

Introduction
Although N-Acetylaspartylglutamate (NAAG) was first reported in the mid 1960s to be present in the mammalian brain and spinal cord at mM concentrations, the critical experiments that established this peptide as a neurotransmitter emerged several decades later (reviewed in Neale et al., 2000). Since NAAG is by far the most prevalent and widely distributed peptide transmitter in the mammalian brain, it could be predicted to have roles in a broad spectrum of neuronal circuits and nervous system functions. A series of studies in animal models of clinical disorders have shown this to be the case (reviewed in Neale et al., 2005; 2011; Tsukamoto et al. 2007). A central element in defining the function of this peptide relates to its selective activation of the type 3 metabotropic “glutamate” receptor (mGluR3), a group II mGluR. This receptor has been localized to presynaptic terminals where it inhibits transmitter release via its negative coupling to cyclic nucleotides (reviewed in Niswender and Conn, 2010). As an endogenous mGluR3 agonist, NAAG has been shown to reduce cAMP and cGMP levels and to inhibit transmitter release (Adedoyin et al., 2010; Wroblewska et al., 1993; 1997; 1998, 2006; Zhao et al., 2001; Zhong et al., 2006). A model of the synaptic function of NAAG has emerged from a substantial literature on this peptide over the past 20 years and from data on other peptide cotransmitter actions in the perisynaptic space (Figure 1).
Figure 1. A model of the role of NAAG peptidase inhibition and its influence on NAAG and NAAG2 in the nervous system (From Neale et al., 2011).
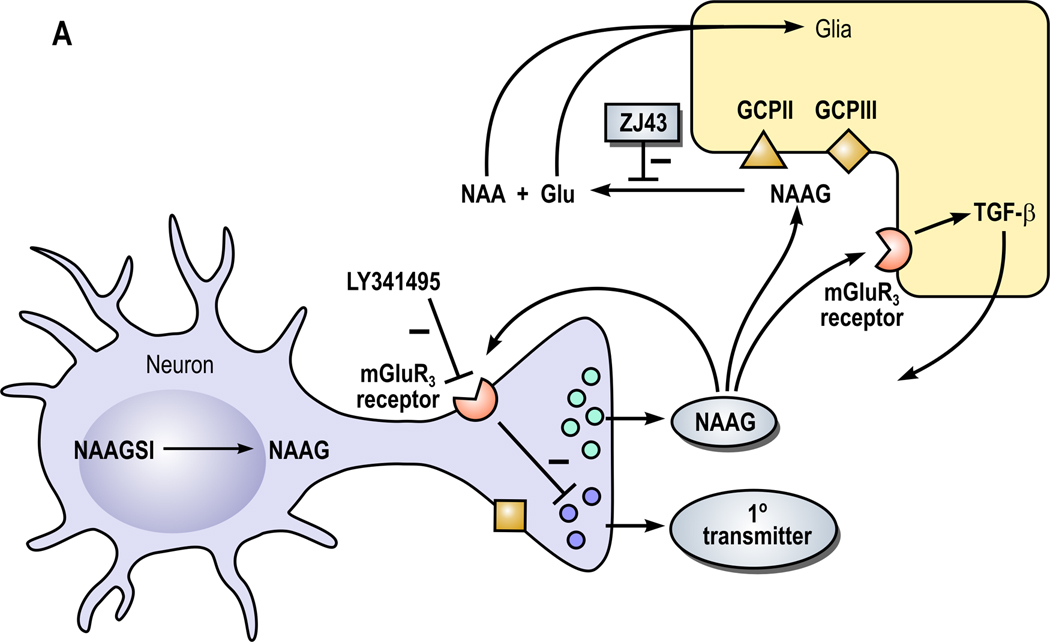
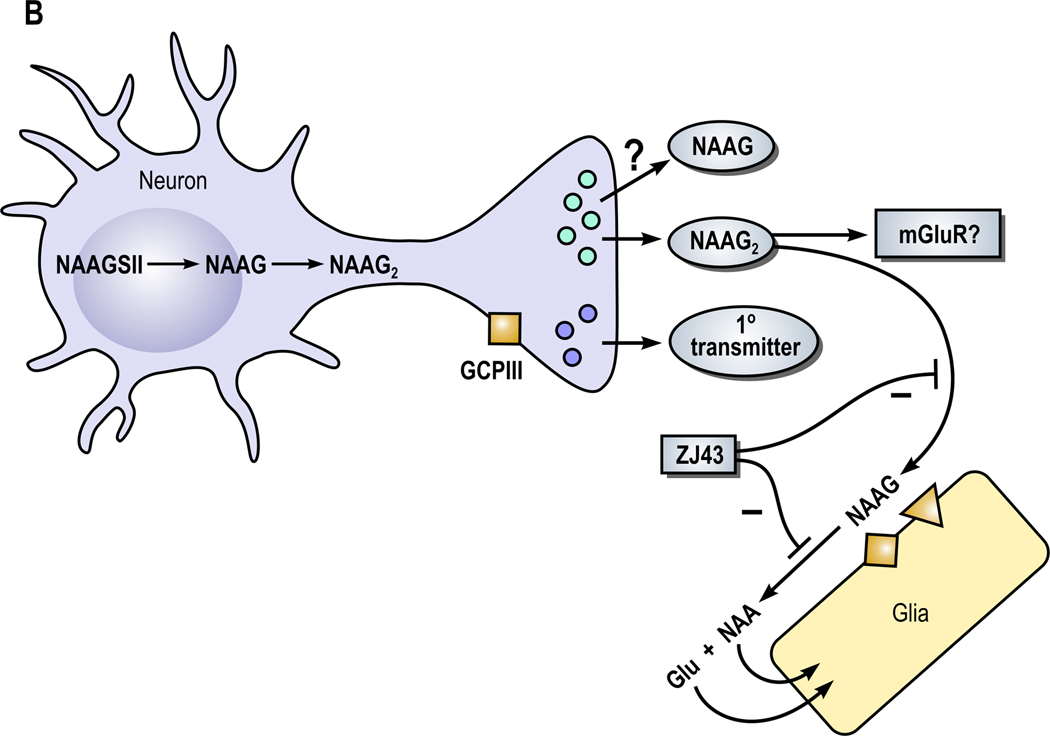
