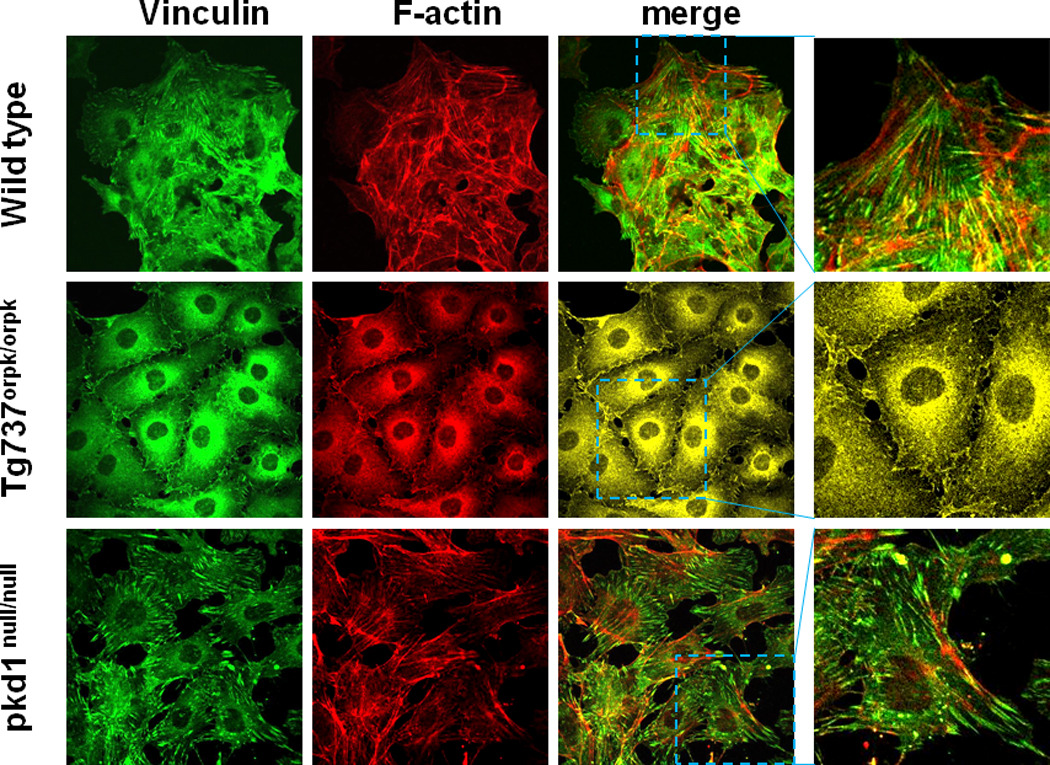
Confocal fluorescence images of wild type, Tg737orpk/orpk, and pkd1null/null endothelial cells immunostained for focal adhesions (vinculin = green) and actin stress fibers (phalloidin = red). Colocalization of stress fibers and focal adhesions (yellow) is shown in the merged image. (Inset: enlarged images). Images shown are representative of at least three independent experiments.
Directional migration is impaired in endothelial cells lacking primary cilia
Cell migration is dependent on dynamic actin remodeling and focal adhesion formation (Gardel et al., 2010). Therefore, we investigated if alterations in the actin cytoskeleton and focal adhesion assembly, induced by impaired primary cilia structure or function, could influence cell migration in a scratch-wound assay. Cells were grown to confluence and directional cell migration was measured by making a scratch-wound. Images were acquired from defined areas demarcated at the start of the experiment. Wild type cells were observed to migrate as an intact monolayer, while Tg737orpk/orpk cells appeared to detach from the monolayer and migrated randomly (Fig.2A). Quantification of the migration (% wound closure) revealed no statistically significant difference between cell types (Fig.2C), though both Tg737orpk/orpk and pkd1null/null cells appeared to migrate faster than the wild type cells. Interestingly, at complete wound closure, we found that the wild type wound closed by orienting its cells in the direction of migration, while Tg737orpk/orpk cells oriented perpendicular to the direction of migration (Fig.2B). On the other hand, the pkd1null/null cells orientated randomly at the edge of the wound closure. These findings suggest that the absence of cilia impairs the directionality of endothelial cell migration.
