

Abstract
Background & Aims
Dopamine and cAMP-regulated phosphoprotein, Mr 32000 (DARPP-32), is overexpressed during gastric carcinogenesis. Gastric tumors can become resistant to gefitinib, an inhibitor of the epidermal growth factor receptor (EGFR). We investigated the role of DARPP-32 in gastric tumor resistance to gefitinib.
Methods
Cell survival was determined by clonogenic survival and ATP-Glo viability assays. Apoptosis was assessed by Annexin-V and immunoblot analyses. The association between DARPP-32 and EGFR was evaluated by immunofluorescence and co-immunoprecipitation assays. Findings were validated in mice with gastric xenograft tumors. DARPP-32 expression was reduced using small hairpin (sh)RNAs in the human gastric cancer cell lines SNU-16 and MKN-45 cells.
Results
Overexpression of DARPP-32 in MKN-28 cells, which do not normally express DARPP-32, blocked gefitinib-induced apoptosis and increased the drug’s IC50 10-fold, compared to that of control cells (P<.01). Reduced expression of DARPP-32 in SNU-16 cells increased the sensitivity to gefitinib (P<.01). DARPP-32 activated PI3K-AKT signaling, increased stability of the EGFR, and suppressed EGF- or gefitinib-induced degradation of the EGFR. DARPP-32 co-localized with EGFR on the cell membrane in a complex with EGFR and the EGF receptor ERBB3. DARPP-32-mediated resistance to gefitinib resulted from increased phosphorylation of and interaction between EGFR and ERBB3, which led to phosphorylation of AKT (at serine 473). Knockdown of DARPP-32 in gastric cancer cells reduced the mean size of tumors in mice and increased their response to gefitinib.
Conclusions
DARPP-32 promotes resistance of gastric cancer cells to gefitinib by promoting interaction between EGFR and ERBB3 and activating PI3K-AKT signaling.
Keywords: stomach cancer, treatment, PI3K, AKT, drug resistance, PPP1R1B, dopamine signaling
Introduction
Gastric cancer is one of the most common cancers worldwide1. Moreover, the prognosis for gastric cancer patients remains poor, especially in more advanced stages2. Although several chemotherapeutic drugs have been used to treat gastric cancer patients, drug resistance remains a challenging clinical problem3, 4.
Dopamine and cAMP-regulated phosphoprotein, Mr 32000 (DARPP-32), is abundantly expressed in spiny neurons of the neostriatum5. We have previously shown that DARPP-32 was amplified and overexpressed in approximately two-thirds of gastric cancers6, 7. Additionally, DARPP-32 was shown to be overexpressed in several types of cancers including breast and colorectal adenocarcinomas8, 9. The expression of DARPP-32 is associated with a potent anti-apoptotic advantage for gastric cancer cells through a p53-independent mechanism that involves preservation of mitochondrial membrane potential and increased BCL2 levels10.
The human epidermal growth factor receptor (EGFR) plays a prominent role in tumor growth and metastasis11. It has become increasingly evident that EGFR works as an oncogene in concert with other ERBB family members, particularly ERBB2 and ERBB3. Upon ligand binding, these receptors homodimerize or heterodimerize leading to autophosphorylation and subsequent activation of downstream MAPK and PI3K/AKT signaling cascades, which regulate cell proliferation, angiogenesis, invasion and metastasis12, 13. Aberrant expression or activity of EGFR resulting from amplification and mutations are identified in many human epithelial cancers. Today, many EGFR-targeted agents are either available or in clinical development, including the small-molecule inhibitor gefitinib (Iressa, AstraZeneca Pharmaceuticals). Gefitinib disrupts EGFR kinase activity by binding the ATP pocket within the catalytic domain, and selectively inhibits EGF-stimulated tumor cell growth14. Despite clinical success the vast majority of patients receiving these agents eventually develop resistance.
There are two major forms of drug resistance; primary resistance and acquired resistance. One characterized mechanism of primary resistance to gefitinib is the insertion point mutations in exon 20 of the EGFR gene that render transformed cells less responsive to gefitinib15. Other markers for primary resistance to gefitinib include EGFR mutations (T790M, D761Y, L747S, T854A), c-Met amplification and PI3K/AKT activation16, 17. The first identified mechanism of acquired resistance to gefitinib was the EGFR (T790M) mutation in non-small cell lung carcinomas (NSCLC)18. MET (met proto-oncogene) amplification or activation of IGF1R was reported as an alternative mechanism for acquired resistance to gefitinib19, 20.
In absence of frequent mutations of EGFR in gastric cancer21, little is known about the mechanisms of gefitinib resistance in this cancer. In the present study, we have uncovered a novel mechanism of gefitinib resistance and report, for the first time, that overexpression of DARPP-32 enhances survival of gastric cancer cells and increases resistance to gefitinib. We have demonstrated that DARPP-32 promoted EGFR protein stability, thus enhancing the EGFR/ERBB3 interaction leading to increased activation of the PI3K/AKT pathway.
Materials and Methods
Cell Culture and Reagents
Human gastric cancer cell lines including AGS, MKN-28, MKN-45, SNU-16, and the immortalized human embryonic kidney epithelial cell line (HEK-293) were maintained in culture using either F12 medium or Dulbecco’s modified Eagle’s medium (GIBCO, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen Life Technologies, Carlsbad, CA) and 1% penicillin/streptomycin (GIBCO). Gefitinib, LY294002 and EGF were purchased from LC laboratories and Gibco. Mouse anti-DARPP-32 and Rabbit anti-DARPP-32 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and Abcam (Cambridge, MA), respectively. Horseradish peroxidase-conjugated mouse and rabbit secondary antibodies; ERBB3, EGFR, pEGFR(Y845), AKT, pAKT (S473), GSK-3β, pGSK-3β (S9), and β-actin antibodies were obtained from Cell Signaling Technology (Danvers, MA).
DARPP-32 expression and shRNA vectors
The flag-tagged coding sequence of DARPP-32 was cloned in pcDNA3.1 mammalian expression construct (Invitrogen). AGS and MKN-28 cells stably expressing cells were developed as previously described10. Flag-tagged DARPP-32 was cloned into the adenoviral (pACCMV) shuttle vector, and the adenovirus was generated by co-transfecting HEK-293 cells with the shuttle and (pJM17) backbone adenoviral plasmids using the Calcium Phosphate Transfection Kit (Applied Biological Materials Inc., Richmond, BC). EGFR siRNA and scrambled siRNA were obtained from Santa Cruz Biotechnology. Lentivirus particles expressing DARPP-32 shRNA or control shRNA were produced by GeneCopoeia (Rockville, MD) and then utilized to transduce cells.