A. Neurons expressing NAAG synthetase I (NAAGS I) mediate the synthesis of NAAG but not NAAG2. In this cell, NAAG is co-released with a primary amine transmitter, such as glutamate, under conditions of elevated neuronal activity. NAAG is released into the perisynaptic space where it activates presynaptic and glial type 3 metabotropic glutamate receptors (mGluR3). NAAG is inactivated by glutamate carboxypeptidases II (GCPII) and III (GCPIII), forming N-acetylaspartate (NAA) and glutamate (Glu), which are transported into glial cells. Inhibition of the peptidases GCPII and GCPIII by a NAAG peptidase inhibitor, such as ZJ43, reduces inactivation of NAAG and raises perisynaptic NAAG level. In animal models, the NAAG peptidase inhibitor-mediated elevation of peptide levels increases the activation of mGluR3 receptors on axon endings, inhibiting transmitter release and reducing pathology. In a second neuroprotective pathway, NAAG activation of mGlu3 receptors on glial cells stimulates the release of a trophic factor, transforming growth factor β (TGF-β). B. Neuron synthesizing NAAG2 via NAAG synthetase II (NAAGS II) may release both NAAG and NAAG2.
The significance of NAAG in nervous system function has been most effectively studied through the development of inhibitors of the extracellular enzymes that inactivate it (Tsukamoto et al. 2007; Zhou et al., 2005). These inhibitors elevate extracellular levels of NAAG and consistent with NAAG activation of mGluR3, they reduce the release of other transmitters (Slusher et al., 1999; Zhong et al., 2006; Adedoyin 2010). The application of these inhibitors in vivo revealed that elevating the synaptic level of this peptide has considerable therapeutic potential in clinical conditions ranging from stroke and traumatic brain injury to inflammatory pain and schizophrenia (reviewed in Neale et al., 2005; 2011). NAAG peptidase inhibitors also may inhibit the extracellular hydrolysis of NAAG’s newly discovered sister peptide N-acetylaspartylglutamylglutamate (NAAG2) if it is synaptically released (Lodder-Gadaczek et al., 2011). Importantly, in these preclinical studies, the heterotropic mGluR2/3 antagonist LY341495 blocks the efficacy of NAAG peptidase inhibition (Neale et al., 2005; 2011).