Figure 2. Directional migration is impaired in primary cilia deficient Tg737orpk/orpk endothelial cells.
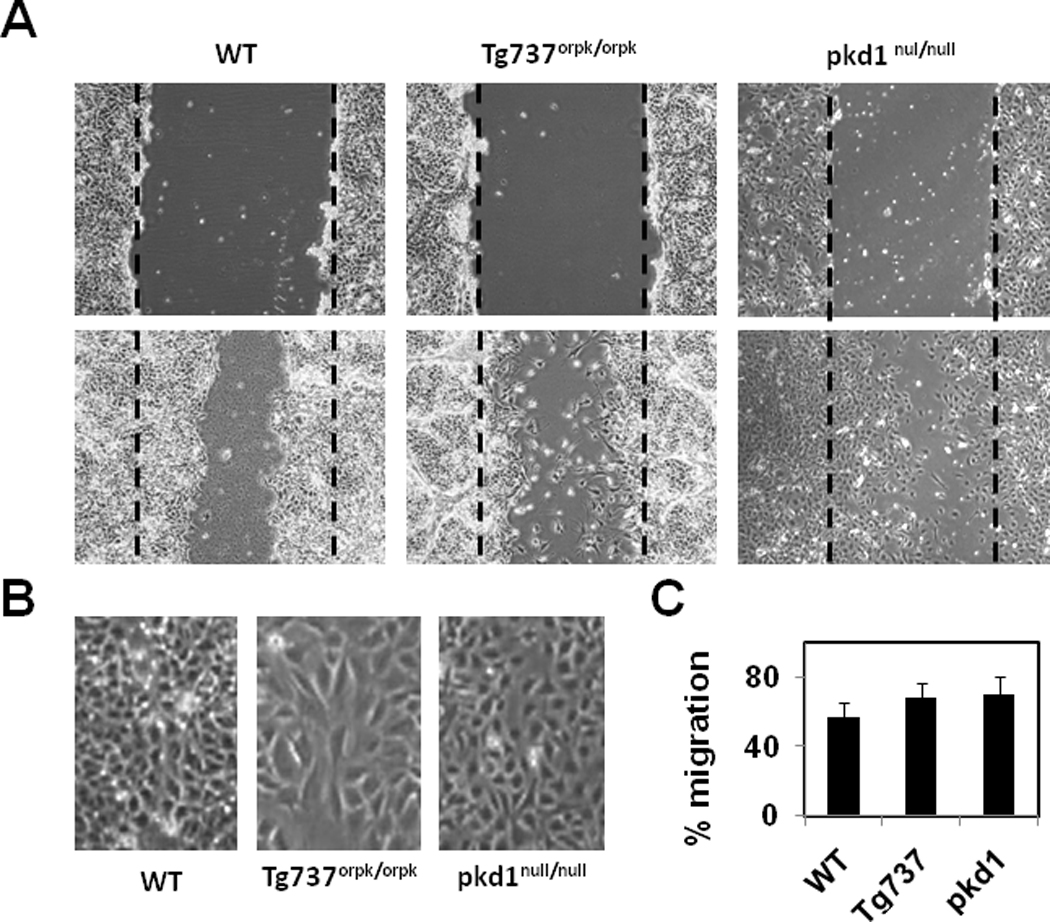
A) Photmicrographs of endothelial cells at 0 and 24 hours after wounding. The dashed line denotes the wound edge. B) Images of endothelial cells showing the orientation at the wound closure. Note: Wild type endothelial cells migrated and closed the wound by orienting perpendicular to the direction of the wound where as Tg737orpk/orpk cells oriented abnormally (parallel) to the wound. C) Quantitative analysis of the wound closure presented as % migration. The results shown are mean ± SEM from 3 independent experiments.
Tg737orpk/orpk endothelial cells exhibit high cell permeability compared to wild type and pkd1null/null cells
To determine if altered primary cilia structure or function could influence cell permeability, we assayed the basal permeability of Tg737orpk/orpk, pkd1null/null and wild type cells. Permeability was determined by the diffusion of 3000 MW dextran Alexa Fluor-488 through fluroblock transwell inserts. We found that Tg737orpk/orpk endothelial permeability to the dextran was significantly higher compared to the wild type endothelial cells (Fig.3). Interestingly, treatment with the vasoactive agonist thrombin had little effect on Tg737orpk/orpk and pkd1null/null cells but significantly increased cell permeability in WT cells (Fig.3).