Clonogenic survival and ATP-Glo cell viability assay
Cells were rinsed with PBS, trypsinized, and harvested in single cell suspension. Cells (1000 cells per well) were seeded in 6-well plates. The next day, cells were treated overnight with gefitinib or vehicle. After incubation for 1–2 weeks, colonies were fixed with 2% paraformaldehyde and stained with 0.05% crystal violet. Colonies with ≥50 cells were counted. For the quantitative estimation of cell growth and survival, we have used the CellTiter-Glo Cell Viability Assay (Promega, Madison, WI). Cells were plated in 96-well microplates at 4000 cells per well, and vehicle (DMSO) or various concentrations of gefitinib were administered for 24h. Measurements using a Luminometer (Turner Designs, Sunnyvale, CA) were conducted following the manufacturer’s protocol.
Apoptosis analysis
MKN-28 cells were treated with vehicle or gefitinib (25 μM) overnight. Cells were then collected and stained with Annexin V-FITC and PI (BioVision, Exton, PA). The samples were washed with PBS and resuspended in binding buffer for subsequent fluorescence-activated cell sorting (FACS) analysis. Apoptotic cell death was assessed by counting the numbers of cells that stained positive for Annexin V-FITC and negative for propidium iodide.
Immunofluorescence and Immunohistochemistry
Following cell fixation and permeabilization, immunofluorescence was performed with both DARPP-32 (1:400) and EGFR (1:400) antibodies. The cells were then washed with cold PBS three times for 3 min each, and incubated with both rabbit FITC-conjugated and mouse TRITC-conjugated secondary antibodies (1:800) (Invitrogen) at room temperature for 1h. The cells were examined by fluorescence microscopy (Olympus America Inc., Center Valley, PA).
Paraffin-embedded tissue was pretreated at 65°C for 2h, followed by deparaffinization using standard procedures. Antigen retrieval was performed using a solution of 10 mmol/L Tris, 1 mmol/L EDTA, pH 9.0 before applying the primary Ki-67 antibody (Invitrogen). Slides were then incubated for 2h at room temperature followed by extensive washes with PBS plus 0.1% Tween 20, and further incubated for 1h at room temperature with the secondary antibody conjugated with horseradish peroxidase. Horseradish peroxidase activity was detected using a Histostain Plus kit (Invitrogen) according to the manufacturer’s instruction. Tumor tissue sections were counterstained with hematoxylin and mounted.
Human EGFR phosphorylation antibody array
Human EGFR Phosphorylation Antibody Array (RayBiotech Inc., Norcross, GA) was subjected to total lysates from MKN-28/pcDNA or MKN-28/DARPP-32 stable cells following treatment with gefitinib (25 μM) overnight. The detection and analysis of protein signals were performed according to the manufacturer’s instructions.
Immunoprecipitation and Western blotting
Cells were lysed with 0.5% Triton X-100, 150 mM NaCl, 5 mM EDTA, and 50 mM Tris supplemented with protease and phosphatase inhibitors. Immunoprecipitations of equivalent total protein amounts were performed at room temperature for 1h by using a primary antibody previously bound to 50 μl of Protein A Dynabeads per sample. The beads were washed four times with wash buffer. The beads in each tube were heated to 100°C for 5 min in 20 μl of sample buffer, and then clarified by magnet. Proteins were separated on 12.5% SDS-PAGE and transferred to Immobilon PVDF membrane (Millipore, Billerica, MA). Membranes were probed with specific antibodies, and proteins were visualized by using horseradish peroxidase (HRP)-conjugated secondary antibodies and Immobilon Western Chemiluminescent HRP Substrate detection reagent (Millipore). Gel loading was normalized for equal β-actin.
Real-time RT-PCR analysis
Total RNA was isolated from cell lines by using the RNeasy Mini Kit (Qiagen, Valencia, CA). Total RNA (1 μg) was reverse transcribed by an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). The qRT-PCR was performed in an iCycler (Bio-Rad), with the threshold cycle number determined by iCycler software version 3.0. Reactions were performed in duplicate and results of three independent experiments were subjected to statistical analysis. cDNA (100 ng) was amplified and relative mRNA expression levels were quantified using the ΔΔC(t) method22. The results of the EGFR gene were normalized to HPRT1.
In vivo experiments
Five-week-old female Sprague Dawley nude mice were purchased from Harlan Laboratories, Inc (Frederick, MD) and maintained under specific pathogen-free conditions. SNU-16 cells stably expressing lentiviral DARPP-32-shRNA or scrambled-shRNA control were injected s.c. (2×106 cells per site) into the flanks. After 2 weeks, the mice were randomized into two groups (10 xenografts per group) and given gefitinib (50 mg/kg/d) or vehicle (0.1% Tween 80) thrice weekly for 18 days by oral gavage. To determine tumor volume by external caliper, the greatest longitudinal diameter (length) and the greatest transverse diameter (width) were measured. Tumor volume was calculated by the formula: Tumor volume = 1/2 (length × width2). All mice were sacrificed on day 24, and tumors were collected. The Vanderbilt Institutional Animal Care and Use Committee approved all animal work.
Statistical analyses
Data were expressed as mean ± SD of three independent experiments. Statistical significance of the in vitro studies was analyzed by a Student’s t test and ANOVA. Differences with p values ≤0.05 are considered significant.
