

Abstract
VILIP-1 (visinin-like protein 1) is downregulated in various human squamous cell carcinoma. In a mouse skin SCC model VILIP-1 expression is reduced in aggressive tumor cells, accompanied by reduced cAMP levels. Overexpression of VILIP-1 in aggressive SCC cells led to enhanced cAMP production, in turn causing a reduction in invasive properties. Moreover, in primary neurons and neuronal tumor lines VILIP-1 enhanced cGMP-signaling. Here, we set out to determine whether and how cAMP and cGMP-signaling contribute to the VILIP-1 effect on enhanced SCC model cell migration, and thus most likely invasiveness in vivo. We found stronger increase in cGMP levels in aggressive, VILIP-1-negative SCC cells following stimulation of guanylyl cyclases NPR-A and -B with the natriuretic peptides ANP and CNP, respectively. Incubation with ANP or 8Br-cGMP to increase cGMP levels further enhanced the migration capacity of aggressive cells, whereas cell adhesion was unaffected. Increased cGMP was caused by elevated expression levels of NPR-A and NPR-B. However, the expression level of VILIP-1 did not affect cGMP signaling and guanylyl cyclase expression in SCC. In contrast, VILIP-1 led to reduced migration of aggressive SCC cells depending on cAMP levels as shown by use of adenylyl cyclase inhibitor 2′,3′-dideoxyadenosine. Involvement of cAMP-effectors PKA and EPAC play a role downstream of adenylyl cyclase activation. VILIP-1-positive and -negative cells did not differ in mRNA expression of adenylyl cyclases, but an effect on enhanced protein expression and membrane localization of ACs was shown to underlie enhancement of cAMP production and, thus, reduction in cell migration by VILIP-1.
Keywords: squamous cell carcinoma, cAMP-, cGMP-signaling, cell migration, calcium-binding protein, tumor invasion suppressor
INTRODUCTION
Squamous cell carcinoma (SCC) is a common histological type in head and neck, esophageal, cervical, lung, and skin cancer. We have recently shown that expression of neuronal calcium sensor VILIP-1 (visinin-like protein 1) (1, 2) was lost in human esophageal cancer and in non-small cell lung carcinoma (3, 4). In esophageal cancer VILIP-1 expression was completely lost or significantly reduced compared with normal squamous epithelium. The protein expression level significantly correlated with invasive features, such as depth of tumor invasion and local lymph node metastasis (3). In non-small cell lung carcinoma statistical analysis of expression and promoter methylation showed significant relationship between promoter methylation and protein expression downregulation as well as between survival and downregulation of VILIP-1 expression (4). These findings suggest that VILIP-1 acts as a tumor invasion suppressor in human epithelial tumors such as esophageal cancer and in non-small cell lung carcinoma and that its reduction or loss may serve as a predictor of short-term survival in cancer patients (3, 4). Moreover, in an experimental animal model, chemically-induced murine skin cancer cells, VILIP-1 was studied for its possible regulation of tumor cell invasiveness in more detail. Initially, VILIP-1 was identified in this model to be differentially expressed in skin cancer cells of high and low invasive ability (5). VILIP-1 expression in normal basal epidermal keratinocytes was increased in less aggressive SCCs and decreased or lost in aggressive and invasive SCCs. Ectopic overexpression of VILIP-1 resulted in a cAMP-mediated decrease of in vivo and in vitro growth and invasiveness of SCC cells (5). Enforced expression of VILIP-1 led to inhibition of cell adhesion and migration by down-regulating fibronectin receptors, suggestive of a tumor suppressor function for VILIP-1 (6). In the SCC model cells reduced invasiveness and elevated cAMP levels were accompanied by decreased MMP-9 as well as lowered RhoA activity. Recently, this was confirmed in in vivo studies in transgenic mice expressing VILIP-1 under the control of the keratinocyte-specific K5 promoter, where we have demonstrated that overexpression of VILIP-1 in keratinocytes decreases cell proliferation, susceptibility to skin tumor formation and levels of MMP-9 (7).
The cAMP-signaling pathway is an important modulator of tumor cell properties such as proliferation, differentiation and cell migration. Intracellular cAMP levels are regulated by the activity of adenylyl cyclases (ACs) producing cAMP from ATP and of phosphodiesterases (PDEs) hydrolyzing cAMP to AMP. cAMP signaling molecules target cyclic nucleotide-gated channels (CNGs), exchange protein activated by cAMP (EPAC) and cAMP-dependent protein kinase A (PKA) (9, 10). By activating Rap, a small GTPase of the Ras family, EPAC can influence cell migration (10) and integrin-mediated cell adhesion (11). PKA can inhibit proliferation, and influence differentiation and apoptosis (12). For tumor invasion the effect of the cAMP-signaling pathway via protein kinase A (PKA) on changes in cell motility, e.g. via inhibition of the small GTPase RhoA, is particularly important (13, 14). The Rho family of small GTPases, such as RhoA und Rac, promote reorganisation of the actin cytoskeleton during migration of cancer cells (15, 16). RhoA via Rho-associated kinase ROCK and LIM-kinase influences the actin cytoskeleton, stress fibers and contractility of the actin-myosin complex during tumor invasion (17–19). Pharmacological blockage of ROCK function leads to inhibition of terminal differentiation as well as enhancement of proliferation in keratinocytes.
In addition the PKA-effector RhoA is a substrate of cGMP-dependent protein kinase (PKG), linking also cGMP-signaling to cytoskeleton re-arrangements and cell motility (20). cGMP is synthesized from intracellular GTP either by soluble (sGC) or by particulate guanylyl cyclases (NPR-A and NPR-B) and affects hydrolyzing cGMP-specific PDEs, CNGs and cGMP-dependent protein kinases (PKGs) (21). cGMP signaling exhibits a diverse and contrary role in cancer cell migration. Growth inhibitory and apoptotic functions of sGC/cGMP signaling pathway have been shown in colon cancer cells (22), whereas proliferation of ovarian cancer cells was promoted (23). Migration capacity of glial cells and non-small cell lung cancer cells is increased by PKG activity (20, 24), whereas in colon cancer cells induction of PKG inhibits cell migration (24).
Interestingly in this context, VILIP-1 was not only shown to enhance cAMP-, but also cGMP-signaling in glioma tumor cell lines and primary neurons (20–24). Thus, we were interested to investigate how SCC cell lines respond to modulations of cAMP- versus cGMP-signaling regarding cell adhesion and cell migration, and whether the tumor invasion suppressing effect of VILIP-1, which has previously been correlated with an effect of VILIP-1 on enhanced cAMP production, may also be linked to cGMP levels in SCC cell lines.
MATERIAL AND METHODS
Material
Natriuretic peptides ANP, CNP (guanylyl cyclase activators), soluble guanylyl cyclase activator SNOG (S-nitrosoglutathione); adenylyl cyclase activator Forskolin, 8Br-cAMP, 8Br-cGMP, H89 (protein kinase A inhibitor), DDA (2′,5′-dideoxyadenosine, general AC inhibitor), KT5823 (protein kinase G inhibitor), 8CPT-2Me-cAMP (EPAC activator ) for cell stimulation experiments were obtained from Sigma (St. Louis, MO), Tocris (Bristol, UK) and Calbiochem (San Diego, CA). Cell culture reagents were obtained from Gibco-Invitrogen (San Diego, CA). Unless otherwise specified, all other reagents were purchased from Sigma and Roth (Karlsruhe, Germany).
Antibodies
Rabbit polyclonal antibodies, raised against recombinant VILIP-1 fusion protein, were affinity-purified on corresponding glutathion-S-transferase (GST)-tagged fusion proteins immobilized on N-hydroxysuccinimide ester coupled agarose colums (Bio-Rad, Hercules, CA) as previously described (26). Rabbit polyclonal antibodies detecting adenylyl cyclase isoforms, were raised against a 14-amino acid peptide conserved in all adenylyl cyclase isoforms (AC-comm) (26, 31). Isoform-specific polyclonal antibodies against ACIII (sc-32113), V/VI (sc-590), VII (sc-32120), IX (sc-20765), monoclonal antibodies against beta-actin (sc-81178) and HRP-labeled secondary antibodies were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). Cy3 labeled secondary antibodies were purchased from Dianova (Hamburg, Germany).
