

Abstract
Numerous studies report splicing alterations in a multitude of cancers by using gene-by-gene analysis. However, understanding of the role of alternative splicing in cancer is now reaching a new level, thanks to the use of novel technologies allowing the analysis of splicing at a large-scale level. Genome-wide analyses of alternative splicing indicate that splicing alterations can affect the products of gene networks involved in key cellular programs. In addition, many splicing variants identified as being misregulated in cancer are expressed in normal tissues. These observations suggest that splicing programs contribute to specific cellular programs that are altered during cancer initiation and progression. Supporting this model, recent studies have identified splicing factors controlling cancer-associated splicing programs. The characterization of splicing programs and their regulation by splicing factors will allow a better understanding of the genetic mechanisms involved in cancer initiation and progression and the development of new therapeutic targets.
1. Introduction
Each cellular program results from the expression of gene networks or transcriptional programs that are under the control of transcription factors. However, human genes can no longer be considered as simple functional units producing a single transcript. Rather, human genes are an assemblage of exons that can be differentially selected through the use of alternative promoters, alternative polyadenylation sites, and alternatively spliced exons (Figure 1). Genome-wide analyses of splicing based on ESTs (expressed sequence tags), splicing sensitive microarrays, or deep sequencing data sets have revealed that most, if not all human genes can generate different transcripts with different exon content and there are at least 10 times more mRNAs than genes [1–4]. It is now widely accepted that different cell types not only differ because they express different sets of genes but also because genes produce different splicing variants depending on cell type [3, 5–10]. Furthermore, coordinated regulation of alternative splicing of gene products within gene networks plays a key role during differentiation [11–13]. Therefore, an emerging model is that each cell type at a specific developmental stage is characterized by splicing programs that together, with other layers of gene expression programs (e.g., transcriptional programs), determine the precise nature of their transcriptome and therefore their proteome.
Figure 1.
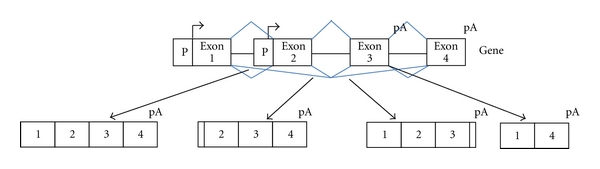
Genes are an assemblage of exons that can be differentially selected through the use of alternative promoters (P), alternative polyadenylation sites (pA), and alternatively spliced exons.
Tumor cells are able to adapt and evolve. Indeed, tumor cells that proliferate develop mechanisms to escape control by their environment. Some tumor cells stimulate angiogenesis or degrade the extracellular matrix, migrate and colonize other tissues to form metastasis (Figure 2) [14]. Gene expression regulation is obviously playing a critical role in this phenotypic plasticity. It is now widely accepted that many transcription factors are altered during tumor initiation and progression. Alterations can occur at the gene level (mutations, misexpression, etc.) or because signaling pathways controlling the activity of transcription factors are altered [14]. Collectively, these alterations result in the changes in transcriptional programs and therefore cellular programs. For example, it has been shown that misregulation of transcription factors, like TWIST that are involved in embryonic development, can induce the epithelial-mesenchymal transition (EMT) that is implicated in the conversion of early-stage tumors into invasive malignancies [15].
Figure 2.
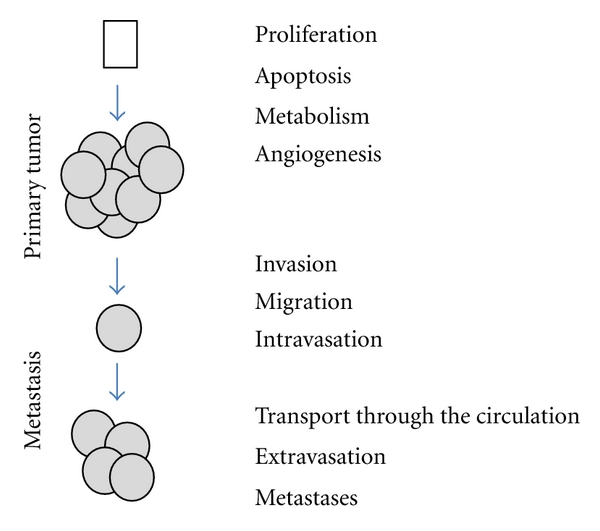
Tumor cells are able to adapt and to evolve. Tumor cells that proliferate develop mechanisms to escape apoptosis and control of their environment. Some tumor cells stimulate angiogenesis or degrade the extracellular matrix, migrate and colonize other tissues to form metastasis.