Results
DARPP-32 inhibits gefitinib-induced cell death
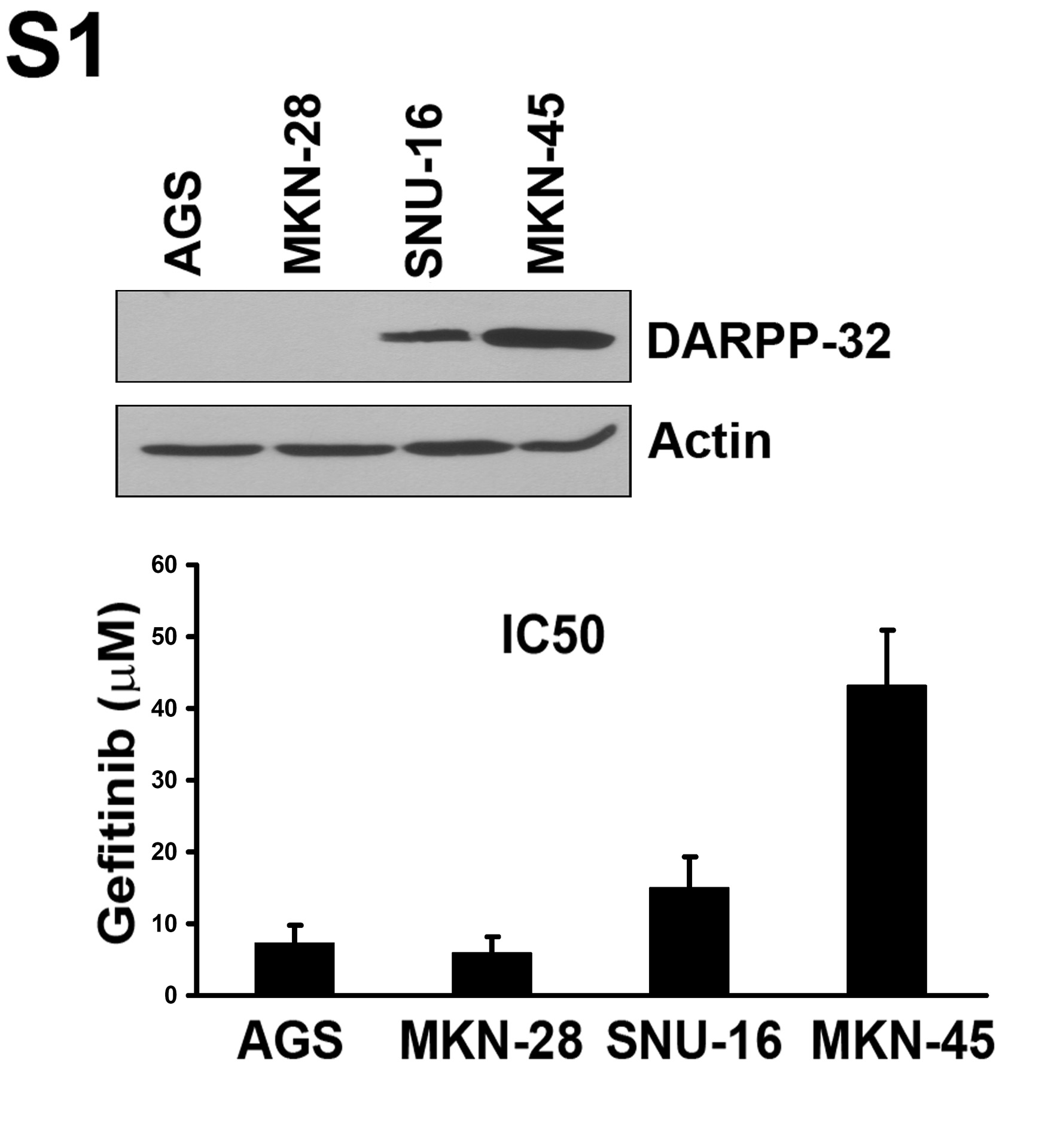
We first evaluated the IC50 and DARPP-32 protein expression in a panel of 4 gastric cancer cell lines. The results indicated that the cell lines that have a high level of DARPP-32 are more resistant to gefitinib than the cell lines that have a low level of DARPP-32 (Sup Figure 1). The ATP-Glo cell viability assay results revealed a 10-fold increase in the gefitinib IC50 in MKN-28 cells stably expressing DARPP-32 (10 μM) as compared to empty vector control (1 μM) (Figure 1A). For increased stringency, we used gefitinib (25 μM) for an overnight treatment and long-term (14 days) clonogenic survival assay. The results indicated that MKN-28 cells stably expressing DARPP-32 were more resistant to gefitinib (3-fold survival increase, p<0.01) as compared to control cells (Figure 1B). Using the SNU-16 cells that are resistant to gefitinib, the knockdown of endogenous DARPP-32 by lentiviral shRNA system led to a 4-fold reduction in the IC50 from 20 μM in scrambled shRNA cells to 5 μM in DARPP-32 shRNA cells (Figure 1C). The cell survival was decreased by 70% relative to scrambled shRNA control cells (p<0.01) (Figure 1D). Consistent with these results, the Annexin V-FITC apoptosis assay showed that overexpression of DARPP-32 inhibited gefitinib-induced apoptosis by approximately 2.5-fold relative to control cells (p<0.01) (Figure 2A). Western blot analysis indicated that DARPP-32 expression in MKN-28 cells blocked activation of caspases 3 & 9 and cleavage of PARP (Figure 2B). In contrast, the knockdown of endogenous DARPP-32 in SNU-16 cells increased activation of caspases 3 & 9 and cleaved PARP (Figure 2C). Taken together, these results have established an important role of DARPP-32 in gefitinib resistance in gastric cancer cells, raising the question about the mechanism by which DARPP-32 suppresses gefitinib-induced apoptosis.
Figure 1. DARPP-32 counteracts gefitinib-induced gastric cancer cell death.

A) Western blot analysis showing the levels of DARPP-32 in MKN-28 cells stably expressing DARPP-32 or pcDNA (upper panel). The relative cell viability following gefitinib treatment demonstrates a 10-fold increase in the IC50 in DARPP-32-expressing cells (lower panel). B) The clonogenic survival assay demonstrates a significant increase in relative cell survival in the MKN-28 cells stably expressing DARPP-32. The data were normalized to untreated cells (right panel); error bars indicate SD; **, p<0.01 (ANOVA test). C) Western blot analysis showing the levels of DARPP-32 in the SNU-16 cells following the DARPP-32 knockdown using shRNA expressing lentiviruses (10 MOI) (upper panel). The relative cell viability following gefitinib treatment is shown in the lower panel. D) The knockdown of DARPP-32 in the SNU-16 cells lead to a significant reduction in cell survival following gefitinib treatment (25 μM) overnight; error bars indicate SD; **, p<0.01 (t test).
Figure 2. DARPP-32 blocks gefitinib-induced apoptosis in gastric cancer cells.