Figure 3. Absence of primary cilia increases endothelial monolayer permeability.
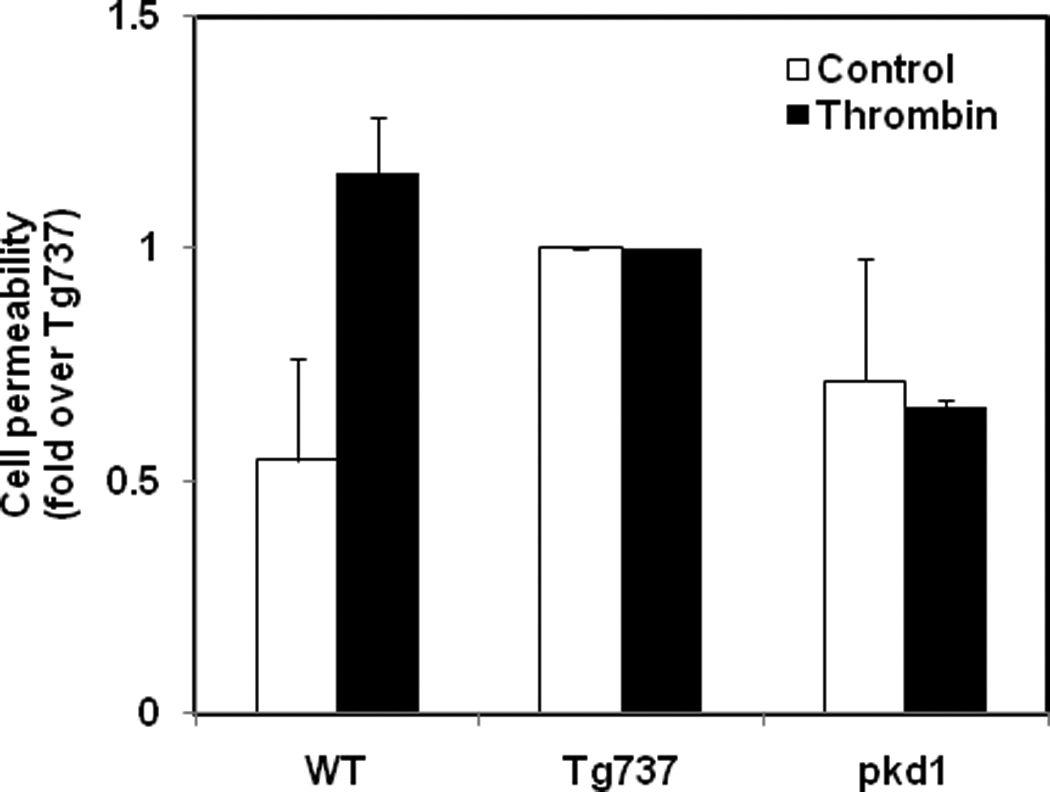
Serum starved confluent monolayers of wild type (WT), Tg737 (Tg737orpk/orpk) and pkd1 null (pkd1null/null) endothelial cells in 24 well fluoroblock tranwell plates (BD Biosciences) were incubated with 1ug/ml Alexa Fluor-488- dextran (3 kDa). The transfer of dextran into the bottom chamber was measured overtime in the presence or absence of thrombin (1U/ml). The graph represents the maximal change (at 11 min) in the fluorescence. The results shown are mean ± SEM from 3–4 independent experiments.
Absence of primary cilia suppresses heat shock protein 27 expression
To identify a molecular mechanism responsible for the observed cytoskeletal changes and increased permeability in these cells, we focused our attention on the small heat-shock protein 27 (hsp27). Hsp27 has been previously shown to regulate endothelial cell permeability by altering the actin cytoskeleton and focal adhesion assembly (Liu et al., 2009). Western blot analysis showed a suppression of hsp27 protein expression by almost 90% in Tg737orpk/orpk cells compared to wild type and pkd1null/null cells (Fig. 4A, B). However, the expression of another heat shock protein, Hsp90 was unchanged (Fig. 4C, D) demonstrating the specific down regulation of hsp-27 expression.