Because alternative splicing permits the generation of protein isoforms having different biological activities, it is likely that alterations of splicing regulation participate in the phenotypic plasticity of tumor cells. In this context, many splicing variants have been found to be misregulated in cancers [16–24]. However, one major challenge now is to better characterize the splicing programs contributing to specific cancer-associated phenotypes and to identify the splicing factors that control such splicing programs. Indeed, the recognition of exons and introns relies on degenerated sequences at the boundaries between them (splicing sites) that are recognized by the spliceosome, as well as on splicing regulatory sequences located within exons and introns that are recognized by accessory factors or splicing factors (e.g., SR and hnRNP proteins) [16, 17, 19, 20, 25–27]. Depending on their nature and the position of their binding sites, splicing factors can either strengthen or inhibit the splice sites recognition by the spliceosome and can therefore enhance or repress the inclusion of alternative exons. Like transcription factors control transcriptional programs by controlling the expression of gene networks, splicing factors control splicing programs by controlling alternative splicing of gene networks (Figure 3). While several excellent reviews on splicing and cancer have been recently published [16–24, 28–32], our aim in this paper was to discuss recent genome-wide analyses of alternative splicing in cancer indicating that splicing programs controlled by splicing factors play a major role in cellular programs and tumor progression.
Figure 3.
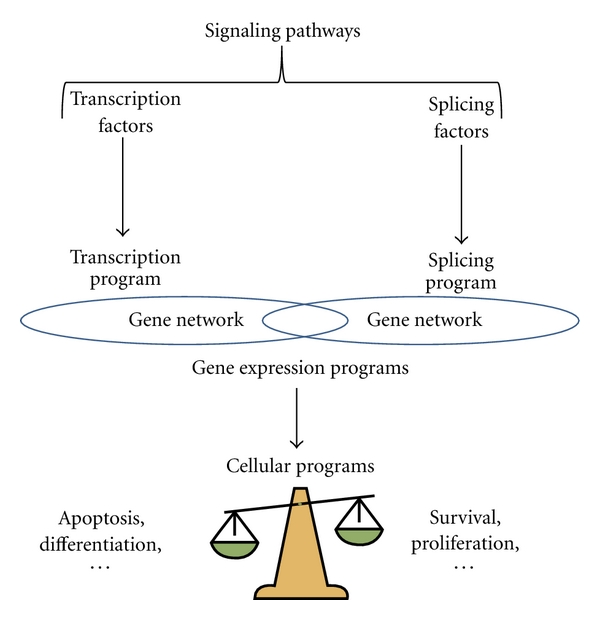
Cellular programs depend on gene expression programs that result from both transcriptional and splicing programs. Transcriptional and splicing programs are under the control of transcription and splicing factors, respectively. Mutations or gene expression alteration of transcription and/or splicing factors can contribute to tumor initiation and progression. The activity of transcription and splicing factors is also under the control of signaling pathways that can be altered in tumor initiation and progression.
2. Large-Scale Analyses of Alternative Splicing in Cancer
There are now numerous studies reporting on splicing alterations in many cancers (see above). However, understanding of the role of alternative splicing in cancer is now reaching a novel level, thanks to the use of new tools including splicing-sensitive microarrays, allowing the analysis of splicing variant expression at a large-scale level. As summarized in Table 1, tumors from breast, lung, digestive tract, and brain have been extensively analyzed thanks to these tools. Although the number of splicing alterations depends on the study design and cancer type, and even though extensive validations using different approaches have generally not been done, it appears that splicing alterations affect gene networks participating in key cellular programs.
Table 1.