A) The MKN-28 cells stably expressing DARPP-32 (DP01 and DP02) or empty vector were analyzed using the Annexin V-FITC and propidium iodide (PI) following the treatment with gefitinib (25 μM) or vehicle overnight. The early apoptotic cells, typically Annexin V positive and PI negative were indicated in the bottom right quadrant. Summary of the data is shown on the right panel. B) Western blot analyses of β-Actin, PARP, caspase 9 and caspase 3 proteins. C) The results in (A) and (B) were validated using the SNU-16 cells following knockdown of DARPP-32 with lentiviral particles (10 MOI) expressing DARPP-32 shRNA or control shRNA.
DARPP-32 induces EGFR-regulated PI3K-AKT pathway
The results showed that stable and transient overexpression of DARPP-32 led to increased p-AKT(S473) and its downstream substrate p-GSK-3β (S9) protein levels in MKN-28 cells (Figure 3A & 3B). In contrast, knockdown of endogenous DARPP-32 expression by shRNA resulted in decreased p-AKT (S473) and p-GSK-3β (S9) protein levels in SNU-16 cells (Figure 3C). These findings indicate that DARPP-32 positively regulates the PI3K/AKT survival pathway in gastric cancer cells. Because of the role of ERBB family members of growth factor receptors in regulating the PI3K/AKT pathway, we next utilized a human EGFR antibody array, which comprises spotted antibodies specific for total and phosphorylated proteins of EGFR, ERBB2, ERBB3, and ERBB4 receptors. Following the treatment with gefitinib (25 μM) overnight, MKN-28 cells stably expressing DARPP-32 had significantly higher levels of both total EGFR (5-fold) and p-EGFR(Y845) (4-fold) as compared to empty vector controls (Figure 3D). Western blot analysis confirmed these findings, following the treatment with gefitinib (25 μM) overnight (Figure 3E). In contrast, knockdown of endogenous DARPP-32 resulted in a significant decrease of total EGFR and p-EGFR (Y845) levels in SNU-16 cells (Figure 3F). These results suggested that DARPP-32 expression can significantly up-regulate expression and phosphorylation of EGFR receptor, which may be involved in activating the PI3K/AKT survival pathway in response to gefitinib in gastric cancer cells.
Figure 3. DARPP-32 regulates EGFR-mediated AKT survival pathway in gastric cancer cells.

A) Western blot analysis of AKT, pAKT(S473), GSK-3β, and pGSK-3β(S9) in MKN-28 stably expressing DARPP-32 (DP01 and DP02) or empty vector. B) The same as in (A) following transient infection with different titers of adenoviruses expressing DARPP-32 or control. C) The same as in (A) following lentiviral shRNA knockdown of DARPP-32 or control shRNA in SNU-16. D) Human EGFR phosphorylation antibody array using lysates from MKN-28 cells stably expressing DARPP-32 or empty vector, cultured in the presence of gefitinib (25 μM) overnight, demonstrates up-regulation of total and phosphor-EGFR protein in DARPP-32 expressing cells. Quantitative data are shown on the right panel. E–F) Western blot analysis, following overexpression (E) or knockdown of DARPP-32 (F), confirms that results in (D).
DARPP-32 enhances EGFR/ERBB3 heterodimerization in a protein complex
We next investigated whether the DARPP-32 protein could bind EGFR and ERBB3 thereby affecting downstream PI3K/AKT signaling. We transiently expressed exogenous DARPP-32 and EGFR or DARPP-32 and ERBB3 in HEK-293 cells. We found that both EGFR and ERBB3 co-immunoprecipitated independently with the DARPP-32 protein (Figure 4A). In order to confirm that endogenous DARPP-32, EGFR, and ERBB3 proteins are present in the same complex, we carried out a 3-way immunoprecipitation with specific antibodies in MKN-45 cells that express high levels of endogenous DARPP-32. The results indicated that DARPP-32 co-exists in the same protein complex with both EGFR and ERBB3 (Figure 4B). Using immunofluorescence, we confirmed the co-localization of DARPP-32 and EGFR on the cell membrane (Figure 4C). We next tested the possibility that DARPP-32 enhances the interaction between EGFR and ERBB3, thereby promoting the activation of the PI3K-AKT pathway. The co-immunoprecipitation experiments showed a stronger interaction between EGFR and ERBB3 in DARPP-32-expressing cells than in control cells (Figure 4D & 4E). These results were validated using shRNA knockdown of endogenous DARPP-32 in the SNU-16 cell model (Figure 4F). This enhanced interaction in DARPP-32 expressing cells could explain the increase in AKT phosphorylation and resistance to gefitinib in DARPP-32 expressing cells.
Figure 4. DARPP-32 associates with EGFR/ERBB3 and enhances their interaction.

A) HEK-293 cells were transiently transfected with the indicated constructs, and the interaction between DARPP-32 and ERBB3 or DARPP-32 and EGFR was examined by co-immunoprecipitation using specific antibodies. B) The protein interaction of endogenous DARPP-32, EGFR, and ERBB3 was evaluated by a three-way co-immunoprecipitation in MKN-45 cells. C) Immunofluorescence analysis using MKN-28 cells, following adenoviral infection with DARPP-32 (5 MOI), demonstrates co-localization of EGFR and DARPP-32 proteins on the cell membrane. D) Using immunoprecipitation of EGFR (left panel) or ERBB3 (right panel), the EGFR-ERBB3 interaction was examined in MKN-28 cells stably expressing DARPP-32 or empty vector. E) The same as in (D), using adenoviral infection of DARPP-32. F) The same as in (D), following lentiviral shRNA DARPP-32 knockdown in SNU-16 cells.