NAAG as an mGluR3 agonist
Central to this model of the physiology of NAAG are reports that it activates mGluR3, in neurons in culture and brain slices and in transfected cells (Adedoyin et al., 2010; Bischofberger and Schild, 1996; Ghose et al., 1997; Lea et al., 2001; Wroblewska et al., 1993; 1997; 1998; 2006). However, two recent papers reported that under some conditions NAAG does not activate mGluR3 and these data have been interpreted to suggest that NAAG is not an mGluR3 agonist. A full review of the relevant data directly refutes this suggestion.
In a well designed and executed set of experiments, commercial NAAG containing 0.3–0.5% glutamate, but not NAAG that was repurified to remove glutamate, activated G-protein-coupled K+ channels in HEK-293 cells that had been transiently cotransfected with the ion channel and mGluR3 (Fricker et al., 2009). While leaving open other possibilities, this paper suggested that glutamate contamination of commercial NAAG might have been responsible for the activation of mGluR3 by NAAG that was reported in some earlier papers in which high concentrations of NAAG were used.
A second paper titled, “The Neuroactive Peptide N-Acetylaspartylglutamate is Not an Agonist at the Metabotropic Glutamate Receptor Subtype 3 of Metabotropic Glutamate Receptor” (sic), was less circumspect. This paper reported that purified NAAG also failed to activate this same potassium channel in Xenopus oocytes that had been cotransfected with mGluR3 and the channel, in contrast to glutamate and the unpurified peptide (Chopra et al., 2009). The ill-chosen title of this paper reflects an inadequately vetted review of the relevant literature and has the potential to set back this field for years to come despite the very substantial evidence, discussed below, that clearly refutes it.
First, it should be noted that glutamate contamination of commercial NAAG is not new to the literature but was reported in 2004 (Losi et al.). The author’s research group detected this contamination in 1996 and routinely repurified NAAG after that time using ion exchange chromatography. Glutamate levels in the repurified NAAG used in studies since that time have been ≤ 0.1% as verified in precolumn derivitized, HPLC resolved samples.
While the Chopra and Fricker papers demonstrate that glutamate contamination of commercial NAAG has the potential to activate glutamate receptors, the data fail to demonstrate that NAAG does not activate mGluR3 as asserted by the title of the Chopra paper. At best, this conclusion could only be applied to data in an early report in which high levels of NAAG were used to activate mGluR3 in cerebellar granule cells in culture (Wroblewska et al., 1993) only if it is assumed that the peptide used at that time did, in fact, contain sufficiently high levels of glutamate to activate mGluR3 in this specific assay system. In support of their conclusion, Chopra and colleagues cite estimates of the affinity of NAAG and glutamate for mGluR3 that are obtained from in vitro competitive radioligand binding studies to membranes prepared from transfected cells (Schweitzer et al., 2000), data that bare little resemblance to results from physiological studies of the relative efficacy of these two transmitters for mGluR3 (see below). More critically, these papers on mGluR3/K-channel cotransfected cells fail to account for the considerable published data that directly contradict the conclusion that 0.3–0.5% glutamate in NAAG, rather than the peptide itself, is responsible for reports of the peptide’s activation of mGluR3. These contrary data include: 1) Bischofberger and Schild (1996) reported that NAAG and glutamate exhibited very similar dose responses for inhibition of voltage-dependent calcium currents in olfactory mitral cells via a group II mGluR; 2) Wroblewska et al. (1997) demonstrated that 100 uM NAAG was 75% as effective as 30 uM glutamate in elevation of intracellular calcium concentrations in HEK cells transfected with an mGluR3/mGluR1 chimeric receptor, while being inactive at an mGluR2/mGluR1 chimeric receptor. In this same study, 1 mM NAAG elicited a calcium response that was substantially larger than that produced by100 uM glutamate in the same cell, results inconsistent with the contamination theory. Confirming the lack of significant glutamate contamination of the NAAG used in this study, 100 uM NAAG failed to activate mGluR4 expressed in stably transfected CHO cells while as little as 100 nM glutamate maximally activated this receptor; 3) Wroblewska et al. (1998), reported