Summary of studies using different approaches (1: high throughput RT-PCR, 2: Affymetrix junctions arrays; 3: Affymetrix exon arrays; 4: exonhit arrays; 5: custom arrays) to identify misregulated gene at the splicing level in cancer. Bioinformatic gene pathway analyses.
| Samples | # | Genes functions | References | |
|---|---|---|---|---|
| Pooled human normal and tumor samples | 1 | *Cellular architecture, plasticity and movement | [33] | |
| 26 human breast cancer cell lines and 5 nonmalignant immortalized cell lines | 2 | *Signaling (axon guidance, ephrin receptor, integrin, and tight junctions), cytoskeleton organization, biogenesis, and cell signaling | [34] | |
| Breast | Nonmetastatic (67NR, 168FARN) and metastatic (4T07, 4T1) mouse primary tumors | 3 | *Cellular morphology, and cellular movement | [35] |
| 168FARN, 4T07 and 4T1 mouse primary tumors | 3 | *Cell growth, cell interactions, cell proliferation, cell migration, cell-to-cell signaling, cell death | [36] | |
| 120 human breast tumors and 45 benign lesions | 4 | [37] | ||
|
| ||||
| 18 paired samples of human lung tumors and normal adjacent tissues | 3 | Remodeling of the cytoskeleton and cell movement | [38] | |
| Lung | 20 paired of human primary lung tumors and adjacent normal tissues | 3 | *Tissue development, cellular growth and proliferation, tissue morphology, and immune response | [39] |
| 20 paired of human primary lung tumors and adjacent normal tissues | 5 | Cell adhesion, differentiation, proliferation, adhesion, migration, cytoskeleton, trafficking | [40] | |
| 29 paired of human primary lung tumors and adjacent normal tissues | 5 | Cell signaling, cell proliferation, angiogenesis, cytoskeleton | [41] | |
|
| ||||
| 10 paired of human colon primary tumors and adjacent normal tissues | 3 | *Cell motility and organization of the actin cytoskeleton, cell adhesion, and matrix organization | [42] | |
| Digestive tract | 20 human colon adenocarcinoma and 10 normal samples | 3 | Cancer-related, cytoskeleton, matrix organization, Wnt signaling | [43] |
| 12 samples of isolated cells from 10 patients | 3 | [44] | ||
| 83 human colorectal tissue samples | 3 | [45] | ||
|
| ||||
| 14 pediatric medulloblastomas and 5 samples of normal cerebellum | 3 | Neuronal differentiation, cancer progression: cytoskeleton remodeling, cell morphology regulation, and cell-to-cell interaction | [46] | |
| Brain | 47 human neuroblastoma samples in stage 1 and stage 4 with normal or amplified MYCN copy number | 3 | *Nervous system development, cell adhesion, synaptic transmission, and cytoskeleton organization and biogenesis | [47] |
| 24 human glioblastoma and 12 nontumor samples | 3 | Splicing, intracellular transport and cell migration, central nervous system, notch signaling, cell adhesion, apoptosis, cell growth | [48] | |
| 26 human glioblastoma, 22 oligodendrogliomas and 6 nontumor samples | 3 | [49] | ||
For example, we recently used the exon arrays from Affymetrix to search for potential splicing variants associated with different metastatic properties in the clinically relevant 4T1 mouse model of spontaneous breast cancer metastasis [35]. The 4T1 mouse model comprises four syngenic tumor lines (67NR, 168FARN, 4T07, and 4T1) that have differential metastatic behavior: the 67NR cell line forms primary carcinomas when implanted into the mouse mammary fat pad, and no tumor cells are detected at distant tissue; the 168FARN cell line forms primary carcinomas with extensions to local lymph nodes; the 4T07 and 4T1 cell lines generate micrometastases and macroscopic metastases, respectively, in the lungs. By comparing the transcriptome at the exon level in primary tumors generated from each cell line, we identified 679 splicing variants that were differentially expressed in primary tumors with different metastatic abilities. Many of the splicing variations identified in the 4T1 model affected genes involved in cellular morphology and movement, which suggests that splicing may play a role in these cellular activities that are highly relevant to tumor progression. In addition, many splicing events identified in the primary tumors were conserved during evolution and were found in normal tissues, demonstrating that at least a large subset of splicing variations during tumor progression are not due to aberrant splicing. Importantly, some of the splicing variants identified in mouse tumors giving rise to metastases were linked with poor prognosis (shorter metastasis-free survival) in a large cohort of breast cancer patients. Using the primary tumors generated from the 168FARN, 4TO7, and 4T1 cell lines, Bemmo and collaborators also identified genes that are differentially spliced in association with the metastatic ability of tumors [36]. The misregulated genes were involved in cell growth and proliferation, cell death, cellular development, and cellular movement.