Figure 4. Loss of primary cilia suppresses heat shock protein 27 expression.
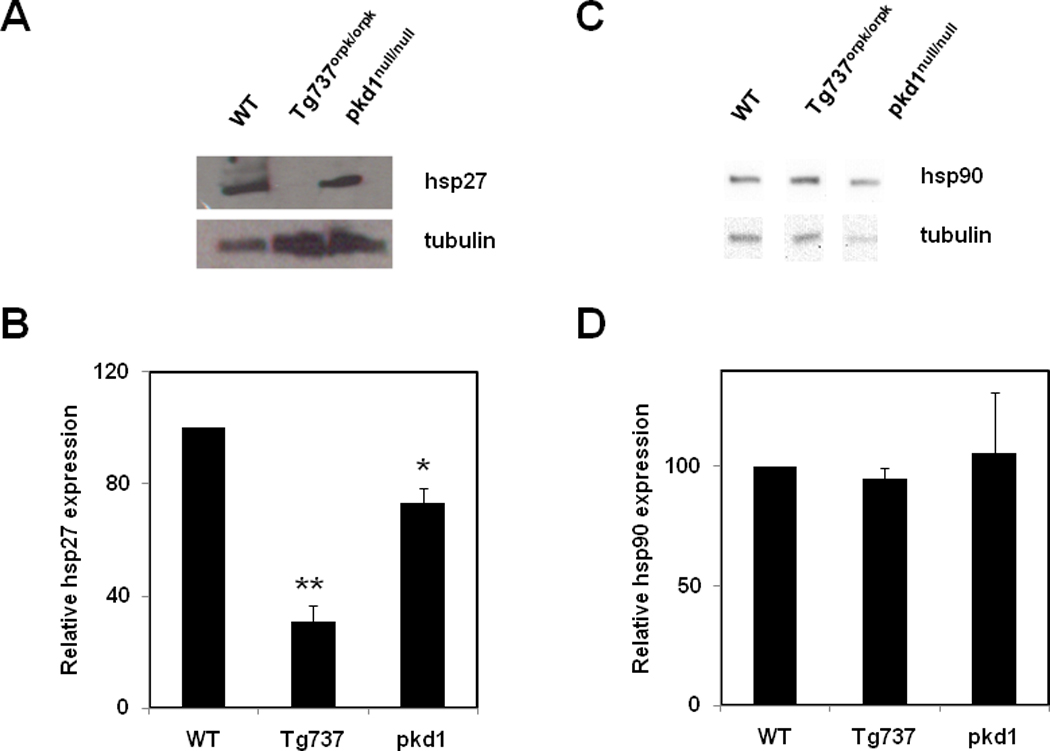
Representative Western blots showing expression of hsp27 (A) and hsp90 (C) in wild type (WT), Tg737 (Tg737orpk/orpk) and pkd1 null (pkd1null/null) endothelial cells. Quantitative analysis of the relative expression of hsp27 (B) and hsp90 (D), normalized to tubulin. The results shown are mean ± SEM from 3 independent experiments.
FAK phosphorylation is inhibited in Tg737orpk/orpk cells
To further explore the signaling of hsp27 in these cells we measured the phosphorylation of focal adhesion kinase (FAK), a down-stream mediator of hsp27 dependent modulation of actin cytoskeleton dynamics (Lee et al., 2008). To investigate this, endothelial cells cultured on cover glasses were fixed and stained with a phospho-specific antibody against FAK-tyr397. Tyrosine 397 phosphorylation of FAK is an indirect indicator of activation (Lee et al., 2008) and is known to be regulated by hsp-27 (Lee et al., 2008). Immunofluorescence analysis revealed a robust staining of the focal adhesions and stress fibers in both wild type and pkd1null/null cells (Fig.5) indicating that FAK was activated and colocalized with the stress fibers. However, phospho-FAK staining was significantly reduced in Tg737orpk/orpk cells and presented as a punctuate pattern throughout the cell with some co-localization to peripheral focal adhesions (Fig.5). Finally, we asked if overexpression of hsp27 would rescue focal adhesion formation and FAK phosphorylation. To achieve this, we transfected the Tg737orpk/orpk cells with an hsp27-GFP construct and evaluated its effects on FAK phosphorylation, focal adhesions, and directed migration. As shown in Fig.5B, we found that overexpression of hsp27-GFP in Tg737orpk/orpk cells increased FAK phosphorylation (Fig.5B) as well as focal adhesion formation (not shown) and decreased cell migration (Fig.5C) compared to transfection with GFP alone. These findings support that hsp27 is an important modulator of primary cilia-dependent EC cytoskeletal remodeling.