DARPP-32 enhances EGFR protein stability and activates ERBB3 and downstream signaling
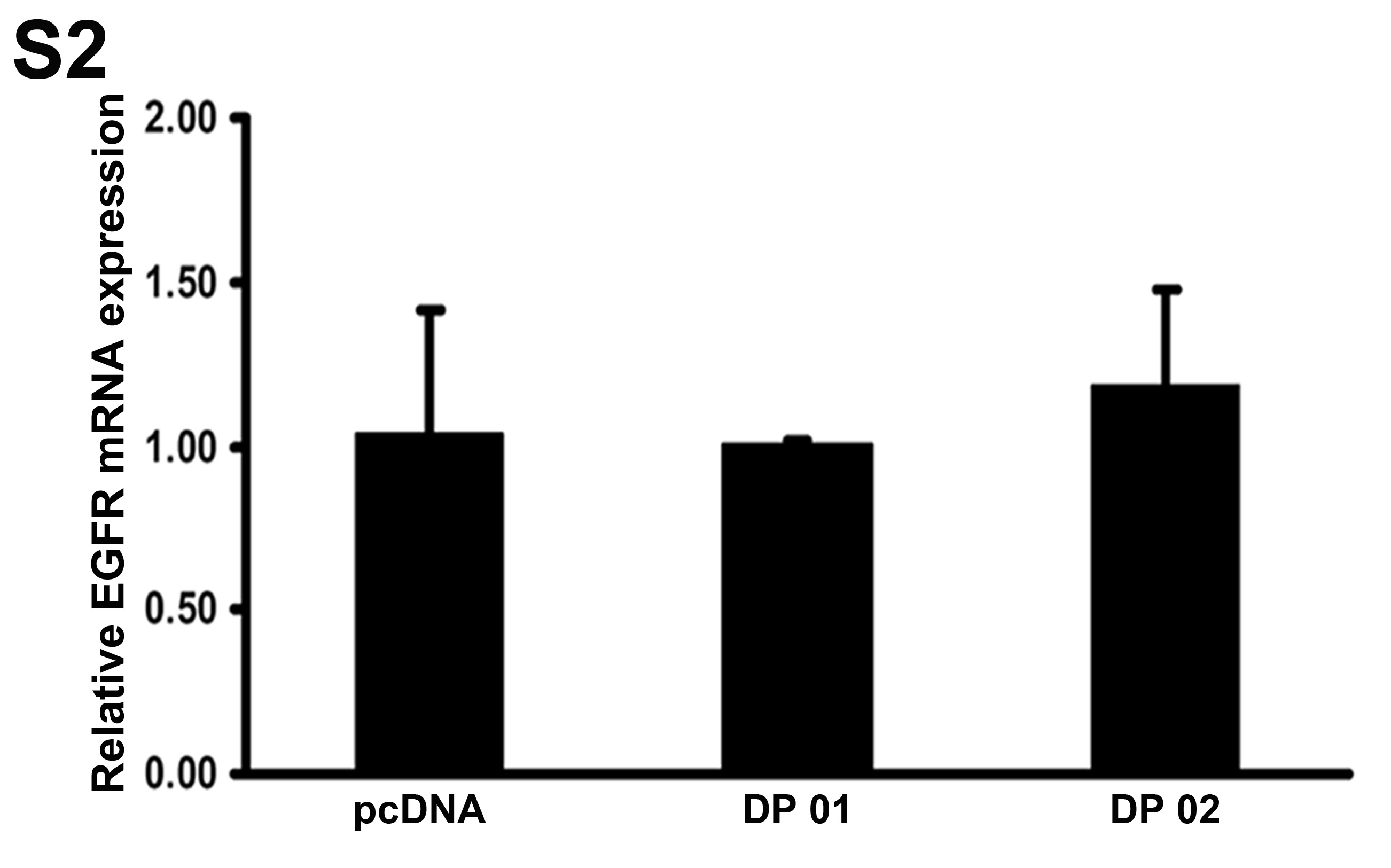
Using quantitative real-time RT-PCR, we could not detect a significant difference in EGFR mRNA levels between DARPP-32-expressing cells and control cells (Sup Figure 2). This suggested that DARPP-32-mediated up-regulation of EGFR protein expression could be due to increased protein stability. To analyze the effects of DARPP-32 on EGF-induced EGFR degradation, MKN-28/DARPP-32 or MKN-28/pcDNA stable cells were treated with EGF (100 ng/ml) for 30 and 45 min, and EGFR protein levels were determined by Western blot analysis. The data indicated that DARPP-32 significantly delayed the EGF-induced degradation of EGFR protein in MKN-28 cells (Figure 5A). In addition, stable and transient expression of DARPP-32 significantly blocked degradation of EGFR protein after treatment with 25 μM gefitinib overnight (Figures 5B). The results were validated using knockdown of DARPP-32 in the SNU-16 cell model (Figure 5C). Using immunoprecipitation and Western blot results, we have shown that DARPP-32 stable expression significantly enhanced EGFR phosphorylation after treatment with EGF (100 ng/ml) for 1h in MKN-28 cells (Figure 5D). Moreover, knockdown of endogenous DARPP-32 expression significantly reduced EGFR phosphorylation following treatment with EGF (100 ng/ml, 1h) or gefitinib (25 μM) overnight in SNU-16 cells (Figures 5E & 5F).
Figure 5. DARPP-32 promotes EGFR protein stability and tyrosine phosphorylation.

A) MKN-28 cells stably expressing DARPP-32 or empty vector were treated with EGF (100 ng/ml) for the indicated time points, and total EGFR protein levels were determined by Western blot analysis. Relative EGFR expression levels are presented, time=0 is shown as 1.0. B) Western blot analysis using lysates from MKN-28/DARPP-32 stable cells (left panel) or infected with adenoviral DARPP-32 (right panel), following treatment with gefitinib (25μM) or vehicle (DMSO) overnight. C) Western blot analysis as in (B), following lentiviral shRNA DARPP-32 knockdown in SNU-16 cells. D-F) Immunoprecipitation with anti-p-Tyr(102) antibody followed by Western blot analysis of EGFR protein in MKN-28/DARPP-32 stable cells, treated with EGF (100 ng/ml) for 1h (D) or following lentiviral shRNA DARPP-32 knockdown in SNU-16 cells and treatment with EGF (E) or gefitinib (25μM) overnight (F).
To determine if DARPP-32-mediated EGFR protein stability increases phosphorylation of ERBB3, we stably or transiently expressed DARPP-32 in MKN-28 cells followed by EGF (100 ng/ml, 1h) treatment, p-Tyr (102) immunoprecipitation, and Western blot analysis of ERBB3 protein. The results clearly showed that DARPP-32 substantially increased phosphorylated ERBB3 protein levels in response to treatment with EGF (Figures 6A, left panel, & Sup Figure 3A). The results were validated using shRNA knockdown of endogenous DARPP-32 in SNU-16 and MKN-45 cells (Figures 6A, right panel, & Sup Figure 3B). We also investigated if DARPP-32 is important for the regulation of gefitinib resistance through ERBB3/PI3K/AKT signaling. Following gefitinib (25 μM) and/or LY294002 (20 μM) treatment overnight, stable and transient DARPP-32 expression blocked gefitinib-induced dephosphorylation of AKT (Figures 6D & 6E, Sup Figure 4A) whereas knockdown of endogenous DARPP-32 sensitized cells to gefitinib and reduced the levels of phospho-AKT (Figure 6F); knockdown of endogenous EGFR reduced the levels of phospho-AKT (Sup Figure 4B).