Cell culture
Murine skin squamous cell carcinoma cell lines CC4A and CC4B, CH72 and CH72T3 were described previously (5). CC4A and CC4B were derived from the same tumor. When injected s.c. into nude mice, CC4A gave rise to a high-grade SCC or spindle cell carcinoma (or SCC IV), whereas CC4B gave rise to a well-differentiated, less aggressive, and low-grade SCC (SCCII). CH72 also gave rise to a low-grade SCC after s.c. inoculation, and CH72T3 is a subcloned cell line obtained by in vivo passaging of CH72 into nude mice that resulted in a high-grade SCC. Cells were grown in DMEM (GIBCO) plus FCS (10%), L-glutamine (2mM) and penicillin/streptomycin (100 μg/ml). CC4A and CH72T3 were transfected with VILIP-1-GFP-vector or empty-GFP-vector (32) whereas CC4B and CH72 were transfected with VILIP-1-siRNA or scrambled siRNA using Optimem and lipofectamin (Invitrogen) following the manufacturers instructions. VILIP-1-siRNA (antiVILIP1_1: sense r(AGC CGU UAG UCU GAA UUA A)dTdT, antisense r(UUA AUU CAG ACU AAC GGC U)dAdA; antiVILIP1_2: sense r(CAA AGA UGA CCA GAU UAC A)dTdT, antisense r(UGU AAU CUG GUC AUC UUU G)dAdA; antiVILIP1_3: sense r(GUG CGA CAU UCA GAA AUG A)dTdT, antisense r(UCA UUU CUG AAU GUC GCA C)dAdA) was used as a cocktail of three siRNA oligos (150 ng of each per transfection) directed against the coding region of VILIP-1 and was purchased from Qiagen (Hilden, Germany).
Measurement of intracellular cAMP- and cGMP-production
Squamous cell carcinoma (1 × 105 cells) were transfected with VILIP-1-GFP construct, VILIP-1-siRNA or corresponding controls for 72h and cultured to confluence in 24 well tissue culture dishes (Biochrom, Berlin). Prior to cyclic nucleotide measurements, cells were pre-incubated with DMEM containing 1 mM IBMX for 30 min and incubated afterwards with the effectors (Forskolin, ANP, CNP and SNOG) for 20 min at 37 °C. Measuring of intracellular cGMP or cAMP concentrations was performed with enzyme immunoassay kits (EIA, RPN 226 and 225, Amersham, Upsala).
Western blot analysis
Cultured cells were homogenized in an appropriate volume homogenization buffer (25 mM Tris, 150 mM NaCl, pH 7.5, containing the protease inhibitors benzamidine (1 mM), phenylmethylsulfonylfluoride (0.1 mM)). Nuclei and debris were removed by centrifugation at 1.000g for 5 min, protein concentrations were measured using BCA assay (Pierce, Rockford, IL) and 40μg protein of each sample was applied to 5–20 % gradient SDS-PAGE. To analyze the expression level of VILIP-1 or adenylyl cyclase isoforms, separated proteins were blotted on a PVDF membrane. The membrane was blocked with 5% milk powder in TBST (25 mM Tris, 150 mM NaCl, pH 7.5, 1% Tween 20) for 1h at RT and afterwards incubated with the primary anti-VILIP-1, anti-AC-comm or ACIII, V/VI, VII, IX antibodies at 4°C overnight as previously described (26, 31). After washing three times with TBST, secondary antibodies were applied for one hour at RT. Unbound antibodies were removed and detected protein was visualized in a dark chamber using Western Lightning reagents (PerkinElmer Life Sciences, Boston, MA USA) and Hyperfilm (Amersham, UK).
RT-PCR
PCR primers were developed that selectively amplify cDNA encoding adenylyl cyclase isoforms I-X and guanylyl cyclase isoforms A and B. Primers for AC isoforms (Table 1) have been designed to be species-independent, working in mouse and rat cell lines or tissue. RT-PCR experiments were performed 3 times for each cyclase isoform, using total RNA from murine testis or brain or from SCC-lines. Total RNA was extracted using RNeasy Mini-Kit (Quiagen, Hilden, Germany) and reverse transcribed using Oligo(dT) primers and SuperScript III First-Strand-Kit (Invitrogen). PCR was performed using 0.2 μM of each primer, PCR buffer, 0.2 mM dNTP-Mix, 2mM MgCl2, and 1U taq polymerase (Invitrogen) and DEPCH2O in a 50μl reaction mix. 35 cycles of amplification were performed for each sample. For each primer pair the reaction was also carried out in absence of reverse transcriptase to ensure that there is no DNA contamination.
Table 1.
Primer sets used for RT-PCR (bp, base pairs; s, sense; as, antisense)
| cDNA | accession number | primer sequence in 5′ --> 3′ | size of amplicon in bp |
|---|---|---|---|
| AC1 | XM_001053224.1 | s CTG AAS AGG TGC TCA TAC CAG TT as GGC TTC CTT CCC TGC TGC |
826 |
| AC2 | NM_031007.1 | s GGA GGA AGG AGA YGG TGA GA as CTC CAA AGG AGA AGC CAA GGA |
577 |
| AC3 | NM_130779.1 | s TTG CYG ACT TCT ACA CTG AGG as CGTG TCC TTC ATG GCT ART GC |
283 |
| AC4 | NM_019285.2 | s GCGT YCT CTG TGG AGT CAT YGG as GGA GAA RGA AGA GAA GGA AGG |
650 |
| AC5 | NM_022600.1 | s GAG GCA GTT CYG TGG CGT CAG as TGG GMG TTG GTG TGC AGA GA |
546 |
| AC6 | NM_012821.2 | s GGT TCC CAA AGT GGA TGA ACG as CCT CCT GAT GAA GGC TTC ATC |
400 |
| AC7 | BC115833.1 | s ATG GAG AAT GTG AAC CGC CTC C as CTT GCT CAT CAG GGC CAT GCT AA |
401 |
| AC8 | NM_017142.1 | s CAG GCC AAA GAG GAG ATC AAC G as TGA TCT CAT TCA GCA AGC GCA |
249 |
| AC9 | XM_001077332.1 | s CTG GAG CAT CTA TTT CGC TGT C as CGA ACA CCA GCA GGG TGA G |
166 |
| AC10 | NM_021684.1 | s ACC ACC TAC ATC ACA CTC G as TCA ATC ACC TCT CCT TCC |
688 |
| NPR-A | NM_008727 | s TGC AGA ATG AGC ACT TGA CC as GGC AAT TTC CTG AAG GAT GA |
431 |
| NPR-B | NM_173788 | s TTG CTG TAT CTG GAT GCT CG as GAT TTG GGG GTT CTC GGT AT |
409 |
| VILIP-1 | NM_012038 | s ATG GGG AAR CAG AAT AGC AAA C as TCA TTT CTG MAT GTC KCA CTG CA |
550 |
| GAPDH | NM_008084 | s ACC ACA GTC CAT GCC ATC AC as TCC ACC ACC CTG TTG CTG TA |
590 |
Immunocytochemistry and fluorescence intensity measurement
Cells (3 × 104 cells/well) were plated on glass coverslips coated with fibronectin, poly-D-lysine in PBS. Following fixation of the cells with 4% paraformaldehyde in PBS, pH 7.4 for 15 min at RT, cells were washed twice with 25 mM glycine in PBS and subsequently permeabilized and blocked in 0.2% Triton X-100, 3% bovine serum albumine, 10% horse serum in PBS for 30 min. Cells were incubated with primary antibodies at 4°C overnight. After washing three times with PBS secondary antibodies were applied to the cells for one hour at RT. Unbound antibodies were removed and coverslips with the cells were mounted on slides with Moviol (Calbiochem). Cy3 fluorescence images were recorded digitally with fixed settings using a Leica SP laser scanning microscope (Leica DM LFSA or DM 2500; Wetzlar, Germany) with Argon-ion (488 nm), Helium-Neon (543 nm) and Helium-Neon (633 nm) or solid-state (488, 532, 635 nm) laser excitation, respectively. The emitted light was collected in sequential scans in the range of 550 nm to 600 nm for Cy3. The pinhole was set airy 1. Images were processed using Photoshop 7.0 (Adobe Systems, San Jose, CA) and Image J v1.33 (NIH, USA). Average fluorescence intensity was measured within 20-pixel-wide (approximately 5 μm) elliptic bands sparing the cell lumen and comprising the cell surface as illustrated in figure 8. For each cell line at least 28 cells from three independent experiments have been analyzed.
Figure 8.

Subcellular localization of AC proteins in VILIP-1-negative CC4A, CH72T3 and VILIP-1-positive CC4B, CH72 SCC cells. A. Images from confocal immunofluorescence microscopy of CC4A and CC4B cells using AC-comm antibody against all AC isoforms were analyzed in Image J (outer rings show whole cell area, outer minus inner rings show membrane areas used for quantification). B. Quantitative analysis of the signal intensity of AC fluorescence show that the whole cell fluorescence intensity, as well as intensity at the cell surface membrane is significantly increased in VILIP-1-positive cell lines CC4B and CH72 compared to more aggressive VILIP-1-negative cell lines CC4A and CH72T3 (p< 0.0001, p< 0.001, N = 3, n > 28). Error bars indicate standard deviations. C. Relative fluorescence intensity (ratio of surface and whole cell fluorescence). D. Western blot analysis using AC-comm antibody against all AC isoforms in VILIP-1-positive cell lines CC4B and CH72 compared to more aggressive VILIP-1-negative cell lines CC4A and CH72T3. Molecular weights of marker proteins are indicated in the left margin, arrows indicate adenylyl cyclase isoforms showing differential expression. E. Western blot analysis using AC isoform specific antibodies against ACIII, ACV/VI, ACVII and ACIX in VILIP-1-positive cell lines CC4B and CH72 compared to more aggressive VILIP-1-negative cell lines CC4A and CH72T3. Protein markers are indicated in the left margins, arrows indicate adenylyl cyclase isoforms showing differential expression and β-actin bands are shown as loading control.