Figure 5. FAK phosphorylation is reduced in Tg737orpk/orpk cells.
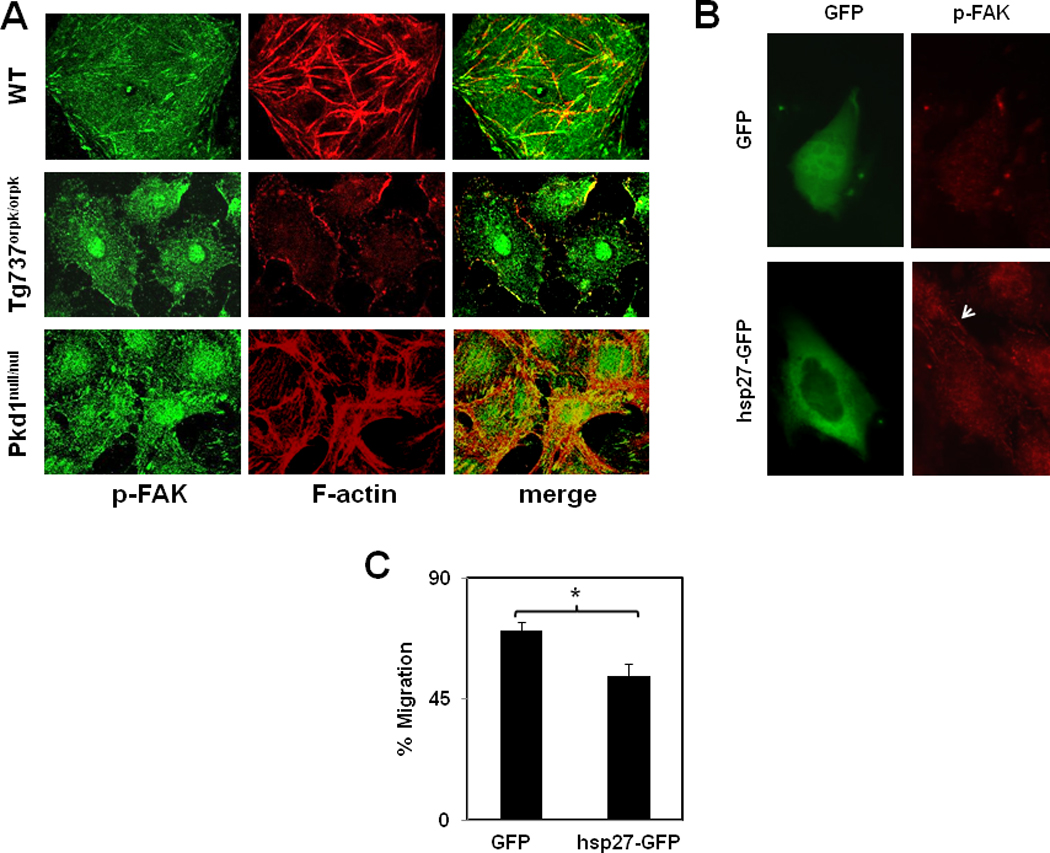
A) Confoncal fluorescence images of endothelial cells immunostained for phospho-FAK (green) and actin stress fibers (red). Colocalization of stress fibers with p-FAK (yellow) is shown in the merged images. Images shown are representative from three independent experiments. Fluorescent images showing an increase in FAK phosphorylation (B) in Tg737orpk/orpk cells transfected with hsp27-GFP compared to GFP alone. C) Quantitative analysis of the wound closure presented as % migration.
Discussion
In the present study, we show that the absence of primary cilia alters actin cytoskeleton organization through the reduction of stress fibers and focal adhesions with a concomitant impairment of directional migration and endothelial barrier integrity. Further, we provide the first evidence, that the primary cilium modulates hsp27 signaling pathway(s) through the regulation of its expression. Primary cilia themselves are predominantly known for their mechanosensing of fluid shear inducing calcium influx; however, in this study we show for the first time a direct role for primary cilia in the regulation of the actin cytoskeleton and focal adhesion assembly.