Figure 6. DARPP-32 regulates phosphorylation of ERBB3 and downstream PI3K-AKT pathway.

A–C) MKN-28 cells stably expressing DARPP-32 or empty vector and SNU-16 cells infected with lentiviral shRNA DARPP-32 or control (10 MOI) were treated with EGF (100 ng/ml) for 1h (A) or gefitinib overnight (B-C), followed by immunoprecipitation with an anti-p-Tyr(102) antibody and Western blot analysis for DARPP-32 and ERBB3. D–E) MKN-28 cells stably expressing DARPP-32 (D) or infected with DARPP-32 adenovirus (5 MOI) (E) were treated with gefitinib (25 μM) or vehicle overnight. Cell lysates were subjected to immunoblotting for p-AKT(S473), AKT, and DARPP-32. F) SNU-16 cells were infected with DARPP-32 shRNA or scrambled shRNA lentiviruses (10 MOI), treated with gefitinib (25 μM) overnight and analyzed as in (D).
Knockdown of DARPP-32 inhibits tumor growth and increases response to gefitinib treatment in the xenografted carcinoma mouse model
The results demonstrated that tumors derived from SNU-16 cells stably expressing DARPP-32 shRNA grew substantially slower than the scrambled shRNA control tumors (p<0.05, Figure 7A and B). Western blot analysis confirmed the lower levels of EGFR and p-AKT in the shRNA DARPP-32 xenografts (Figure 7C). Immunostaining with the anti-Ki-67 antibody indicated that the reduced tumor growth is likely due to a lower proliferation rate caused by knockdown of DARPP-32, as Ki-67 staining was much weaker in DARPP-32 shRNA than the scrambled shRNA control tumors (p<0.01, Figure 7C). We also evaluated the effects of gefitinib in combination with knocking down DARPP-32 in SNU-16 cells. Treatment with gefitinib significantly inhibited relative growth in DARPP-32 shRNA tumors as compared to scrambled shRNA control tumors (p<0.01, Figure 7D).
Figure 7. Knockdown of DARPP-32 inhibits tumor growth and enhances response to gefitinib treatment.

A) SNU-16 cells stably expressing DARPP-32 shRNA or scrambled shRNA were injected s.c. (2×106 cells per site) into nude mice. Tumor volume was measured at the indicated times; each data point represents the mean ±SD for 10 xenografts. B) Representative xenograft tumors of sacrificed mice (left panel) and quantification of tumor weight at the end of experiment (right panel). The tumor weight is indicated by mean ±SD (p<0.05). C) Immunohistochemical staining (upper panel) and evaluation of Ki-67 protein expression (lower panel). Tumors derived from DARPP-32 knockdown SNU-16 cells revealed a lower level of Ki-67 antigen than the scrambled control. Expression of EGFR, ERBB3, and p-AKT in tumors derived from SNU-16 cells stably expressing DARPP-32 shRNA or scrambled shRNA were analyzed by Western blot (lower panel). D) Xenografts were prepared as in (A). After 2 weeks, the mice were given gefitinib (50 mg/kg/d) or vehicle thrice weekly by oral gavage for 18 d. The knockdown of DARPP-32 enhanced the response to gefitinib treatment (p<0.01).
Discussion
Resistance to chemotherapeutics is a major obstacle in clinical practice leading to an overall poor outcome and reduced patient survival. Understanding the mechanisms of chemoresistance aims to identify possible strategies to overcome this challenging clinical problem. Overexpression of DARPP-32 has been identified in more than two-thirds of gastric cancers6, 7 as well as in several other malignancies such as esophageal, colon, and breast cancers8, 9. In this study, we demonstrate a novel signaling mechanism for DARPP-32 in mediating chemotherapeutic resistance to EGFR inhibitor and gefitinib in gastric cancer.
Gefitinib, a selective EGFR-tyrosine kinase inhibitor that blocks the PI3K/AKT signaling pathway, has shown anti-tumor activity in clinical trials against cancers such as NSCLC and gastric cancer4, 23. However, clinical trials using gefitinib in patients with advanced refractory gastric tumors revealed modest clinical response due to resistance3. In lung cancer, the percentage of lung tumor cells expressing EGFR, p-EGFR, p-MAPK, p-AKT, and Ki-67 were evaluated immunohistochemically before and after treatment with gefitinib. A decreased proliferation was observed only in those tumors with low levels of p-AKT, suggesting a critical role for the PI3K/AKT pathway in gefitinib resistance24. On the other hand, little is known about molecular mechanism(s) governing gefitinib resistance in gastric cancer patients. Our findings demonstrate that DARPP-32 promotes gefitinib resistance by maintaining activated AKT pathways as indicated by increased phosphorylation of AKT and its classical downstream substrate, GSK-3β. Earlier studies have shown rare incidence of mutations of EGFR in gastric cancer21. Conversely, EGFR expression seems to be a significant predictor of poor prognosis in gastric cancer25, 26. Consistent with this report, relative expressions of EGFR and ERBB3 are higher in gastric cancer compared with normal stomach27. Therefore, we tested our hypothesis that gefitinib resistance mediated by DARPP-32 in gastric cancer cells might involve sustained signaling via EGFR/ERBB3-dependent activation of PI3K/AKT pathway. Our findings demonstrate that DARPP-32 expression enhanced protein stability of EGFR and phosphorylation of EGFR and ERBB3 in several gastric cancer cell models. Indeed, we have shown that DARPP-32 expression blocked gefitinib-induced EGFR protein degradation and maintained phosphorylation of ERBB3. Furthermore, we demonstrated that DARPP-32 up-regulated both total and p-EGFR(Y845) protein levels in response to gefitinib treatment. This finding is in line with earlier reports that have shown that a phosphorylated form of EGFR (Tyr845) is critical for EGFR/PI3K signaling28.