similar dose response curves for NAAG and glutamate with respect to negative coupling to cAMP levels in astrocytes, cells that expressed high levels of mGluR3 message and no detectable levels of mGluR2. It is notable that these cultured astrocytes efficiently remove glutamate from their medium (Silva et al., 1999) and thus required high levels of bath applied glutamate to activate their mGluRs; 4) Acting via a group II mGluR, NAAG, glutamate and the group II agonist ACPD had very similar dose response profiles in inducing expression of the alpha 6 subunit of the GABA-A receptor in cerebellar granule cells (Ghose et al., 1997); 5) Less than 10 uM NAAG (<50 nM glutamate if NAAG had been contaminated at 0.5% level) significantly reduced cAMP levels in cerebellar granule cells and mGluR3 transfected BHK cells with both effects being blocked by an mGluR3 antagonist (Lea et al., 2001). In this same study, 200 uM NAAG failed to induce currents via NMDA receptors in hippocampal granule cells demonstrating the absence of significant glutamate in the peptide used in this study, while 200 uM NAAG blocked LTP in an mGluR3-dependent manner; 6) FN6, a rigid analogue of NAAG that was synthesized without glutamate and highly purified via HPLC, selectively activated mGluR3 but not mGluR2 (Nan et al., 2000); 7) 1 uM re-purified NAAG, as well as the NAAG peptidase inhibitor ZJ43, reduced excitatory postsynaptic currents in amygdaloid neurons via presynaptic inhibition of glutamate release, an effect that was blocked by a group II mGluR antagonist (Adedoyin et al., 2010). These data clearly and unequivocally demonstrate that NAAG directly activates mGluR3.
A series of studies on the efficacy of NAAG peptidase inhibitors in animal models of stroke, peripheral nerve injury, inflammatory and neuropathic pain, traumatic brain injury, cocaine abuse and schizophrenia further substantiate the conclusion that NAAG selectively activates a group II mGluR in vivo while the peptide has repeatedly been shown to be inactive at mGluR2 (reviewed in Neale et al., 2005; 2011). Importantly, group II mGluR antagonists block the in vivo effects of peptidase inhibitors ZJ43 and 2-PMPA and these inhibitors fail to interact directly with mGluRs (Yamamoto et al, 2004; 2007; 2008). Fricker et al. (2009) suggested that, “Further investigation is required to elucidate the involvement of NAALADase (original term for NAAG peptidase activity) and NAAG in the pathophysiology of schizophrenia.” Consistent with this suggestion and additionally documenting the conclusion that the effects of NAAG peptidase inhibition is mediated specifically by mGluR3 in the animal models of schizophrenia, the NAAG peptidase inhibitor 2-PMPA reduced the motor activation effects of PCP, a model of schizophrenia, in mGluR2 but not in mGluR3 knockout mice (submitted, Olszewski et al., 2011).
The recent discovery that NAAG is synthesized in neurons by NAAG synthetase I (NAAGS I) while a sister peptide N-acetylaspartylglutamylglutamate (NAAG2) is synthesized from NAAG and glutamate at much lower levels in other neurons by a second enzyme, (NAAGS II), suggests that this sister peptide also is neuroactive (Lodder-Gadaczek et al., 2011). While not discounting the role of NAAG that is synthesized and released from a separate set of neurons, the expression of this second member of the NAAG peptide family opens new possibilities. Since both NAAG and NAAG2 are hydrolyzed by GCPII (Lodder-Gadaczek et al., 2011), some of the effects of NAAG peptidase inhibitors also could result from increasing extracellular levels of NAAG2 if it is synaptically released and activates a receptor (Figure 1B).
While the paper by Chopra et al. (2009) dismissed the data on the efficacy of NAAG peptidase inhibitors as well as other data that were contrary to this paper’s title, the report by Fricker et al. (2009) suggested that the efficacy of the inhibitors was mediated “through modification of NAAG catabolism, consequently modulating baseline glutamate levels and group II mGluR activation.” However, the consequences of NAAG peptidase inhibitors on NAAG metabolism have been demonstrated in vivo. They inhibit NAAG hydrolysis with a consequent elevation of extracellular levels of NAAG and a reduction in extracellular glutamate levels via activation of a presynaptic group II mGluR (Adedoyin et al., 2010; Slusher et al., 1999; Zhong et al., 2006; Diaying Zuo et al., manuscript in preparation 2011). Clearly, this reduced glutamate release would not result in an increase in group II mGluR activation.