Although, primary cilia currently appear to mediate most of their cellular effects through the hedge hog signaling pathways (Satir et al., 2010), the mechanotransduction pathways activated downstream of primary cilia are not well understood. Primary cilia not only have the ability to sense and respond to shear but can also sense mechanical cues from the ECM. This response is evident in the preferential orientation of primary cilia in the direction of the collagen fibers (Donnelly et al., 2010) and the triggering of calcium influx by contact with fibronectin (Praetorius et al., 2004). In general, endothelial cells sense the mechanical forces applied to the ECM through integrin receptors (Alenghat and Ingber, 2002; Ingber, 2006; Katsumi et al., 2004). These receptors transduce extracellular mechanical signals in to biochemical responses through its intracellular interactions with the actin cytoskeleton forming a continous signaling path (Alenghat and Ingber, 2002). Similar to integrins, primary cilia also transduce mechanical signals into the cell through its interactions with the actin cytoskeleton (Berbari et al., 2009). Moreover, the mechanotransduction ability of ciliary appears to be governed by the actin cytokeleton (Alenghat et al., 2004). Our results show that primary cilia can regulate the organization of actin cytoskeleton and focal adhesion in part, through the modulation of hsp27 expression. Hsp27 was previously shown to regulate actin cytoskeleton reorganization and contractility by modulating FAK activity (Lee et al., 2008). In agreement with the hsp27 effects on FAK, we found that cells lacking cilia exhibited poor focal adhesion assembly and stress fiber formation as well as reduced FAK tyr397 phosphorylation (Lee et al., 2008). To accomplish cell migration, cells require the dynamic ability of actin cytoskeleton remodeling and focal adhesion assembly (Gardel et al., 2010) as well as the capability to orientate the cilia in the direction of migration (Christensen et al., 2008; Schneider et al., 2010; Schneider et al., 2009). We found that wild type cells, following scratch wounding, migrated in a uniform direction (perpendicular to the wound) with the cilia in the leading edge of cells oriented in the direction of cell migration (Fig.S4 and Fig.2). In contrast, Tg737orpk/orpk cells deficient in primary cilia structure or pkd1null/null cells with impaired cilia function migrated randomly. Specifically, Tg737orpk/orpk cells oriented parallel to the scratch at complete wound closure (Fig2 B). The pkd1null/null cells, on the other hand, oriented themselves randomly at complete wound closure with the cilia in the leading edge exhibiting a random orientation (not shown). Primary cilia themselves have been implicated in directional cell migration in response to the soluble mediator PDGF (Schneider et al., 2010). Here, for the first time, we show that cilia are required for the directional cell migration of endothelial cells. Further, we show that cilia dependent organization of the actin cytoskeleton is critical for endothelial cell barrier integrity. This was evident in the Tg737orpk/orpk cells lacking primary cilia, which exhibited the highest cell membrane permeability (lowest integrity) correlating with impaired actin organization and focal adhesion assembly. Importantly, consistent with the alterations in the actin cytoskeletal organization, treatment with thrombin had little effect on Tg737orpk/orpk and pkd1null/null cells but significantly increased cell permeability in WT cells. Since the actin cytoskeleton and hsp27 are important regulators of endothelial cell barrier integrity (Liu et al., 2009), it is conceivable that the effect of primary cilia is mediated through hsp27-dependent modulation of the actin cytoskeleton. Indeed, we found that the overexpression of hsp27 increased FAK phosphorylation and focal adhesion formation while reducing migration in Tg737orpk/orpk cells.
In summary, this study shows that deficits in primary cilia structure or function result in alterations in the endothelial actin cytoskeleton and focal adhesion assembly leading to impaired directional migration and cell permeability. These findings have significant implications in unraveling novel mechanisms underlining endothelial dysfunction in PKD. Alterations in the endothelial cell cytoskeleton caused by changes to primary cilia structure or function could trigger endothelial dysfunction (i.e. orientation in response to flow and alteration in permeability) by participating in and contributing to the early occurrence of a hypertension state in the progression of PKD.