Lapuk and collaborators also identified 156 genes being differentially spliced when comparing 26 breast cancer cell lines representing the luminal, basal, and claudin-low subtypes of primary breast tumors, and five nonmalignant breast cell lines [34]. Functional annotation analyses of the regulated genes showed preferential enrichment of biological processes related to cytoskeleton and actin. In addition, this study revealed that many of these splicing events are regulated by the FOX2 (RBM9) splicing factor. This is of particular interest as Venables and collaborators also identified a large number of differentially spliced genes when comparing human breast and ovarian tumors to normal tissues that are under the control the FOX2 splicing factor that the authors showed to be downregulated in ovarian cancer and to be altered at the splicing level in breast cancer samples [33]. Interestingly, several studies suggest that FOX2 is a critical regulator of a splicing network. Indeed, by integrating binding specificity with phylogenetic conservation and splicing microarray data from 47 tissues and cell lines, Zhang and collaborators found thousands of FOX tissue-specific targets [50]. Yeo and collaborators also constructed an RNA map of FOX2-regulated alternative splicing via CLIP-seq in human embryonic stem cells and found a large cohort of targets [51]. Supporting further FOX2 function as a critical regulator of splicing networks, many cancer- and FOX-regulated splicing events reported by Venables and collaborators affected genes associated with actin filaments, myosin dynamics, kinesins, and microtubule binding and trafficking complexes [33]. Therefore, it appears that splicing alteration in breast cancer may have a marked effect on genes involved in cellular architecture, plasticity, and movement.
This observation is likely relevant to other types of cancers as a large number of differentially spliced genes when comparing lung, digestive tract, and brain tumors to normal tissues were associated with cell motility and organization of the actin cytoskeleton (Table 1). These observations suggest that genes involved in cellular architecture, plasticity, and movement are particularly prone to alternative splicing and/or that tumor progression results in or requires changes in the splicing patterns of genes involved in these cellular programs. It must be underlined that genes involved in other cellular programs, such as cell proliferation, are also often found to be differentially spliced in tumors (Table 1). Although further genome-wide analyses of alternative splicing are still required, an emerging concept is that cellular programs altered during tumor initiation and progression depend on splicing programs (Figure 3).
3. Cellular Programs Involve Splicing Programs Controlled by Splicing Factors
The first demonstration that splicing programs participate in cellular programs came from the study of the neuronal splicing factor, Nova-1, that was shown to regulate splicing events of a network of genes involved in synapse function [52, 53]. In the context of cancer-related cellular programs, the best example illustrating this concept was provided by a recent study of Warzecha and collaborators, who uncovered a network of alternative splicing changes that are under the control of the ESRP1 (RBM35A) and ESRP2 (RBM35B) splicing factors and that contribute to the epithelial-mesenchymal transition (EMT) [54]. Epithelial splicing regulatory proteins 1 and 2 (ESRP1 and ESRP2) have been identified to be regulators of the epithelial to mesenchymal splicing pattern of FGFR2, CD44, CTNND1 (p120-Catenin), and ENAH transcripts [55]. Warzecha and collaborators demonstrated that ESRPs are components of an epithelial gene signature and demonstrated a downregulation of these proteins in cells that undergo EMT. Knockdown of ESRPs results in loss of characteristic morphological features of epithelial cells with an increased motility and expression of invasive markers, concomitant with changes in expression of several prototypical EMT markers [54]. To know if ESRPs are master regulators of epithelial cell-specific splicing program, they analyzed splicing profiles derived from ectopic expression of ESRP1 in mesenchymal cells, as well as from knockdown of ESRPs in epithelial cells using splicing sensitive microarrays [54, 56]. They identified hundreds of alternative splicing events within numerous genes with functions in cell-cell adhesion, polarity, and migration, and many events showed reciprocal changes in the two experimental conditions. Components of this global ESRP-regulated epithelial splicing program could be valuable molecular markers to characterize the EMT and could have potential clinical applications, as cancer cells undergoing EMT present more aggressive tumor phenotypes.