While the cumulative data provide direct evidence that NAAG is an endogenous agonist at mGluR3 receptors, they do not explain the reported failure of NAAG to activate the G-protein coupled potassium channel via transiently cotransfected mGluR3. A more parsimonious interpretation of these data is that glutamate and NAAG allosterically regulate the receptor in different ways consistent with the substantial differences that their structures likely induce when interacting with the active site. It is noteworthy that both papers defined mGluR3 activation via conductance through this transfected G-protein-dependent potassium channel while the other NAAG literature assessed the relationship between the receptor and cyclic nucleotide second messenger cascades. Such differential agonist-specific activation mechanisms for G-protein coupled receptors are well documented (reviewed in Ambrosio et al., 2011). Indeed, Fricker et al., (2009) pointed out this alternative interpretation of their data by concluding, “the differential activity of NAAG in the two assays systems could reflect agonist dependent stimulus trafficking such that NAAG, unlike glutamate, couples to group II mGluR intracellular signaling pathways including modulation of cAMP/cGMP but not to GIRK.” Whether this is a physiologically relevant effect with respect to mGluR3 receptors or simply the artificial consequence of cotransfecting cells with this combination of cDNAs remains to be determined. As a first step to testing this hypothesis, Fricker et al. (2009) reported that high concentrations of purified NAAG decrease mGluR3 but not mGluR2 activation by 100 uM glutamate, a result that supports the interaction of NAAG with the mGluR3 but not mGluR2 ligand binding site in these cells. This concept warrants further analysis as it may reveal a new, physiologically relevant distinction between NAAG and glutamate at mGluR3. While Group II antagonists have been shown to block activation of this potassium channel postsynaptically (Lee and Sherman, 2009), mGluR3 specific coupling to the channel in neurons has yet to be described.
NAAG and mGluR3 are a widely expressed in the mammalian nervous system where they have been associated via NAAG peptidase inhibitors with novel therapeutic approaches to a series of clinical conditions. These confirmations of the role of NAAG as an mGluR3 agonist are central to providing a foundation for understanding the mechanism of action of this new class of drugs.
Conclusion
NAAG (and likely its sister peptide NAAG2) is by far the most prevalent peptide transmitter in the mammalian nervous system. Data from a series of studies demonstrate that the levels of glutamate in NAAG were clearly insufficient to have been responsible for the NAAG-mediated mGluR3 activation that was reported. Several other studies found the levels of glutamate and NAAG that activated mGluR3 were very similar, a fact inconsistent with the assertion that low levels of glutamate in NAAG was responsible for its activity. Additionally, drugs that inhibit the inactivation of NAAG following its synaptic release elevate the peptide level and inhibit the subsequent release of other transmitters, including glutamate, via a process that is blocked by a group II mGluR antagonist. Thus, a substantial body of data, in vitro and in vivo, supports the conclusion that a major role of NAAG is activation of presynaptic mGluR3 receptors and unequivocally contradicts the suggestion that glutamate contamination of NAAG was responsible for these results. In the face of these data, the two reports demonstrating that glutamate but not NAAG activates mGluR3 coupling to a cotransfected potassium channel cannot be explained by the conclusion that NAAG is not an agonist at mGluR3. Rather, these data are consistent with the suggestion of Fricker et al., (2009) that “agonist dependent stimulus trafficking” is responsible for this differential activity in cells that were cotransfected with mGluR3 and the potassium channel.
Supplementary Material
Acknowledgment
JHN is supported by NIH grants NS38080 and MH79983 and by generous gifts from Nancy and Daniel Paduano.