Cell adhesion assay
Fibronectin (10μg/ml, Biochrom, Berlin) in phosphate-buffered saline (PBS, Biochrom, Berlin) was spread over the surface of 24 well plates (200μl/well) and the protein was allowed to absorb for 1 h at 37°C. After saturation of the wells with 1% BSA (fraction, V, Roth, Karlsruhe) cells (1 × 105 cells/well) were seeded on the plates in unsupplemented medium containing 0.1% BSA. After 1 h incubation at 37°C non-adherent cells were removed with PBS and adherent cells were fixed with 4% PFA in PBS for 20 min and washed twice with PBS thereafter. Cells were stained with 0.1% Crystal Violet, 0.5% ethanol in destilled water for 1.5h, washed three times and de-stained in 300 μl of 50% ethanol and 1% glacial acetic acid in aqua dest.. The optical density of each well was measured at 570 nm in triplicate. A blank value corresponding to fibronectin-coated, BSA-blocked well without cell suspension was subtracted. Relative adhesion was calculated within each experiment from experimental and control value by determining the mean of duplicate measurements for each condition. The mean and standard deviation were calculated from n adhesion experiments. The mean of all control experiments was set to 100% and the experimental values from n experiments were expressed as percent of control. For all experimental conditions n was 3. Statistical significance of the mean value from experimental values in % of control from n experiments was calculated by t-test for a mean with unknown variance.
Cell wounding assay
Cells were grown in standard medium. 2 × 105 cells/well were plated in 24 well plates. If indicated, they were transfected with VILIP-1-siRNA, VILIP-1-GFP or the corresponding controls, grown to confluence and placed in low growth factor medium (1% FCS) in order to minimize cell proliferation. 8Br-cAMP, 8Br-cGMP, forskolin, ANP, H89 (protein kinase A inhibitor), DDA (2′,5′-dideoxyadenosine, general AC inhibitor), KT5823 (protein kinase G inhibitor) and EPAC activator (8CPT-2Me-cAMP) were added 1h before scratch/wounding of monolayers using a sterile 200 μl pipette tip. The wound was marked, if indicated 12h after, in general 24h after wounding cells were fixed and pictures were taken at a 200× magnification with a Leica inverted microscope and at least eight representative fields for each condition were analyzed. Migration was quantified by counting the number of cells/field.
Statistical analysis
Statistical analysis was performed using unpaired, two-sided Student’s t-test for samples of unequal variance (Welch test). Values were accepted as significant when p was less than 0.05 (*), less than 0.01 (**) or less than 0.001 (***). All error bars represent standard deviations.
RESULTS
Evaluation of cGMP-levels in cultured skin SCC cell lines with or without expression of the tumor invasion suppressor VILIP-1
To investigate whether the second messenger cGMP might be involved in tumor suppressor-signaling of VILIP-1, we employed an established squamous cell carcinoma (SCC) tumor model. In aggressive and invasive SCC cells, CC4A and CH72T3 cells, little VILIP-1 expression and low cAMP levels are detectable, whereas less aggressive SCCs, CC4B and CH72, show high expression of VILIP-1 protein, high cAMP levels and cAMP-mediated decrease of in vivo and in vitro growth and invasiveness of SCC cells (5). Thus, we now measured basal levels of cellular cGMP and found no correlation with the known VILIP-1 expression levels (Fig. 1A). Next we measured cGMP levels in response to activators of the three different guanylyl cyclases. Stimulation of soluble guanylyl cyclase with the NO donor SNOG resulted in moderately increased cGMP levels, again without any correlation with VILIP-1 expression levels. However, following stimulation of natriuretic peptide receptors NPR-A and B with natriuretic peptides ANP and CNP, respectively, resulting in strongly increased cGMP levels, a negative correlation with VILIP-1 expression was observed. These data indicate that high cGMP levels in response to ANP and CNP correlated positively with aggressiveness, but negatively with the VILIP-1 expression level in SCC cell lines.
Figure 1.

Measurement of cGMP levels in SCC cell lines of different aggressiveness A. Basal intracellular cGMP concentrations of aggressive VILIP-1 negative cells, CC4A and CH72T3, and less-aggressive VILIP-1-positive cells, CC4B and CH7, were measured. B. Intracellular cGMP measurement after stimulation of natriuretic peptide receptor (with ANP and CNP) and soluble (with SNOG) guanylyl cyclases for 20 min. The cGMP levels strongly increased after stimulation with ANP and CNP and were positively correlated with aggressiveness and reversely correlated with VILIP-1 expression (CC4A/CC4B p < 0.0001, CH72T3/CH72 p < 0.0001). Stimulation with SNOG did not show a significant effect on cytosolic cGMP concentration. Bars represent the mean of three experiments and error bars indicate standard deviations.
Involvement of cGMP-signaling in cell adhesion of skin SCC with or without expression of the tumor invasion suppressor VILIP-1
To further test whether cGMP-signaling is involved in the VILIP-1-dependent difference in cell adhesion and migration in SCC, we first examined cell adhesion on fibronectin substrates within 1h. As previously shown (6) SCC skin tumor cells with VILIP-1-expression (CC4B, CH72) showed reduced adhesion on fibronectin substrate, compared to cell lines which lost VILIP-1 expression (CC4A, CH72T3) (Fig. 2A). To test the influence of cGMP on cell adhesion, cGMP levels were elevated by different means, such as activating soluble guanylyl cyclase with NO-generating agent SNOG, or by activating natriuretic peptide receptors with the natriuretic peptides ANP and CNP, or by adding membrane-permeable cGMP analogue 8Br-cGMP (Fig. 2B). In all cases no influence on cell adhesion on fibronectin substrate was noticed.
Figure 2.
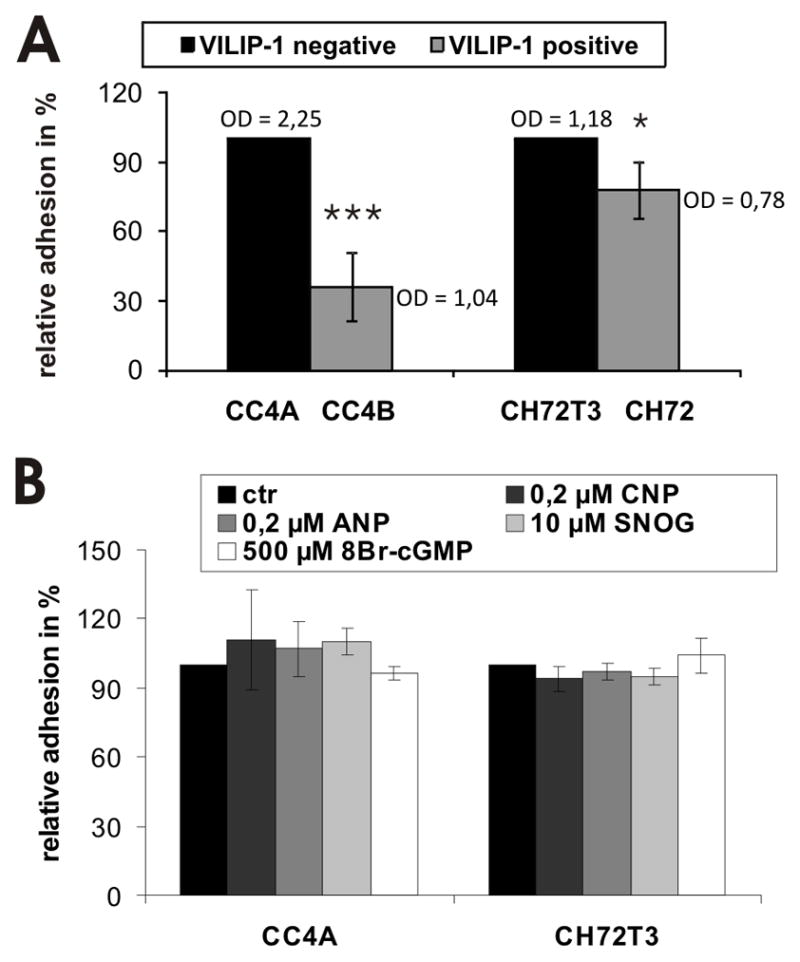
Modulation of cell adhesion by cGMP-signaling in SCC cell lines of different aggressiveness. A. Adhesion of aggressive VILIP-1 negative cells, CC4A and CH72T3, and less aggressive VILIP-1-positive cells, CC4B and CH72, on fibronectin substrate (CC4A/CC4B, p=0.0006; CH72T3/CH72: p=0.014). Optical density (OD) of raw data is shown as mean values. B. Preincubation of VILIP-1-negative cells CC4A and CH72T3 with natriuretic peptides ANP, CNP, with 8Br-cGMP or the soluble guanylyl cyclase activator SNOG, all increasing intracellular cGMP levels, showed no significant effect on cell adhesion on fibronectin. The mean of all control experiments was set to 100% and the experimental values from n experiments were expressed as % of control. Values represent the means from three independent experiments and error bars indicate standard deviations.