Other cellular programs that may involve splicing programs are cell proliferation and apoptosis, two key cellular programs that are altered in cancer [14, 57]. Strikingly, most (if not all) genes involved in apoptosis, can produce splicing variants coding for protein isoforms with either pro- or antiapoptotic activities [16, 58]. Interestingly, an alternative splicing network that links cell cycle and apoptosis has recently been identified [59]. It must be emphasized that several cell cycle regulators, including CDC40 and CDC5L, have been identified as spliceosome components, and that several splicing factors, including SRSF1 (ASF/SF2, see below) affect cell cycle progression [60–62]. In addition, both splicing factors SRSF1 and SRSF2 (SC35) regulate the alternative splicing of several genes involved in apoptosis [59, 63–65]. The SRSF1 factor, a member of the arginine/serine-rich (SR) family of splicing factors, is particularly interesting as it is overexpressed in various human tumors [66, 67], and its overexpression is sufficient to transform immortalized cell lines [66]. Karni and collaborators showed that SRSF1 affects the alternative splicing of the tumor suppressor BIN1, producing an isoform lacking tumor suppressor activity, and of the protein kinases MNK2 and S6K1, leading to an MNK2 isoform promoting MAPK-independent eIF4E phosphorylation and an S6K1 isoform with oncogenic properties [66]. The oncogenic activity of SRSF1 may also be due to its implication in multiple cellular programs, as it regulates the alternative splicing of genes implicated in proliferation (e.g., CyclinD1), apoptosis (e.g., Bcl-x and Mcl1) and cell motility (e.g., Rac1 and Ron) [59, 68]. These studies provide evidence that an abnormally expressed splicing factor can have oncogenic properties by impacting on alternative splicing of cancer-associated genes and genes involved in cancer-related cellular programs.
Also interesting in this context is the Sam68 (Src associated in mitosis, of 68 kDa) factor that is a KH domain RNA-binding protein, whose expression and function have been linked to the onset and progression of tumors, such as prostate and breast carcinomas [69–73]. It was first thought that Sam68 had a tumor suppressor role [74, 75], but direct investigations rather suggest a pro-oncogenic role of the protein (for review [76]). Sam68 regulates splicing events in several genes involved in apoptosis and cell proliferation (e.g., Bcl-x, Cyclin D1, and CD44) [77–80]. Interestingly, Valacca and collaborators suggested that Sam68 could also contribute to the malignant transformation of epithelial cancers by regulating the alternative splicing of SRSF1 and inducing EMT [79].
In addition to the splicing factors mentioned above including FOX2, the polypyrimidine tract-binding protein (PTB/PTBP1), also known as hnRNP I is a splicing regulator that often acts as a repressor, although it can also promote the inclusion of exons [81, 82]. PTB has been shown to be overexpressed in ovarian cancer and in gliomas and may play a role in tumor initiation and progression [83–86]. PTB is expressed throughout development, and then down-regulated in many adult tissues [87]. In ovarian and glioma cell lines, loss of PTB inhibited cell proliferation and cell migration and increased cell adhesion [83, 85]. Cheung and collaborators showed that after removal of PTB in glioma cell lines where the PTB paralog nPTB/PTBP2 is also expressed, a single gene RTN4 had enhanced inclusion of exon 3 and this isoform decreased cell proliferation, migration, and adherence to a similar degree as the removal of PTB [83]. Importantly, it has also been shown recently that PTB, together with two others hnRNP family members, hnRNPA1 and hnRNPA2, controls the alternative splicing of transcripts of the PKM gene coding for the enzyme pyruvate kinase. The expression of these proteins favors switching from PKM1 to PKM2 isoform, which promotes aerobic glycolysis and thus will provide a selective advantage for tumor formation [84, 88]. Further studies will be required to assess the potential relevance of other PTB-regulated exons in the context of cancer.