References
- Adedoyin MO, Vicini S, Neale JH. Endogenous N-acetylaspartylglutamate (NAAG) inhibits synaptic plasticity/transmission in the amygdala in a mouse inflammatory pain model. Mol. Pain. 2010;6:60. doi: 10.1186/1744-8069-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosio M, Zurn A, Lohse M. Sensing G protein-coupled protein activation. Neuropharmacology. 2011;60:45–51. doi: 10.1016/j.neuropharm.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Bischofberger J, Schild D. Glutamate and N-acetylaspartylglutamate block HVA calcium currents in frog olfactory bulb interneurons via a mGluR2/3-like receptor. J. Neurophysiol. 1996;76:2089–2092. doi: 10.1152/jn.1996.76.3.2089. [DOI] [PubMed] [Google Scholar]
- Chopra M, Yao Y, Blake TJ, Hampson DR, Johnson EC. The neuroactive peptide N-acetylaspartylglutamate is not an agonist at the metabotropic glutamate receptor subtype 3 of metabotropic glutamate receptor. J. Pharmacol. Exp. Ther. 2009;330:212–219. doi: 10.1124/jpet.109.152553. [DOI] [PubMed] [Google Scholar]
- Fricker AC, Mok MH, de la Flor R, Shah AJ, Woolley M, Dawson LA, Kew JN. Effects of N-acetylaspartylglutamate (NAAG) at group II mGluRs and NMDAR. Neuropharmacology. 2009;56:1060–1067. doi: 10.1016/j.neuropharm.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Ghose S, Wroblewska B, Corsi L, Grayson D, DeBlas AL, Vicini S, Neale JH. NAAG stimulates mGluR3 to regulate expression of the GABA-A alpha-6 subunit in cerebellar granule cells. J. Neurochem. 1997;69:2326–2335. doi: 10.1046/j.1471-4159.1997.69062326.x. [DOI] [PubMed] [Google Scholar]
- Lea PM, Wroblewska B, Sarvey JM, Neale JH. β-NAAG rescues LTP from blockade by NAAG in the rat dentate gyrus via the type 3 metabotropic glutamate receptor. J. Neurophys. 2001;85:1097–1106. doi: 10.1152/jn.2001.85.3.1097. [DOI] [PubMed] [Google Scholar]
- Lee CC, Sherman SM. Glutamatergic inhibition in sensor neocortex. Cereb. Cortex. 2009;19:2281–2289. doi: 10.1093/cercor/bhn246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodder-Gadaczek J, Becker I, Gieselmann V, Wang-Eckhardt L, Eckhardt M. N-Acetylaspartylglutamate synthetase-II synthesizes N-acetylaspartyl-glutamylglutamate. J. Biol. Chem. 2011 Mar 25; doi: 10.1074/jbc.M111.230136. (e-pub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losi G, Vicini S, Neale J. NAAG fails to antagonize synaptic and extrasynaptic NMDA receptors in cerebellar granule neurons. Neuropharmacology. 2004;46:490–496. doi: 10.1016/j.neuropharm.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Nan F, Bzdega T, Pshenichkin S, Wroblewski JT, Wroblewska B, Neale JH, Kozikowski AP. Dual function glutamate-related ligands: Discovery of a novel, potent inhibitor of glutamate carboxypeptidase II possessing mGluR2 agonist activity. J. Med. Chem. 2000;43:772–774. doi: 10.1021/jm9905559. [DOI] [PubMed] [Google Scholar]
- Neale JH, Bzdega T, Wroblewska B. N-Acetylaspartylglutamate: the most abundant peptide neurotransmitter in the mammalian central nervous system. J. Neurochem. 2000;75:443–452. doi: 10.1046/j.1471-4159.2000.0750443.x. [DOI] [PubMed] [Google Scholar]
- Neale JH, Olszewski RT, Gehl LM, Wroblewska B, Bzdega T. The neurotransmitter N-acetylaspartylglutamate in models of pain, ALS, diabetic neuropathy, CNS injury and schizophrenia. Trends Pharmacol. Sci. 2005;26:477–484. doi: 10.1016/j.tips.2005.07.004. 2005. [DOI] [PubMed] [Google Scholar]
- Neale JH, Olszewski RT, Zuo D, Janczura KJ, Lavin KM, Madore JC, Profaci CP, Bzdega T. Advances in Understanding the Peptide Neurotransmitter NAAG and Appearance of a New Member of the NAAG Neuropeptide Family. J. Neurochem. 2011 doi: 10.1111/j.1471-4159.2011.07338.x. E-pub, June 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski RT, Bzdega T, Neale JH. The metabotropic glutamate receptor type 3 (mGluR3), not mGluR2, mediates the efficacy of NAAG peptidase inhibition in a PCP model of schizophrenia. 2011 Submitted. [Google Scholar]