Role of cGMP- and cAMP-signaling for cell migration of skin SCC cell lines
We next tested how cGMP-signaling in comparison to cAMP-signaling will influence migratory behavior of SCC. We chose an in vitro wounding assay. Wound repair after scratching occurred in low growth factor medium (1% FCS) (33) and was therefore solely dependent on cell migration, as confirmed by proliferation assays (data not shown). Only a slight inhibitory effect (<10%) of 8Br-cAMP and Forskolin as well as of 8Br-cGMP and ANP on proliferation was observed under these assay conditions. First we evaluated the migratory capacity of aggressive (CC4A, CH72T3) versus non aggressive SCC (CC4B, CH72) cells without and with VILIP-1-expression, respectively (Fig. 3) and confirmed the results previously shown in transwell chambers (5). When VILIP-1 non-expressing cell lines CC4A and CH72T3 were compared to VILIP-1-expressing cells CC4B and CH72 (Fig. 3A), following quantification a clear difference of the capability for migration, and therefore, aggressiveness was observed 24h after scratching the confluent cell layer (Fig. 3B). When we next treated the aggressive CC4A and CH72T3 non-expressing cells with 500 μM 8Br-cAMP to elevate intracellular cAMP levels, reduced migration by −52% and −51%, was observed (Fig. 3B, 8Br-cAMP), similar to the results observed with CC4B cells expressing VILIP-1 (Fig. 3A, table 2). Interestingly, when CC4A and CH72T3 cells were treated with 500 μM 8Br-cGMP to elevate intracellular cGMP levels, we found the opposite effect, with an enhanced migration of these cells by +40% and +36% (Fig. 3B, table 2). To further show specificity of the effect we also stimulated cAMP- and cGMP-pathways by application of the adenylyl cyclase activator forskolin and the guanylyl cyclase activator ANP, which showed similar effects as 8Br-cAMP and 8Br-cGMP, respectively. Quantification of cell migration clearly showed a significant effect of increased cAMP- and cGMP-levels in opposite directions (Fig. 3C, table 2), indicating that cAMP- and cGMP-signaling might play opposite roles in cell migration of skin SCC.
Figure 3.

In vitro migration assay of SCC cells with or without VILIP-1 expression and of VILIP-1-negative SCC following treatment with cGMP or cAMP increasing reagents. A. Confluent monolayers of cells with (CC4B, CH72) or without (CC4A, CH72T3) expression of VILIP-1 were wounded and the migratory capacity of the cells was measured within 24h by counting the number of cells per field in at least 8 fields from three different experiments for each cell line. Representative wounds in monolayers of the different cells are shown. Plot shows the quantification of migration of SCC cells with (CC4B, CH72) or without (CC4A, CH72T3) VILIP-1 expression (CC4A/CC4B: p = 0.0001; CH72T3/CH72: p < 0.0001). B. Confluent monolayers of VILIP-1-non-expressing CC4A and CH72T3 cells were treated with 8Br-cAMP (500 μM cAMP), forskolin (50 μM FSK), 8Br-cGMP (500 μM cGMP) and natriuretic peptide ANP (200 nM ANP) and were wounded to measure the migratory capacity of the cells after 12 and 24 hrs by counting the number of cells per field in at least 8 fields from three different experiments. C. Quantification of the migration of VILIP-1 non-expressing SCC cells, CC4A and CH72T3, after incubation with 8Br-cAMP, Forskolin, 8Br-cGMP and ANP (CC4A: ANP, p = 0,009; 8Br-cAMP, FSK, 8B-rcGMP, p< 0,0001; CH72T3: p< 0,0001 for all conditions). Data represent mean values from at least three independent experiments and error bars indicate standard deviations.
Table 2.
Summary of the effect of compounds activating or inhibiting cyclic nucleotide signaling on cell migration of VILIP-1-negative SCC CC4A, CH72T3 and VILIP-1-positive SCCs CC4B, CH72 in a wound closing assay.
| VILIP-1-negative SCC | ||||
|---|---|---|---|---|
| reagent | CC4A | CH72T3 | ||
| effect in % of ctr | p-value (n,N) | effect in % of ctr | p-value (n,N) | |
| 8Br-cAMP | −52% | < 0,0001 (10,3) | −51% | < 0,0001 (10,3) |
| FSK | −67% | < 0,0001 (10,3) | −40% | < 0,0001 (10,3) |
| 8Br-cGMP | +40 % | < 0,0001 (10,3) | +36% | < 0,0001 (10,3) |
| ANP | +21% | 0,0085 (10,3) | + 48% | < 0,0001 (10,3) |
| Epac-activator (8CPT-2Me-cAMP) | −19 % | 0,017 (5,2) | −23% | 0,0108 (5,2) |
| 8Br-cAMP and 8Br-cGMP | −44% (+17% compared to 8Br-cAMP) |
0,012 (5,2) (0,1539) |
−41% (+22% compared to 8Br-cAMP) |
< 0,0001 (5,2) (0,022) |
| VILIP-1-positive SCC | ||||
| reagent | CC4B | CH72 | ||
| effect in % of ctr | p-value (n,N) | effect in % of ctr | p-value (n,N) | |
| H89 | +17% +21% |
0,007 (8,3) 0,0069 (12,3) |
+68% +55% |
0,003 (8,3) 0,0003 (12,3) |
| DDA | +44% | 0,0042 (6,2) (0,074 vs H89) |
+63% | 0,0002 (6,2) (0,5368 vs H89) |
| KT5823 | −13% | 0,4949 (8,3) | −12% | 0,352 (8,3) |
Possible involvement of natriuretic peptide receptor isoforms in VILIP-1 effects as putative tumor invasion suppressor gene
Loss of VILIP-1 expression appears to correlate with enhanced cGMP accumulation following guanylyl cyclase stimulation in SCC cells (Fig. 1). However, we have previously shown that only enhanced VILIP-1 expression enhances cGMP production by natriuretic peptide receptors NRP-A and B in neural cells (27, 28). Thus, due to the loss of VILIP-1-expression in SCC cells a positive modulatory effect of VILIP-1 on natriuretic peptide receptors appears unlikely. Another possibility would be that VILIP-1 expression suppresses NPR-A and B expression levels, and a loss of VILIP-1 would enhance NPR-A and B expression levels and explain the observed enhanced cGMP levels. In order to test this hypothesis, we first evaluated the efficacy of siRNA specific for VILIP-1 on down-regulation of VILIP-1 protein expression in Western Blot analysis (Fig. 4A, B). Transfection with siRNA specific for VILIP-1 for 48h led to significantly reduced VILIP-1 expression by 30% in CC4B cells, and 72 h of siRNA incubation decreased VILIP-1 protein level significantly in CC4B and CH72 cell lines by about 70% (Fig. 4B). Next we evaluated the expression level of NPR-A and -B mRNA in the four cell lines with or without VILIP-1 expression using RT-PCR (Fig. 5A). NPR-A and -B mRNA were both found to be higher in the VILIP-1 negative cell lines CC4A and CH72T3 in comparison to the VILIP-1-positive cell lines CC4B and CH72, where only very few or no transcripts were detectable. The densitometric analysis of different RT-PCR experiments confirmed that the NPR-A and -B mRNA signal was significantly lower in VILIP-1-positive cells compared to VILIP-1 negative cells (Fig. 5B), indicating that VILIP-1 expression and NPRA and -B expression correlated negatively. However, following over-expression of VILIP-1-GFP in the VILIP-1 negative CC4A and CH72T3 cells (Fig. 5C, CC4A and CH72T3, compare lane 1 and 2), or down-regulation of VILIP-1 by VILIP-1 siRNA in VILIP-1 positive CC4B and CH72 SCC cell lines (Fig. 5C, CC4B and CH72, compare lane 3 and 4), neither NPR-A nor NPR-B mRNA levels were affected by expression levels of VILIP-1. Thus, we found mRNA expression levels to be upregulated in aggressive SCCs independently from the expression level of VILIP-1 (Fig. 3 and 4).
Figure 4.