4. Conclusions
Genome-wide analyses of alternative splicing allow us to propose a model whereby splicing programs, together with transcription programs participate in the corruption of cellular programs during tumor initiation and progression (Figure 3). It is important to underline that recent proteomic analyses confirm the presence of alternative protein isoforms in tumor samples [89]. Because of the diversity generated by alternative splicing and because of its highly dynamic regulation, it is likely that this process plays a central role in the phenotypic plasticity of tumor cells. Therefore, the identification of splicing programs that impact on key cellular programs will allow a better understanding of the genetic programs involved in cancer initiation and progression. This concept likely applies to other cancer-related cellular programs in addition to EMT, migration, proliferation, and death. For example, angiogenesis (blood vessel formation) favors tumor progression by improving tumor cell feed [14]. Several alternative splicing events induce a switch from pro- to antiangiogenic functions [90, 91]. Likewise, there is increasing evidence that primary metabolism is altered in tumor cells, and the pyruvate kinase M1 and M2 splicing isoforms control the balance between aerobic and anaerobic glycolysis during tumor progression [92, 93]. Supporting a model where cellular metabolism could depend on splicing programs, it has been recently shown that several key regulators of cholesterol biosynthesis and uptake are regulated by alternative splicing in a coordinated manner by the splicing factor PTB/hnRNP I [94].
A current challenge raised by large-scale analyses of alternative splicing is the functional analysis of large sets of splice variants that are identified as misregulated in cancer. Most studies so far have relied on the functional annotation of genes and on the analysis of functional features encoded by alternative exons (e.g., protein domains or premature stop codons). However, there is a need for midscale functional analyses of splice variants using experimental screens. Such an approach was used in a recent study, where 41 splice variants previously associated with breast and/or ovarian cancer were functionally analyzed by transfecting cells with either isoform-specific siRNAs or bifunctional-targeted oligonucleotides allowing to reprogram alternative splicing, followed by analysis of cell growth, viability, and apoptosis assays [95]. In this pioneering study, about 10% of the analyzed splice variants were found to play a role in cell viability in the growth conditions that were used. While it is possible that only a subset of cancer-associated splice variants play a role in cancer cell phenotypes, it is likely that unraveling the functions of cancer-associated splice variants will require using more varied growth conditions and phenotypic screens involving various cellular programs.
Understanding the mechanisms leading to splicing program alteration in cancer will require identification of the splicing factors that govern these splicing programs, knowing that many of them are misregulated in cancer. While several splicing factors have recently emerged as good candidates, further studies are needed to identify the whole sets of splicing events they regulate, their precise cellular functions or their potential alterations in cancer. Moreover, it is likely that many other splicing factors will be involved in oncogenesis and tumor progression.
Finally, the identification of the splicing factors controlling cancer-associated splicing programs will allow developments of new therapeutic targets [96, 97]. In this context, several strategies targeting splicing regulators are currently being developed. The recent identification of small molecules that can inhibit the activity of splicing factors will allow improved targeted cancer therapies [98, 99]. However, as splicing factors are RNA-binding proteins that are often involved in other aspects of RNA metabolism (e.g., mRNA stability and translation, or processsing of noncoding RNAs), it will be important to determine whether these different activities cooperate in the induction of a given cellular program or can be selectively targeted to induce distinct phenotypes.
Acknowledgments
The authors' sincere apologies to researchers whose articles could not be cited due to length constraints of this paper. The authors thank Mark Mc Carron for helpful reading of the paper. This work was supported by EC (NoE EURASNET), Institut National du Cancer, Association pour la Recherche sur le Cancer, Ligue Nationale Contre le Cancer, and Agence Nationale de la Recherche. S. Germann, was supported by Association pour la Recherche sur le Cancer; L. Gratadou by Agence Nationale de la Recherche.