- Schweitzer C, Kratzeisen C, Adam G, Lundstrom K, Malherbe P, Ohresser S, Stadler H, Wichmann J, Woltering T, Mutel V. Characterization of [(3)H]-LY354740 binding to rat mGlu2 and mGlu3 receptors expressed in CHO cells using Semliki forest virus vectors. Neuropharmacology. 2000;39:1700–1706. doi: 10.1016/s0028-3908(99)00265-8. [DOI] [PubMed] [Google Scholar]
- Silva R, Mata LR, Gulbenkian S, Brito MA, Tiribelli C, Brites D. Inhibition of glutamate uptake by unconjugated bilirubin in cultured cortical rat astrocytes: Role of concentration and pH. Biochem. Biophys. Res. Comm. 1999;265:67–72. doi: 10.1006/bbrc.1999.1646. [DOI] [PubMed] [Google Scholar]
- Slusher BS, Vornov JJ, Thomas AG, Hurn PD, Harukuni I, Bhardwaj A, Traystman RJ, Robinson MB, Britton P, Lu XC, Tortella FC, Wozniak KM, Yudkoff M, Potter BM, Jackson PF. Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat. Med. 1999;5:1396–402. doi: 10.1038/70971. [DOI] [PubMed] [Google Scholar]
- Tsukamoto T, Wozniak KM, Slusher BS. Progress in the discovery and development of glutamate carboxypeptidase II inhibitors. Drug Discov. Today. 2007;12:767–776. doi: 10.1016/j.drudis.2007.07.010. 2007. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Santi MR, Neale JH. N-acetylaspartylglutamate activates cyclic-AMP coupled metabotropic glutamate receptors in cerebellar astrocytes. Glia. 1998;24:172–180. doi: 10.1002/(sici)1098-1136(199810)24:2<172::aid-glia2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wegorzewska IN, Bzdega T, Olszewski RT, Neale JH. Differential negative coupling of type 3 metabotropic glutamate receptor to cyclic GMP levels in neurons and astrocytes. J. Neurochem. 2006;96:1071–1077. doi: 10.1111/j.1471-4159.2005.03569.x. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Pshenichkin S, Surin A, Sullivan SE, Neale JH. N-Acetylaspartylglutamate selectively activates mGluR3 receptors in transfected cells. J. Neurochem. 1997;69:174–182. doi: 10.1046/j.1471-4159.1997.69010174.x. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Saab O, Neale JH. N-acetylaspartylglutamate inhibits forskolin-stimulated cyclic AMP levels via a metabotropic glutamate receptor in cultured cerebellar granule cells. J. Neurochem. 1993;61:943–948. doi: 10.1111/j.1471-4159.1993.tb03606.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Hirasawa S, Wroblewska B, Zhou J, Grajkowska E, Kozikowski A, Wroblewski J, Neale JH. Antinociceptive effects of N-acetylaspartylglutamate (NAAG) peptidase inhibitors ZJ-11, ZJ-17 and ZJ-43 in the rat formalin test and in the rat neuropathic pain model. Eur. J. Neuroscience. 2004;20:483–494. doi: 10.1111/j.1460-9568.2004.03504.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Kozikowski A, Zhou J, Neale JH. Intracerebroventricular administration of N-acetylaspartylglutamate (NAAG) peptidase inhibitors is analgesic in inflammatory pain. Mol. Pain. 2008;4:31. doi: 10.1186/1744-8069-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Saito O, Aoe T, Bartolozzi A, Sarva J, Zhou J, Kozikowski A, Wroblewska B, Bzdega T, Neale JH. Local administration of N-acetylaspartylglutamate (NAAG) peptidase inhibitors is analgesic in peripheral pain in rats. Eur. J. Neurosci. 2007;25:147–158. doi: 10.1111/j.1460-9568.2006.05272.x. [DOI] [PubMed] [Google Scholar]
- Zhong C, Zhao X, Van KC, Bzdega T, Smyth A, Zhou J, Kozikowski AP, Jiang J, O'Connor WT, Berman RF, Neale JH, Lyeth BG. NAAG peptidase inhibitor increases dialysate NAAG and reduces glutamate, aspartate and GABA levels in the dorsal hippocampus following fluid percussion injury in the rat. J. Neurochem. 2006;97:1015–1025. doi: 10.1111/j.1471-4159.2006.03786.x. [DOI] [PubMed] [Google Scholar]
- Zhou J, Neale JH, Pomper MG, Kozikowski AP. NAAG peptidase inhibitors and their potential for diagnosis and therapy. Nature Rev. Drug Discovery. 2005;4:1015–1026. doi: 10.1038/nrd1903. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.