Down-regulation of VILIP-1-expression in the VILIP-1-expressing SCC cells, CC4B and CH72, using a VILIP-1 specific siRNA cocktail. A. Western Blot analysis of VILIP-1-expression in CC4B and CH72 cells with and without transfection of siRNA specific for VILIP-1 (siRNA) or control siRNA (scr). Beta-actin (Actin) was used as internal control protein. B. Quantification of VILIP-1 expression in Western blots after 48 h and 72 h incubation with 450 nM siRNA cocktail (CC4B: p < 0.0001; CH72: p = 0.002). Values represent the means from three independent experiments and error bars indicate standard deviations.
Figure 5.
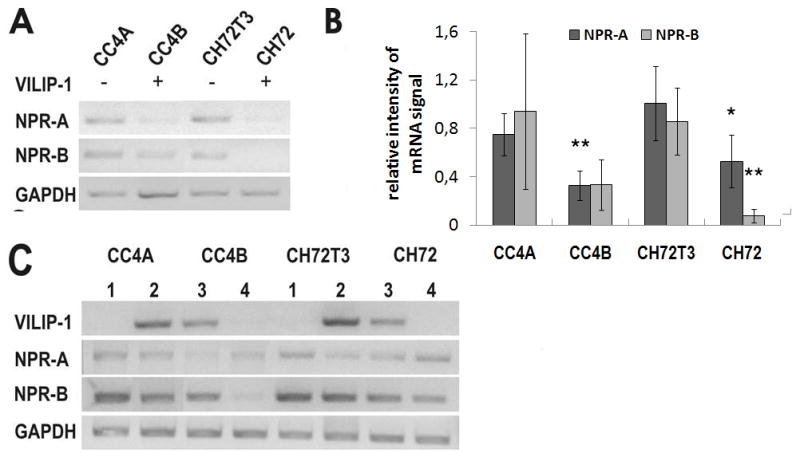
Effect of VILIP-1 on expression of natriuretic peptide receptor A and B (NPR-A, NPR-B) in SCC cell lines of different aggressiveness. A. RT-PCR analysis of NPR-A and NPR-B in VILIP-1-negative SCC cell lines CC4A and CH72T3 and in VILIP-1-positive cell lines CC4B and CH72. GAPDH was used as internal control. B. Densitometry of RT-PCR analysis in A. Intensity of guanylyl cyclase NPR-A and NPR-B PCR bands was normalized to the intensity of GAPDH control PCR bands. Bars represent the mean of three experiments (NPR-A: CC4A p= 0.009, CH72: 0.048; NPR-B: CH72 p= 0.009). Error bars indicate standard deviations. C. Effect of VILIP-1 expression on mRNA expression levels of NPR-A and NPR-B. Up- or down-regulation of VILIP-1 by overexpressing VILIP-1 in VILIP-1-negative SCC cell lines CC4A and CH72T3 (lane 2) or suppressing VILIP-1 by siRNA treatment of VILIP-1-positive cell lines CC4B and CH72 (lane 4). The RT-PCR shown is a representative experiment out of three different experiments.
Involvement of VILIP-1 and cAMP-signaling in cell migration of skin squamous cell carcinoma
We next addressed the mechanisms how cell migration is affected via VILIP-1 and cAMP-signaling in scratch assays in SCC lines of different aggressiveness with or without VILIP-1-expression. Over-expression of VILIP-1-GFP versus GFP control in CC4A and CH72T3 cells led to significant reduction in cell migration of about −31% for CC4A and −23% for CH72T3 cells (Fig. 6A, table 2), whereas application of VILIP-1 siRNA compared to scrambled control siRNA in VILIP-1-expressing CC4B and CH72 cells significantly enhanced cell migration by about +46% for CC4B and +93% for CH72T3 cells (Fig. 6B, table 2). Although SCC cells are somewhat more difficult to transfect than other cell lines with transfection efficiencies of about 50% for VILIP-1-GFP and 60–70% for VILIP-1 siRNA, as measured with BLOCK-iT (Invitrogen), significant and highly significant effects were observed for VILIP-1-overexpression and knock down, respectively (Fig. 6ABC, table 2). Thus, these data are confirming that VILIP-1 expression is partially responsible for the differences in aggressiveness of SCC lines. Importantly, the observed negative effect of VILIP-1 overexpression on cell migration (Fig. 6AC) was completely abolished by treatment of the cells with the general adenylyl cyclase inhibitor 2′,3′-dideoxyadenosine (DDA) compared to control (Fig. 6C), indicating that the cAMP-pathway is acting downstream of the VILIP-1 effect on cell migration. Additionally we tested EPAC activator 8CPT-2Me-cAMP and the combination of 8Br-cAMP and 8Br-cGMP in wounding assays with VILIP-1 negative cells CC4A and CH72T3 as well as the general inhibitor of adenylyl cyclases DDA and the PKA inhibitor H89 in wounding assays with VILIP-1 positive cells CC4B and CH72. Table 2 is a summary of all treatments in the wounding assays with the different SCC lines. Of note, the EPAC activator 8CPT-2Me-cAMP also reduced cell migration, indicating that not only PKA-dependent cAMP signaling might play a role downstream of VILIP-1. Moreover, when 8Br-cAMP and 8Br-cGMP were applied in parallel, the net effect on cell migration was inhibition by −44% and −41%, however, the level of inhibition was still +17% and +22% above the level of treatment with 8Br-cAMP alone. Blocking the cAMP pathway with the protein kinase A inhibitor H89 partially enhanced migratory activity of VILIP-1-positive CC4B and CH72 cells by +21 and +55%, however, the general AC inhibitor had a much more pronounced effect at +44% and + 63% (table 2). Complementary, KT5823, an inhibitor of cGMP-pathway, reduced cell migration by −13% and −12% as predicted by the opposite effect of cGMP activator experiments in less aggressive SCC lines (table 2).
Figure 6.

Effect of VILIP-1 expression level and cAMP-signalling on in vitro migration of SCC cells. Confluent monolayers of cells with (CC4B, CH72) or without (CC4A, CH72T3) expression of VILIP-1 were wounded and the migratory capacity of the cells was measured within 24 hrs by counting the number of cells per field in at least 8 fields from three different experiments. A. Quantification of the effect of VILIP-1-GFP and GFP-overexpression on cell migration of CC4A and CH72T3 cells. B. Quantification of the effect of VILIP-1 suppression by treatment with VILIP-1 siRNA (siRNA) and control siRNA (scr) on cell migration of CC4B and CH72 cells (CC4A: p = 0.002; CC4B: p = 0.0002; CH72T3 p= 0.019; CH72: p <0.0001). C. Quantification of migration after transfection of CC4A and CH72T3 cells with GFP (Ctr) and GFP-VILIP-1 (+VILIP-1) with and without incubation with DDA, a general adenylyl cyclase inhibitor (CC4A: +VILIP-1 p =0,0013, +VILIP-1 + DDA p =0,75; CH72T3: +VILIP -1 p =0,017, +VILIP-1 + DDA p = 0,49). D. Confluent monolayers of VILIP-1-expressing CC4B and CH72 cells were treated with PKA inhibitor H89 (10 μM), general AC inhibitor DDA (500 μM), and the selective inhibitor of cGMP-dependent protein kinase (PKG) KT5823 (1 μM) and were wounded to measure the migratory capacity of the cells after 24 hrs by counting the number of cells per field in at least 8 fields from three different experiments. E. Quantification of migration after H89 (CC4B: p =0,0069, CH72: p =0,0003), DDA (CC4B: p =0,0042, CH72: p =0,0002) and KT5823 (CC4B: p =0,4949, CH72: p =0,352) treatment in D. Data represent mean values from at least three independent experiments and error bars indicate standard deviations.
Possible involvement of distinct adenylyl cyclase isoforms in the VILIP-1-effect as a putative tumor invasion suppressor gene
Since VILIP-1 appears to act via cAMP-, but not cGMP-signaling on tumor cell migration in the SCC model, we were interested in the adenylyl cyclase isoforms, which might be involved in the tumor suppressor effect of VILIP-1. To this end, an effect of VILIP-1 on adenylyl cyclases in glioma cells expressing the isoforms ACVI und III has been observed (26). Therefore, we have examined whether ACVI, ACIII or any other AC-isoform is expressed in SCCs using RT-PCR analysis (Fig. 7). In the positive control tissue from mouse brain or testis (Fig. 7, pc) all isoforms were amplified using adenylyl cyclase-specific primers, whereas in SCC cell lines only isoforms ACIII, V, VI, VII and IX were found. Interestingly, two different mRNA splice forms of the ACV isoform seem to exist in mouse brain samples, with only one isoform expressed in peripheral SCC cell samples (Fig. 7, ACV). Unlike the regulation seen for guanylyl cyclases (Fig. 5A), none of the adenylyl cyclase isoforms showed up- or down-regulation in the more aggressive SCC tumor lines.
Figure 7.