The one critically important and well-established signaling activity of ERBB3 is its unique and potent ability to activate downstream PI3K/AKT pathway signaling29, 30, thus promoting resistance to gefitinib19. Of note, the kinase-dead ERBB3 acquires signaling activity through heterodimerization with the other ERBB receptors31. EGFR is a tyrosine kinase that specifically phosphorylates ERBB332 which enhances the activity of PI3K/AKT signaling pathway29. On the basis of our findings that DARPP-32 activated AKT pathways and regulated EGFR in response to gefitinib, we hypothesized that DARPP-32 interacts with EGFR/ERBB3 and enhances their heterodimerization. Indeed, our results demonstrated that exogenous and endogenous DARPP-32 interacted with EGFR and ERBB3 in several cell models. Additionally, we demonstrated that DARPP-32 enhanced EGFR/ERBB3 heterodimerization. We also confirmed that DARPP-32 maintained phosphorylation of EGFR and ERBB3 in response to treatment with EGF or gefitinib. In addition, DARPP-32 prevented gefitinib-induced down-regulation of the PI3K/AKT pathway. In this context, our findings support the recent observations in breast and lung cancer where persistent ERBB3 phosphorylation and PI3K/AKT activation were associated with gefitinib resistance19, 33,34. Other reported mechanisms of gefitinib resistance included a secondary EGFR(T790M) mutation that alters the physical biochemical properties of the EGFR, and activation of parallel pathways (MET, IGF1R) in which the key downstream targets of EGFR are activated independently of EGFR19, 35. Taken together, overexpression of DARPP-32 provides a plausible explanation for EGFR/ERBB3-dependent activation of the AKT pathway and highlights a novel mechanism for gefitinib resistance in gastric cancer.
Upon EGF ligand binding to the extracellular domain, the EGFR is internalized and targeted to a lysosomal-proteasomal degradation pathway36, 37. Furthermore, some earlier reports suggested that gefitinib-induced EGFR degradation is cell line dependent38. We demonstrated that DARPP-32 significantly delayed EGFR protein degradation in gastric cancer cells. This stabilization of EGFR was maintained following gefitinib treatment. Taken together, our findings suggest that DARPP-32/EGFR binding may lead to EGFR sequestration and block EGFR internalization, thereby preventing its ubiquitination. As reported for the heat shock protein (HSP90)39, it is possible that DARPP-32 may play a similar role in maintaining the functional conformation of EGFR. Another alternative mechanism may involve DARPP-32-mediated up-regulation of recycling of internalized EGFR to the cell surface. However, these possibilities require further investigation to determine the detailed mechanism(s) by which DARPP-32 regulates EGFR protein stability.
In conclusion, our findings suggest that frequent overexpression of DARPP-32 in gastric cancer underlies a chemotherapeutic resistance phenotype; a common clinical feature of gastric cancers. The ability of DARPP-32 to bind to EGFR/ERBB3 and activate PI3K/AKT signaling provide a novel mechanism by which DARPP-32 could modulate the response to gefitinib and possibly several other chemotherapeutics.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abbreviations
- NSCLC
non-small cell lung carcinomas (cancer)
- DARPP-32
Dopamine and cAMP-regulated phosphoprotein, Mr 32000
- EGFR
epidermal growth factor receptor
- MET
met proto-oncogene
- FITC
Fluorescein Isothiocynate
- PI
propidium iodide
- PVDF
Polyvinylidene Fluoride
- HRP
horseradish peroxidase
Footnotes
Shoumin Zhu: study concept and design; experimental design and acquisition of data; analysis and interpretation of data; drafting of the manuscript; statistical analysis; obtained funding; technical and material support
Abbes Belkhiri: analysis and interpretation of data; experimental troubleshooting; drafting of the manuscript; critical revision of the manuscript
Wael El-Rifai: study concept and design; study supervision; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content
Conflict of Interest: All authors indicated “no conflict of interest”.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN. Int J Cancer. 2008 doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Catalano V, Labianca R, Beretta GD, Gatta G, de Braud F, Van Cutsem E. Gastric cancer. Crit Rev Oncol Hematol. 2009;71:127–64. doi: 10.1016/j.critrevonc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Doi TWK, Siena S, Cascinu S, Ohtsu A, Michael M, Takiuchi H, Swaisland H, Gallagher N, Van Cutsem E. Efficacy, tolerability and pharmacokinetics of gefitinib (ZD1839) in pretreated patients with metastatic gastric cancer. Proc Am Soc Clin Oncol. 2003;22 (abstr 1036) 2003. [Google Scholar]
- 4.Becker JC, Muller-Tidow C, Serve H, Domschke W, Pohle T. Role of receptor tyrosine kinases in gastric cancer: new targets for a selective therapy. World J Gastroenterol. 2006;12:3297–305. doi: 10.3748/wjg.v12.i21.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walaas SI, Aswad DW, Greengard P. A dopamine- and cyclic AMP-regulated phosphoprotein enriched in dopamine-innervated brain regions. Nature. 1983;301:69–71. doi: 10.1038/301069a0. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee K, Peng D, Brifkani Z, Belkhiri A, Pera M, Koyama T, Koehler EA, Revetta FL, Washington MK, El-Rifai W. Dopamine and cAMP regulated phosphoprotein MW 32 kDa is overexpressed in early stages of gastric tumorigenesis. Surgery. 2010;148:354–363. doi: 10.1016/j.surg.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El-Rifai W, Smith MF, Jr, Li G, Beckler A, Carl VS, Montgomery E, Knuutila S, Moskaluk CA, Frierson HF, Jr, Powell SM. Gastric cancers overexpress DARPP-32 and a novel isoform, t-DARPP. Cancer Res. 2002;62:4061–4. [PubMed] [Google Scholar]
- 8.Beckler A, Moskaluk CA, Zaika A, Hampton GM, Powell SM, Frierson HF, Jr, El-Rifai W. Overexpression of the 32-kilodalton dopamine and cyclic adenosine 3′,5′-monophosphate-regulated phosphoprotein in common adenocarcinomas. Cancer. 2003;98:1547–51. doi: 10.1002/cncr.11654. [DOI] [PubMed] [Google Scholar]
- 9.Wang MS, Pan Y, Liu N, Guo C, Hong L, Fan D. Overexpression of DARPP-32 in colorectal adenocarcinoma. Int J Clin Pract. 2005;59:58–61. doi: 10.1111/j.1742-1241.2004.00305.x. [DOI] [PubMed] [Google Scholar]