References
- 1.Hallegger M, Llorian M, Smith CWJ. Alternative splicing: global insights: minireview. FEBS Journal. 2010;277(4):856–866. doi: 10.1111/j.1742-4658.2009.07521.x. [DOI] [PubMed] [Google Scholar]
- 2.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genetics. 2008;40(12):1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 3.Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126(1):37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 5.Castle JC, Zhang C, Shah JK, et al. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nature Genetics. 2008;40(12):1416–1425. doi: 10.1038/ng.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark TA, Schweitzer AC, Chen TX, et al. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biology. 2007;8(4):p. R64. doi: 10.1186/gb-2007-8-4-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Grange P, Gratadou L, Delord M, Dutertre M, Auboeuf D. Splicing factor and exon profiling across human tissues. Nucleic Acids Research. 2010;38(9):2825–2838. doi: 10.1093/nar/gkq008. Article ID gkq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwan T, Benovoy D, Dias C, et al. Genome-wide analysis of transcript isoform variation in humans. Nature Genetics. 2008;40(2):225–231. doi: 10.1038/ng.2007.57. [DOI] [PubMed] [Google Scholar]
- 9.Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods. 2009;6(5):377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 10.Barash Y, Calarco JA, Gao W, et al. Deciphering the splicing code. Nature. 2010;465(7294):53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 11.Bland CS, Wang ET, Vu A, et al. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Research. 2010;38(21):7651–7664. doi: 10.1093/nar/gkq614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson MB, Kawasawa YI, Mason CE, et al. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62(4):494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Kang Y, Massagué J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118(3):277–279. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 16.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes and Development. 2010;24(21):2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dutertre M, Vagner S, Auboeuf D. Alternative splicing and breast cancer. RNA Biology. 2010;7(4):403–411. doi: 10.4161/rna.7.4.12152. [DOI] [PubMed] [Google Scholar]
- 18.Fackenthal JD, Godley LA. Aberrant RNA splicing and its functional consequences in cancer cells. Disease Models and Mechanisms. 2008;1(1):37–42. doi: 10.1242/dmm.000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grosso AR, Martins S, Carmo-Fonseca M. The emerging role of splicing factors in cancer. EMBO Reports. 2008;9(11):1087–1093. doi: 10.1038/embor.2008.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim MY, Hur J, Jeong S. Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB Reports. 2009;42(3):125–130. doi: 10.5483/bmbrep.2009.42.3.125. [DOI] [PubMed] [Google Scholar]
- 21.Pettigrew CA, Brown MA. Pre-mRNA splicing aberrations and cancer. Frontiers in Bioscience. 2008;13(3):1090–1105. doi: 10.2741/2747. [DOI] [PubMed] [Google Scholar]
- 22.Skotheim RI, Nees M. Alternative splicing in cancer: noise, functional, or systematic? International Journal of Biochemistry and Cell Biology. 2007;39(7–8):1432–1449. doi: 10.1016/j.biocel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 23.Srebrow A, Kornblihtt AR. The connection between splicing and cancer. Journal of Cell Science. 2006;119, part 13:2635–2641. doi: 10.1242/jcs.03053. [DOI] [PubMed] [Google Scholar]
- 24.Venables JP. Unbalanced alternative splicing and its significance in cancer. BioEssays. 2006;28(4):378–386. doi: 10.1002/bies.20390. [DOI] [PubMed] [Google Scholar]
- 25.Hertel KJ. Combinatorial control of exon recognition. Journal of Biological Chemistry. 2008;283(3):1211–1215. doi: 10.1074/jbc.R700035200. [DOI] [PubMed] [Google Scholar]
- 26.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136(4):701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14(5):802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Körner M, Miller LJ. Alternative splicing of pre-mRNA in cancer: focus on G protein-coupled peptide hormone receptors. American Journal of Pathology. 2009;175(2):461–472. doi: 10.2353/ajpath.2009.081135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miura K, Fujibuchi W, Sasaki I. Alternative pre-mRNA splicing in digestive tract malignancy. Cancer Science. 2011;102(2):309–316. doi: 10.1111/j.1349-7006.2010.01797.x. [DOI] [PubMed] [Google Scholar]