RT-PCR analysis of the expression pattern of adenylyl cyclase (AC) isoforms 1–9 and soluble AC in SCC cell lines. Mouse brain or testis transcriptom was used as positive control (pc). Negative control was without cDNA (nc). Transcripts of isoforms 3, 6, 7 and 9 were clearly detectable in all SCC cell lines. For isoform AC5 the brain positive control exhibits two bands, with the lower band representing the calculated transcript size. Transcripts found in SCC cells represent the upper band. Representative results from three different RT-PCR experiments for each isoform are shown.
Differences in surface expression of adenylyl cyclase isoforms may account for the cAMP effect on SCC tumor cell migration
Reduced VILIP-1 expression and reduced cAMP levels account for enhanced aggressiveness of SCC cells. Since one of the mechanisms by which VILIP-1 modulates signal transduction is modulation of cell surface/membrane expression of receptors, including receptor guanylyl cyclase NPR-B and the α4β2 nicotinic acetylcholine (28, 33), we hypothesized that reduced surface expression of adenylyl cyclases might account for reduced cAMP levels and in turn reduced aggressive behavior of SCC. Thus, we have examined whether differences in the surface expression of adenylyl cyclases occur in VILIP-1-expressing CC4B and CH72 compared to VILIP-1-negative cell lines CC4A and CH72T3. According to the PCR analysis (Fig. 7) we needed to target several adenylyl cyclases including ACIII, V, VI, VII and IX. Therefore, an antibody directed against all adenylyl cyclase isoforms (AC-comm, 26) was used, which has been previously characterized and validated in detail (31). Confocal immunofluorescence microscopy was used to quantify the fluorescence intensity of the relative adenylyl cyclase fluorescence intensity of the relative adenylyl cyclase fluorescence in VILIP-1-expressing CC4B and CH72 cell lines and VILIP-1-negative cell lines CC4A and CH72T3 (Fig. 8A). Overall immunofluorescence in the cells and particularly at the cell membrane appears to be increased in VILIP-1-positive cell lines (Fig. 8A, compare CC4A and CC4B). Quantification of fluorescence intensities of whole cell and membrane localization revealed a highly significant difference in adenylyl cyclase expression and surface membrane expression levels in the VILIP-1-positive cell lines (Fig. 8B). When the ratio of surface membrane versus whole cell fluorescence staining intensity was calculated, a highly significant increase in VILIP-1-expressing cell lines CC4B and CH72 is noticeable, indicating that even with an enhanced overall expression level the surface membrane localization was increased (Fig. 8C). Moreover, to further investigate the possibility of enhanced AC expression levels, Western blot analysis using the AC-comm antibody was performed (Fig. 8D). The antibody recognized several protein bands in the molecular weight range of 100 to 160kDa in the cell lysates, which most likely reflect expression of AC isoforms III, V/VI, VII and IX as indicated in the PCR analysis (Fig. 7). Several of these bands appear to be higher expressed in less-aggressive VILIP-1-positive SCC lines CC4B and CH72 (Fig. 8D, arrows). We next used isoform specific antibodies against ACIII, IV, V/VI, VII and IX. A clear upregulation of protein bands in the expected molecular weight range and recognized by the isoform-specific antibodies was observed (Fig. 8E, arrows). These results indicate that differences in expression and membrane expression of adenylyl cyclases might account for cAMP-dependent differences in the aggressiveness of skin SCC. However, these initial findings have to be further substantiated by future studies on the VILIP-1 effect on selected AC isoforms.
DISCUSSION
Cyclic nucleotides, such as cAMP and cGMP, are key cellular second messengers implicated in a variety of cellular processes, including cell adhesion and cell migration. Particularly, activation of the cAMP pathway has been reported both to enhance and inhibit cell migration, probably depending on different downstream signaling pathways (34). In HeLa cells, a SCC model, the cAMP-PKA pathway, and phosphodiesterases (PDE) in particular, were identified as drugable targets. The cAMP-dependent PDE2 and PDE3 genes, rather than cGMP-dependent PDEs are clearly overexpressed in HeLa cells, and indicate the potential importance to control cAMP levels by these enzymes in squamous carcinoma (35). Similarly, PDE inhibitors targeting the cGMP-PKG pathway have previously been identified as a potentially useful class of anticancer agents (36, 37).
In the model of murine skin SCC cell lines with different aggressiveness used in this study, increased basal levels of cAMP in less aggressive VILIP-1 expressing tumor cells have been described previously (5). In this study we found increased cGMP levels in aggressive tumor cells in response to guanylyl cyclase receptor NPR-A and NPR-B agonists ANP and CNP activation, respectively. Furthermore, we show that the cGMP analogue 8-Br-cGMP as well as the NPR-A agonist ANP, leading to enhanced intracellular cGMP levels, both have a migration-promoting effect, whereas the cAMP analogue 8-Br-cAMP and the AC activator forskolin, leading to enhanced intracellular cAMP levels, had a migration inhibitory role. Although an unexpected result at first, this phenomenon is supported by data from the literature showing that 8-Br-cGMP has a migration-promoting effect in fibroblasts, mammary tumor and non-small cell human lung cancer cells (24, 38, 39), whereas 8-Br-cAMP had a migration inhibiting role in fibroblasts (40, 41). Similar concentrations of cAMP and cGMP as used in our study have been used in the literature for migration studies (42), and we have not observed an obvious contribution of cell proliferation on the effect on migration. Only negligible effects on proliferation between 5–10% (data not shown) were measured under the chosen experimental conditions. Moreover, cross activation of PKA and PKG by 8-Br-cAMP or 8-Br-cGMP can be excluded, given the opposite effect of the signaling systems on cell migration.
The precise mechanisms and downstream signaling pathways of how cAMP- and cGMP-signaling affect squamous carcinoma cell migration in opposite directions have yet to be determined. Using specific activators and inhibitors for cAMP-downstream signaling in migration assays, such as EPAC, we obtained first hints that other cAMP-pathways beside PKA phosphorylation play a role for the downstream effect of cAMP on cell migration in SCCs. VILIP-1 activated cAMP-signaling may regulate integrin alpha(5)beta(1) signaling via PKA (42), and cAMP-dependent activation of EPAC may modulate focal adhesion dynamics and actomyosin-driven leading edge protrusion during cell migration (43). When we used the PKG inhibitor KT5823 in migration assays we obtained data implying PKG in the enhancing effect of cGMP signaling on cell migration. Furthermore, we found up-regulation of NPR-A and NPR-B mRNA occuring in more aggressive SCC cells. Interestingly, when in another study NPR-A expression was tested on the protein level invarious tumor cells, higher levels were detected in different tumor lines, such as lung carcinoma (A549, LLC1), melanoma (B16), ovarian cancer(SKOV3 and ID8), and prostate cancer cells (DU145), as comparedto normal human epithelial cells (44). Moreover, receptor knockout NPR-A−/− mice were resistant to tumorigenesis and tumor progression of these tumor lines in several C57/BL6 murine models, suggesting that NPR-A represents a new targetfor cancer therapy (44). Our experiments clearly show that increased expression of receptor guanylyl cyclases NPR-A and –B and in turn enhanced cGMP levels influence migratory behavior of SCC cells partly via the PKG pathway. However, further data from a variety of primary tumors is needed to support our results on differential expression of guanylyl cyclases in more aggressive tumors. This issue might also be addressed in the transgenic mice expressing VILIP-1 under the control of the keratinocyte-specific K5 promoter that we have recently generated (7).
As an additional aspect of tumor cell invasion we analyzed fibronectin-mediated cell adhesion. Although other studies showed a reduction of cell adhesion on fibronectin substrate, e.g. in melanocytes (45), we could not detect an effect of increased cGMP levels on adhesion of mouse skin SCC, whereas a reduction of cell adhesion by increased cAMP levels could be confirmed in control experiments. Thus, reduced adhesion of less invasive SCC lines, previously shown to be affected via cAMP-signaling (5, 6), seems to be unaffected by cGMP-signaling.