- 10.Belkhiri A, Zaika A, Pidkovka N, Knuutila S, Moskaluk C, El-Rifai W. Darpp-32: a novel antiapoptotic gene in upper gastrointestinal carcinomas. Cancer Res. 2005;65:6583–92. doi: 10.1158/0008-5472.CAN-05-1433. [DOI] [PubMed] [Google Scholar]
- 11.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 12.Soltoff SP, Carraway KL, 3rd, Prigent SA, Gullick WG, Cantley LC. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994;14:3550–8. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9:463–75. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 14.Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62:5749–54. [PubMed] [Google Scholar]
- 15.Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, Meyerson M. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, Lindeman NI, Murphy C, Akhavanfard S, Yeap BY, Xiao Y, Capelletti M, Iafrate AJ, Lee C, Christensen JG, Engelman JA, Janne PA. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A, Lifshits E, Brown A, Lee C, Christensen JG, Kwiatkowski DJ, Engelman JA, Janne PA. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene. 2010;29:2346–56. doi: 10.1038/onc.2009.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobayashi S, Ji H, Yuza Y, Meyerson M, Wong KK, Tenen DG, Halmos B. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 2005;65:7096–101. doi: 10.1158/0008-5472.CAN-05-1346. [DOI] [PubMed] [Google Scholar]
- 19.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 20.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, Gazdar A, Pass H, Rusch V, Gerald W, Huang SF, Yang PC, Miller V, Ladanyi M, Yang CH, Pao W. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–7. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moutinho C, Mateus AR, Milanezi F, Carneiro F, Seruca R, Suriano G. Epidermal growth factor receptor structural alterations in gastric cancer. BMC Cancer. 2008;8:10. doi: 10.1186/1471-2407-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu JY, Wu SG, Yang CH, Chang YL, Chang YC, Hsu YC, Shih JY, Yang PC. Comparison of gefitinib and erlotinib in advanced NSCLC and the effect of EGFR mutations. Lung Cancer. 2010 doi: 10.1016/j.lungcan.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 24.Rojo F, Tabernero J, Albanell J, Van Cutsem E, Ohtsu A, Doi T, Koizumi W, Shirao K, Takiuchi H, Ramon y Cajal S, Baselga J. Pharmacodynamic studies of gefitinib in tumor biopsy specimens from patients with advanced gastric carcinoma. J Clin Oncol. 2006;24:4309–16. doi: 10.1200/JCO.2005.04.2424. [DOI] [PubMed] [Google Scholar]
- 25.Slesak B, Harlozinska A, Porebska I, Bojarowski T, Lapinska J, Rzeszutko M, Wojnar A. Expression of epidermal growth factor receptor family proteins (EGFR, c-erbB-2 and c-erbB-3) in gastric cancer and chronic gastritis. Anticancer Res. 1998;18:2727–32. [PubMed] [Google Scholar]
- 26.Galizia G, Lieto E, Orditura M, Castellano P, Mura AL, Imperatore V, Pinto M, Zamboli A, De Vita F, Ferraraccio F. Epidermal growth factor receptor (EGFR) expression is associated with a worse prognosis in gastric cancer patients undergoing curative surgery. World J Surg. 2007;31:1458–68. doi: 10.1007/s00268-007-9016-4. [DOI] [PubMed] [Google Scholar]
- 27.Sithanandam G, Anderson LM. The ERBB3 receptor in cancer and cancer gene therapy. Cancer Gene Ther. 2008;15:413–48. doi: 10.1038/cgt.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selvendiran K, Bratasz A, Tong L, Ignarro LJ, Kuppusamy P. NCX-4016, a nitro-derivative of aspirin, inhibits EGFR and STAT3 signaling and modulates Bcl-2 proteins in cisplatin-resistant human ovarian cancer cells and xenografts. Cell Cycle. 2008;7:81–8. doi: 10.4161/cc.7.1.5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prigent SA, Gullick WJ. Identification of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. Embo J. 1994;13:2831–41. doi: 10.1002/j.1460-2075.1994.tb06577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL., 3rd Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc Natl Acad Sci U S A. 1994;91:8132–6. doi: 10.1073/pnas.91.17.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. Embo J. 2000;19:3159–67. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frolov A, Schuller K, Tzeng CW, Cannon EE, Ku BC, Howard JH, Vickers SM, Heslin MJ, Buchsbaum DJ, Arnoletti JP. ErbB3 expression and dimerization with EGFR influence pancreatic cancer cell sensitivity to erlotinib. Cancer Biol Ther. 2007;6:548–54. doi: 10.4161/cbt.6.4.3849. [DOI] [PubMed] [Google Scholar]
- 33.Arteaga CL. HER3 and mutant EGFR meet MET. Nat Med. 2007;13:675–7. doi: 10.1038/nm0607-675. [DOI] [PubMed] [Google Scholar]
- 34.Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, Moasser MM. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–41. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cappuzzo F, Toschi L, Tallini G, Ceresoli GL, Domenichini I, Bartolini S, Finocchiaro G, Magrini E, Metro G, Cancellieri A, Trisolini R, Crino L, Bunn PA, Jr, Santoro A, Franklin WA, Varella-Garcia M, Hirsch FR. Insulin-like growth factor receptor 1 (IGFR-1) is significantly associated with longer survival in non-small-cell lung cancer patients treated with gefitinib. Ann Oncol. 2006;17:1120–7. doi: 10.1093/annonc/mdl077. [DOI] [PubMed] [Google Scholar]
- 36.Ceresa BP, Bahr SJ. rab7 activity affects epidermal growth factor:epidermal growth factor receptor degradation by regulating endocytic trafficking from the late endosome. J Biol Chem. 2006;281:1099–106. doi: 10.1074/jbc.M504175200. [DOI] [PubMed] [Google Scholar]
- 37.Ono M, Hirata A, Kometani T, Miyagawa M, Ueda S, Kinoshita H, Fujii T, Kuwano M. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther. 2004;3:465–72. [PubMed] [Google Scholar]
- 38.Perez-Torres M, Guix M, Gonzalez A, Arteaga CL. Epidermal growth factor receptor (EGFR) antibody down-regulates mutant receptors and inhibits tumors expressing EGFR mutations. J Biol Chem. 2006;281:40183–92. doi: 10.1074/jbc.M607958200. [DOI] [PubMed] [Google Scholar]
- 39.Yang J, Li M, Hong L, Zhang H, Sun L, Wang W. Preparation of a Novel Monoclonal Antibody Specific to DARPP-32. Hybridoma (Larchmt) 2010;29:351–4. doi: 10.1089/hyb.2010.0014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.