Supplementary Material
Acknowledgments
Grants: Contract Grant Sponsor: NEOUCOMP (CKT and TJJ)
Contract Grant Sponsor:NIH; Contract Grant Number : DK080640 (SMN).
Literature Cited
- AbouAlaiwi WA, Takahashi M, Mell BR, Jones TJ, Ratnam S, Kolb RJ, Nauli SM. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res. 2009;104(7):860–869. doi: 10.1161/CIRCRESAHA.108.192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alenghat FJ, Ingber DE. Mechanotransduction: all signals point to cytoskeleton, matrix, and integrins. Sci STKE. 2002;2002(119):pe6. doi: 10.1126/stke.2002.119.pe6. [DOI] [PubMed] [Google Scholar]
- Alenghat FJ, Nauli SM, Kolb R, Zhou J, Ingber DE. Global cytoskeletal control of mechanotransduction in kidney epithelial cells. Exp Cell Res. 2004;301(1):23–30. doi: 10.1016/j.yexcr.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Berbari NF, O'Connor AK, Haycraft CJ, Yoder BK. The primary cilium as a complex signaling center. Curr Biol. 2009;19(13):R526–R535. doi: 10.1016/j.cub.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen ST, Pedersen SF, Satir P, Veland IR, Schneider L. The primary cilium coordinates signaling pathways in cell cycle control and migration during development and tissue repair. Curr Top Dev Biol. 2008;85:261–301. doi: 10.1016/S0070-2153(08)00810-7. [DOI] [PubMed] [Google Scholar]
- Donnelly E, Ascenzi MG, Farnum C. Primary cilia are highly oriented with respect to collagen direction and long axis of extensor tendon. J Orthop Res. 2010;28(1):77–82. doi: 10.1002/jor.20946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecder T, Schrier RW. Hypertension in autosomal-dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol. 2001;12(1):194–200. doi: 10.1681/ASN.V121194. [DOI] [PubMed] [Google Scholar]
- Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM. Mechanical Integration of Actin and Adhesion Dynamics in Cell Migration. Annu Rev Cell Dev Biol. 2010 doi: 10.1146/annurev.cellbio.011209.122036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hierck BP, Van der Heiden K, Alkemade FE, Van de Pas S, Van Thienen JV, Groenendijk BC, Bax WH, Van der Laarse A, Deruiter MC, Horrevoets AJ, Poelmann RE. Primary cilia sensitize endothelial cells for fluid shear stress. Dev Dyn. 2008;237(3):725–735. doi: 10.1002/dvdy.21472. [DOI] [PubMed] [Google Scholar]
- Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB J. 2006;20(7):811–827. doi: 10.1096/fj.05-5424rev. [DOI] [PubMed] [Google Scholar]
- Katsumi A, Orr AW, Tzima E, Schwartz MA. Integrins in mechanotransduction. J Biol Chem. 2004;279(13):12001–12004. doi: 10.1074/jbc.R300038200. [DOI] [PubMed] [Google Scholar]
- Kelleher CL, McFann KK, Johnson AM, Schrier RW. Characteristics of hypertension in young adults with autosomal dominant polycystic kidney disease compared with the general U.S. population. Am J Hypertens. 2004;17(11 Pt 1):1029–1034. doi: 10.1016/j.amjhyper.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Kocaman O, Oflaz H, Yekeler E, Dursun M, Erdogan D, Demirel S, Alisir S, Turgut F, Mercanoglu F, Ecder T. Endothelial dysfunction and increased carotid intima-media thickness in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2004;43(5):854–860. doi: 10.1053/j.ajkd.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Kolb RJ, Nauli SM. Ciliary dysfunction in polycystic kidney disease: an emerging model with polarizing potential. Front Biosci. 2008;13:4451–4466. doi: 10.2741/3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lee JW, Kwak HJ, Lee JJ, Kim YN, Park MJ, Jung SE, Hong SI, Lee JH, Lee JS. HSP27 regulates cell adhesion and invasion via modulation of focal adhesion kinase and MMP-2 expression. Eur J Cell Biol. 2008;87(6):377–387. doi: 10.1016/j.ejcb.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Liu T, Guevara OE, Warburton RR, Hill NS, Gaestel M, Kayyali US. Modulation of HSP27 alters hypoxia-induced endothelial permeability and related signaling pathways. J Cell Physiol. 2009;220(3):600–610. doi: 10.1002/jcp.21773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131(3):911–920. doi: 10.1053/j.gastro.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, Woychik RP. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development. 2000;127(11):2347–2355. doi: 10.1242/dev.127.11.2347. [DOI] [PubMed] [Google Scholar]