VILIP-1 is known to activate cAMP and/or cGMP-signaling pathways depending on the cell type (5, 6, 26–30). In the murine model of skin SCC we investigated whether there is a cGMP-mediated effect on the inhibition of cell adhesion and migration by VILIP-1 in addition to the cAMP-mediated effects and added new data for our recent discovery of VILIP-1 as a tumor invasion suppressor gene. We have found that in the murine SCC model enhanced cGMP levels were associated with aggressiveness of tumor cell lines, whereas enhanced cAMP-signaling correlated with reduced aggressiveness. Although VILIP-1 expression correlated negatively with enhanced cGMP-signaling pathway, there was no causal relation between enhanced or reduced VILIP-1 expression and natriuretic peptide receptor expression or function in SCC. However, since VILIP-1 has been shown to be up-regulated in certain cancer forms (4) and to activate both cAMP- and cGMP-signaling depending on cell type (26–30), we can not exclude that in other cell types VILIP-1 might act as an enhancer of cell migration by activating cGMP-signaling. Thus, it will be interesting to determine whether VILIP-1 via cAMP- and/or cGMP-signaling may also act as tumor invasion modulator in several other cancer cell lines, such as glioblastoma or colon carcinoma cells, where the protein exhibits enhanced expression levels (4). We have previously shown in C6 glioma cells that VILIP-1 expression leads to enhanced cAMP- and cGMP-accumulation which is paralleled by changes in cell morphology, such as cell rounding and differentiation (26–29). In these cells VILIP-1 leads to enhanced surface expression of cGMP-producing NPR receptor via interfering with clathrin-dependent receptor recycling (28). A similar mechanism of enhancing surface expression of adenylyl cyclases might be envisaged to explain the anti-invasive effects of VILIP-1 mediated by elevated cAMP levels. VILIP-1 expression causes the same reducing effect on migration as elevated cAMP levels, as shown by ectopic expression of VILIP-1 in the VILIP-1-negative SCCs. Blocking adenylyl cyclase activity in VILIP-1-transfected SCCs by the general cyclase inhibitor DDA completely prevented the effect on migration, demonstrating that the effect of VILIP-1 on cell migration requires adenylyl cyclase activity. Using an antibody against several AC isoforms, AC-comm, and isoform-specific antibodies in Western Blot analysis and immunofluorescence studies we obtained first evidence that VILIP-1 has an effect on protein expression as well as surface membrane localization of the cAMP-producing enzyme adenylyl cyclase. Thus, the VILIP-1 effect on enhanced cAMP-production by adenylyl cyclases might be mediated via enhanced protein and membrane expression of one or more of the adenylyl cyclases isoforms ACIII, V, VI, VII and IX, which were shown to be expressed in SCC cells. Whether VILIP-1 specifically affects only a subset of adenylyl cyclase isoforms in SCC, or whether the effect occurs via general enhancement of expression or general exocytotic and/or endocytotic mechanisms, as has been previously postulated for the effect of VILIP-1 on guanylyl cyclases in neurons (28), has yet to be determined. Based on our initial observation that aggressive SCC lines show lower expression and membrane expression of adenylyl cyclases, we hypothesize that regulation of surface expression of signaling effector molecules, such as adenylyl cyclases, might be a novel mechanism for influencing cell migration and, thus, regulating aggressiveness of cancer cells.
In summary, based on our results we hypothesize that cAMP- and cGMP-signaling play opposite roles in in vitro cell migration and possibly in vivo invasiveness of SCC. We found no evidence that VILIP-1 acts as a tumor suppressor via direct or indirect modulation of cGMP levels, but instead we demonstrated that enhanced cGMP signaling contributes to enhanced invasive characteristics in SCC cells. The tumor suppressor VILIP-1, which is down-regulated in various SCC, acts via the cAMP-signaling pathway affecting downstream regulators of migration, such as PKA and EPAC. Regulation of expression and membrane expression of different isoforms of the cAMP-producing enzyme adenylyl cyclase by the tumor suppressor protein VILIP-1 in SCC might be responsible for the regulation of aggressiveness of tumor cells. For certain types of aggressive SCCs a combination treatment, simultaneously interfering with cAMP-dependent effects and inhibiting cGMP-signaling, may in the future develop into a possible approach for treatment of highly invasive SCC.
Acknowledgments
This work was supported by the Deutsche Krebshilfe, an intramural research grant of the Charité, Berlin, and from the National Institutes of Health (AJK-RO1, CA107257).
Abbreviations used are
- AC
adenylyl cyclases
- ANP
atrial natriuretic peptide
- cAMP
protein kinase PKA
- cGMP
protein kinase PKG
- CNP
C-type natriuretic peptide
- DDA
2′,3′-dideoxyadenosine
- EPAC
exchange protein activated by cAMP
- SCC
squamous cell carcinoma
- SNOG
S-nitrosylglutathione
References
- 1.Braunewell K-H, Gundelfinger ED. Intracellular neuronal calcium sensor proteins: a family of EF-hand calcium-binding proteins in search of a function. Cell Tissue Res. 1999;299:1–12. doi: 10.1007/s004410051207. [DOI] [PubMed] [Google Scholar]
- 2.Braunewell K-H, Klein-Szanto AJ. Visinin-like proteins (VSNLs): interaction partners and emerging functions in signal transduction of a subfamily of neuronal Ca2+-sensor proteins. Cell & Tissue Res. 2009;335:301–316. doi: 10.1007/s00441-008-0716-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wickborn C, Klein-Szanto AJ, Schlag PM, Braunewell KH. Correlation of visinin-like-protein-1 expression with clinicopathological features in squamous cell carcinoma of the esophagus. Mol Carcinog. 2006;45:572–581. doi: 10.1002/mc.20201. [DOI] [PubMed] [Google Scholar]
- 4.Fu J, Fong K, Bellacosa A, Ross E, Apostolou S, Bassi DE, Jin F, Zhang J, Cairns P, de Caceres II, Braunewell KH, Klein-Szanto AJ. VILIP-1 downregulation in non-small cell lung carcinomas: mechanisms and prediction of survival. PLoS ONE. 2008;3:e1698. doi: 10.1371/journal.pone.0001698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahloogi H, Gonzalez-Guerrico AM, De Cicco L, Bassi DE, Goodrow T, Braunewell KH, Klein-Szanto AJP. Overexpression of the calcium sensor visinin-like protein-1 leads to a cAMP-mediated decrease of in vivo and in vitro growth and invasiveness of squamous cell carcinoma cells. Cancer Res. 2003;63:4997–5004. [PubMed] [Google Scholar]
- 6.Gonzalez Guerrico AM, Jaffer ZM, Page RE, Braunewell K-H, Chernoff J, Klein-Szanto AJP. Visinin-like protein-1 is a potent inhibitor of cell adhesion and migration in squamous carcinoma cells. Oncogene. 2005;24:2307–2316. doi: 10.1038/sj.onc.1208476. [DOI] [PubMed] [Google Scholar]
- 7.Fu J, Jin F, Zhang J, Fong K, Bassi DE, Lopez De Cicco R, Ramaraju D, Braunewell KH, Conti C, Benavides F, Klein-Szanto AJ. VILIP-1 expression in vivo results in decreased mouse skin keratinocyte proliferation and tumor development. PLoS One. 2010;5(4):e10196. doi: 10.1371/journal.pone.0010196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howe AK, Juliano RL. Regulation of anchorage-dependent signal transduction by protein kinase A and p21-activated kinase. Nat Cell Biol. 2000;2:593–600. doi: 10.1038/35023536. [DOI] [PubMed] [Google Scholar]
- 9.Frisch SM. cAMP takes control. Nat Cell Biol. 2000;2:E167–168. doi: 10.1038/35023650. [DOI] [PubMed] [Google Scholar]
- 10.Grandoch M, Rose A, ter BM, Jendrossek V, Rubben H, Fischer JW, Schmidt M, Weber AA. Epac inhibits migration and proliferation of human prostate carcinoma cells. Br J Cancer. 2009;101:2038–2042. doi: 10.1038/sj.bjc.6605439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of cAMP sensor Epac. J Physiol. 2006;577:5–15. doi: 10.1113/jphysiol.2006.119644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen TC, Hinton DR, Zidovetzki R, Hofman FM. Up-regulation of the cAMP/PKA pathway inhibits proliferation, induces differentiation, and leads to apoptosis in malignant gliomas. Lab Invest. 1998;78:165–174. [PubMed] [Google Scholar]
- 13.Laudanna C, Campbell JJ, Butcher EC. Elevation of intracellular cAMP inhibits RhoA activation and integrin-dependent leukocyte adhesion induced by chemoattractants. J Biol Chem. 1997;272:24141–24144. doi: 10.1074/jbc.272.39.24141. [DOI] [PubMed] [Google Scholar]