- Murcia NS, Sweeney WE, Jr, Avner ED. New insights into the molecular pathophysiology of polycystic kidney disease. Kidney Int. 1999;55(4):1187–1197. doi: 10.1046/j.1523-1755.1999.00370.x. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117(9):1161–1171. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauli SM, Rossetti S, Kolb RJ, Alenghat FJ, Consugar MB, Harris PC, Ingber DE, Loghman-Adham M, Zhou J. Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol. 2006;17(4):1015–1025. doi: 10.1681/ASN.2005080830. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Zhou J. Polycystins and mechanosensation in renal and nodal cilia. Bioessays. 2004;26(8):844–856. doi: 10.1002/bies.20069. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Praetorius J, Nielsen S, Frokiaer J, Spring KR. Beta1-integrins in the primary cilium of MDCK cells potentiate fibronectin-induced Ca2+ signaling. Am J Physiol Renal Physiol. 2004;287(5):F969–F978. doi: 10.1152/ajprenal.00096.2004. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184(1):71–79. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. Removal of the MDCK cell primary cilium abolishes flow sensing. J Membr Biol. 2003;191(1):69–76. doi: 10.1007/s00232-002-1042-4. [DOI] [PubMed] [Google Scholar]
- Rasband WS. ImageJ. Maryland, USA: Bethesda: U. S. National Institutes of Health, Bethesda; 1997–2007. [Google Scholar]
- Satir P, Pedersen LB, Christensen ST. The primary cilium at a glance. J Cell Sci. 2010;123(Pt 4):499–503. doi: 10.1242/jcs.050377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Cammer M, Lehman J, Nielsen SK, Guerra CF, Veland IR, Stock C, Hoffmann EK, Yoder BK, Schwab A, Satir P, Christensen ST. Directional cell migration and chemotaxis in wound healing response to PDGF-AA are coordinated by the primary cilium in fibroblasts. Cell Physiol Biochem. 2010;25(2–3):279–292. doi: 10.1159/000276562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Stock CM, Dieterich P, Jensen BH, Pedersen LB, Satir P, Schwab A, Christensen ST, Pedersen SF. The Na+/H+ exchanger NHE1 is required for directional migration stimulated via PDGFR-alpha in the primary cilium. J Cell Biol. 2009;185(1):163–176. doi: 10.1083/jcb.200806019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siroky BJ, Ferguson WB, Fuson AL, Xie Y, Fintha A, Komlosi P, Yoder BK, Schwiebert EM, Guay-Woodford LM, Bell PD. Loss of primary cilia results in deregulated and unabated apical calcium entry in ARPKD collecting duct cells. Am J Physiol Renal Physiol. 2006;290(6):F1320–1328. doi: 10.1152/ajprenal.00463.2005. [DOI] [PubMed] [Google Scholar]
- Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK. Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol Biol Cell. 2001;12(3):589–599. doi: 10.1091/mbc.12.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres VE, Harris PC. Mechanisms of Disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006;2(1):40–55. doi: 10.1038/ncpneph0070. quiz 55. [DOI] [PubMed] [Google Scholar]
- Turgut F, Oflaz H, Namli S, Alisir S, Tufan F, Temiz S, Umman S, Ecder T. Ambulatory blood pressure and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Ren Fail. 2007;29(8):979–984. doi: 10.1080/08860220701641728. [DOI] [PubMed] [Google Scholar]
- Van der Heiden K, Hierck BP, Krams R, de Crom R, Cheng C, Baiker M, Pourquie MJ, Alkemade FE, DeRuiter MC, Gittenberger-de Groot AC, Poelmann RE. Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis. 2008;196(2):542–550. doi: 10.1016/j.atherosclerosis.2007.05.030. [DOI] [PubMed] [Google Scholar]
- Wang D, Iversen J, Strandgaard S. Endothelium-dependent relaxation of small resistance vessels is impaired in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2000;11(8):1371–1376. doi: 10.1681/ASN.V1181371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.