- 14.Mukai M, Nakamura H, Tatsuta M, Iwasaki T, Togawa A, Imamura F, Akedo H. Hepatoma cell migration through a mesothelial cell monolayer is inhibited by cyclic AMP-elevating agents via a Rho-dependent pathway. FEBS Lett. 2000;484:69–73. doi: 10.1016/s0014-5793(00)02129-3. [DOI] [PubMed] [Google Scholar]
- 15.Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 16.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Cell Biol. 2003;5:711–719. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- 17.McMullan R, Lax S, Robertson VH, Radford DJ, Broad S, Watt FM, Rowles A, Croft DR, Olson MF, Hotchin NA. Keratinocyte differentiation is regulated by the Rho and ROCK signaling pathway. Curr Biol. 2003;13:2185–2189. doi: 10.1016/j.cub.2003.11.050. [DOI] [PubMed] [Google Scholar]
- 18.Itoh K, Yoshioka K, Akedo H, Uehata M, Ishizaki T, Narumiya S. An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat Med. 1999;5:221–225. doi: 10.1038/5587. [DOI] [PubMed] [Google Scholar]
- 19.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Borán MS, García A. The cyclic GMP-protein kinase G pathway regulates cytoskeleton dynamics and motility in astrocytes. J Neurochem. 2007;102:216–230. doi: 10.1111/j.1471-4159.2007.04464.x. [DOI] [PubMed] [Google Scholar]
- 21.Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, Chepenik KP, Waldman SA. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–414. [PubMed] [Google Scholar]
- 22.Cook T, Wang Z, Alber S, Liu K, Watkins SC, Vodovotz Y, Billiar TR, Blumberg D. Nitric oxide and ionizing radiation synergistically promote apoptosis and growth inhibition of cancer by activating p53. Cancer Res. 2004;64:8015–21. doi: 10.1158/0008-5472.CAN-04-2212. [DOI] [PubMed] [Google Scholar]
- 23.Fraser M, Chan SL, Chan SS, Fiscus RR, Tsang BK. Regulation of p53 and suppression of apoptosis by the soluble guanylyl cyclase/cGMP pathway in human ovarian cancer cells. Oncogene. 2006;25:2203–2212. doi: 10.1038/sj.onc.1209251. [DOI] [PubMed] [Google Scholar]
- 24.Punathil T, Katiyar SK. Inhibition of non-small cell lung cancer cell migration by grape seed proanthocyanidins is mediated through the inhibition of nitric oxide, guanylate cyclase, and ERK1/2. Mol Carcinog. 2008;48:232–42. doi: 10.1002/mc.20473. [DOI] [PubMed] [Google Scholar]
- 25.Deguchi A, Thompson WJ, Weinstein IB. Activation of protein kinase G is sufficient to induce apoptosis and inhibit cell migration in colon cancer cells. Cancer Res. 2004;64:3966–3973. doi: 10.1158/0008-5472.CAN-03-3740. [DOI] [PubMed] [Google Scholar]
- 26.Braunewell K-H, Spilker C, Behnisch T, Gundelfinger ED. The neuronal calcium-sensor protein VILIP modulates cyclic AMP accumulation in stably transfected C6 glioma cells: amino-terminal myristoylation determines functional activity. J Neurochem. 1997;68:2129–2139. doi: 10.1046/j.1471-4159.1997.68052129.x. [DOI] [PubMed] [Google Scholar]
- 27.Braunewell K-H, Brackmann M, Schaupp M, Spilker C, Anand R, Gundelfinger ED. Intracellular neuronal calcium sensor (NCS) protein VILIP-1 modulates cGMP signalling pathways in transfected neural cells and cerebellar granule neurones. J Neurochem. 2001;78:1277–1286. doi: 10.1046/j.1471-4159.2001.00506.x. [DOI] [PubMed] [Google Scholar]
- 28.Brackmann M, Schuchmann S, Anand R, Braunewell K-H. Neuronal Ca2+ sensor protein VILIP-1 affects cGMP signalling of guanylyl cyclase B by regulating clathrin-dependent receptor recycling in hippocampal neurons. J Cell Sci. 2005;118:2495–2505. doi: 10.1242/jcs.02376. [DOI] [PubMed] [Google Scholar]
- 29.Braunewell K-H, Gundelfinger ED. Low level expression of calcium-sensor protein VILIP induces cAMP-dependent differentiation in rat C6 glioma cells. Neurosci Lett. 1997;234:139–142. doi: 10.1016/s0304-3940(97)00696-4. [DOI] [PubMed] [Google Scholar]
- 30.Dai FF, Zhang Y, Kang Y, Wang Q, Gaisano HY, Braunewell KH, Chan CB, Wheeler MB. The neuronal Ca2+ sensor protein visinin-like protein-1 is expressed in pancreatic islets and regulates insulin secretion. J Biol Chem. 2006;281:21942–2153. doi: 10.1074/jbc.M512924200. [DOI] [PubMed] [Google Scholar]
- 31.Mons N, Harry A, Dubourg P, Premont RT, Iyengar R, Cooper DM. Immunohistochemical localization of adenylyl cyclase in rat brain indicates a highly selective concentration at synapses. Proc Natl Acad Sci U S A. 1995;92:8473–8477. doi: 10.1073/pnas.92.18.8473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao CJ, Noack C, Brackmann M, Gloveli T, Maelicke A, Heinemann U, Anand R, Braunewell KH. Neuronal Ca2+ sensor VILIP-1 leads to the upregulation of functional alpha4beta2 nicotinic acetylcholine receptors in hippocampal neurons. Mol Cell Neurosci. 2009;40:280–292. doi: 10.1016/j.mcn.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmed N, Maines-Bandiera S, Quinn MA, Unger WG, Dedhar S, Auersperg N. Molecular pathways regulating EGF-induced epithelio-mesenchymal transition in human ovarian surface epithelium. Am J Physiol Cell Physiol. 2006;290:C1532–42. doi: 10.1152/ajpcell.00478.2005. [DOI] [PubMed] [Google Scholar]
- 34.Howe AK. Regulation of actin-based cell migration by cAMP/PKA. Biochim Biophys Acta. 2004;1692:159–174. doi: 10.1016/j.bbamcr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 35.Fryknäs M, Rickardson L, Wickström M, Dhar S, Lövborg H, Gullbo J, Nygren P, Gustafsson MG, Isaksson A, Larsson R. Phenotype-based screening of mechanistically annotated compounds in combination with gene expression and pathway analysis identifies candidate drug targets in a human squamous carcinoma cell model. J Biomol Screen. 2006;11:457–468. doi: 10.1177/1087057106288048. [DOI] [PubMed] [Google Scholar]
- 36.Thompson WJ, Piazza GA, Li H, Liu L, Fetter J, Zhu B, Sperl G, Ahnen D, Pamukcu R. Exisulind induction of apoptosis involves guanosine 3′,5′-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res. 200(60):3338–3342. [PubMed] [Google Scholar]
- 37.Soh JW, Mao Y, Kim MG, Pamukcu R, Li H, Piazza GA, Thompson WJ, Weinstein IB. Cyclic GMP mediates apoptosis induced by sulindac derivatives via activation of c-Jun NH2-terminal kinase 1. Clin Cancer Res. 2000;6:4136–4141. [PubMed] [Google Scholar]
- 38.Guo D, Tan YC, Wang D, Madhusoodanan KS, Zheng Y, Maack T, Zhang JJ, Huang XY. A Rac-cGMP signaling pathway. Cell. 2007;128:341–355. doi: 10.1016/j.cell.2006.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jadeski LC, Chakraborty C, Lala PK. Nitric oxide-mediated promotion of mammary tumour cell migration requires sequential activation of nitric oxide synthase, guanylate cyclase and mitogen-activated protein kinase. Int J Cancer. 2003;106:496–504. doi: 10.1002/ijc.11268. [DOI] [PubMed] [Google Scholar]
- 40.Hou Y, Gupta N, Schoenlein P, Wong E, Martindale R, Ganapathy V, Browning D. An anti-tumor role for cGMP-dependent protein kinase. Cancer Lett. 2006;240:60–68. doi: 10.1016/j.canlet.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 41.Chen L, Zhang JJ, Huang XY. cAMP inhibits cell migration by interfering with Rac-induced lamellipodium formation. J Biol Chem. 2008;283:13799–13805. doi: 10.1074/jbc.M800555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim S, Harris M, Varner JA. Regulation of integrin alpha vbeta 3-mediated endothelial cell migration and angiogenesis by integrin alpha5beta1 and protein kinase A. J Biol Chem. 2000;275:33920–8. doi: 10.1074/jbc.M003668200. [DOI] [PubMed] [Google Scholar]
- 43.Lyle KS, Raaijmakers JH, Bruinsma W, Bos JL, de Rooij J. cAMP-induced EPAC-Rap activation inhibits epithelial cell migration by modulating focal adhesion and leading edge dynamics. Cell Signal. 2008;20(6):1104–16. doi: 10.1016/j.cellsig.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 44.Kong X, Wang X, Xu W, Behera S, Hellermann G, Kumar A, Lockey RF, Mohapatra S, Mohapatra SS. Natriuretic peptide receptor a as a novel anticancer target. Cancer Res. 2008;68:249–56. doi: 10.1158/0008-5472.CAN-07-3086. [DOI] [PubMed] [Google Scholar]
- 45.Ivanova K, Lambers B, van den Wijngaard R, Le Poole IC, Grigorieva O, Gerzer R, Das PK. Immortalization of human melanocytes does not alter the de novo properties of nitric oxide to induce cell detachment from extracellular matrix components via cGMP. In Vitro Cell Dev Biol Anim. 2008;44:385–95. doi: 10.1007/s11626-008-9113-1. [DOI] [PubMed] [Google Scholar]