- 30.Pio R, Montuenga LM. Alternative splicing in lung cancer. Journal of Thoracic Oncology. 2009;4(6):674–678. doi: 10.1097/JTO.0b013e3181a520dc. [DOI] [PubMed] [Google Scholar]
- 31.Rajan P, Elliott DJ, Robson CN, Leung HY. Alternative splicing and biological heterogeneity in prostate cancer. Nature Reviews Urology. 2009;6(8):454–460. doi: 10.1038/nrurol.2009.125. [DOI] [PubMed] [Google Scholar]
- 32.Ghigna C, Valacca C, Biamonti G. Alternative splicing and tumor progression. Current Genomics. 2008;9(8):556–570. doi: 10.2174/138920208786847971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Venables JP, Klinck R, Koh C, et al. Cancer-associated regulation of alternative splicing. Nature Structural and Molecular Biology. 2009;16(6):670–676. doi: 10.1038/nsmb.1608. [DOI] [PubMed] [Google Scholar]
- 34.Lapuk A, Marr H, Jakkula L, et al. Exon-level microarray analyses identify alternative splicing programs in breast cancer. Molecular Cancer Research. 2010;8(7):961–974. doi: 10.1158/1541-7786.MCR-09-0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dutertre M, Lacroix-Triki M, Driouch K, et al. Exon-based clustering of murine breast tumor transcriptomes reveals alternative exons whose expression is associated with metastasis. Cancer Research. 2010;70(3):896–905. doi: 10.1158/0008-5472.CAN-09-2703. [DOI] [PubMed] [Google Scholar]
- 36.Bemmo A, Dias C, Rose AAN, Russo C, Siegel P, Majewski J. Exon-Level transcriptome profiling in murine breast cancer reveals splicing changes specific to tumors with different metastatic abilities. PLoS ONE. 2010;5(8) doi: 10.1371/journal.pone.0011981. Article ID e11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.André F, Michiels S, Dessen P, et al. Exonic expression profiling of breast cancer and benign lesions: a retrospective analysis. The Lancet Oncology. 2009;10(4):381–390. doi: 10.1016/S1470-2045(09)70024-5. [DOI] [PubMed] [Google Scholar]
- 38.Langer W, Sohler F, Leder G, et al. Exon Array Analysis using re-defined probe sets results in reliable identification of alternatively spliced genes in non-small cell lung cancer. BMC Genomics. 2010;11(1):p. 676. doi: 10.1186/1471-2164-11-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xi L, Feber A, Gupta V, et al. Whole genome exon arrays identify differential expression of alternatively spliced, cancer-related genes in lung cancer. Nucleic Acids Research. 2008;36(20):6535–6547. doi: 10.1093/nar/gkn697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pio R, Blanco D, Pajares MJ, et al. Development of a novel splice array platform and its application in the identification of alternative splice variants in lung cancer. BMC Genomics. 2010;11(1):p. 352. doi: 10.1186/1471-2164-11-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misquitta-Ali CM, Cheng E, O'Hanlon D, et al. Global profiling and molecular characterization of alternative splicing events misregulated in lung cancer. Molecular and Cellular Biology. 2011;31(1):138–150. doi: 10.1128/MCB.00709-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gardina PJ, Clark TA, Shimada B, et al. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics. 2006;7(325) doi: 10.1186/1471-2164-7-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thorsen K, Sørensen KD, Brems-Eskildsen A, et al. Alternative splicing in colon, bladder, and prostate cancer identified by exon array analysis. Molecular and Cellular Proteomics. 2008;7(7):1214–1224. doi: 10.1074/mcp.M700590-MCP200. [DOI] [PubMed] [Google Scholar]
- 44.Mojica W, Hawthorn L. Normal colon epithelium: a dataset for the analysis of gene expression and alternative splicing events in colon disease. BMC Genomics. 2010;11(1):p. 5. doi: 10.1186/1471-2164-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sveen A, Ågesen TH, Nesbakken A, Rognum TO, Lothe RA, Skotheim RI. Transcriptome instability in colorectal cancer identified by exon microarray analyses: associations with splicing factor expression levels and patient survival. Genome Medicine. 2011;3(5):p. 32. doi: 10.1186/gm248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Menghi F, Jacques TS, Barenco M, et al. Genome-wide analysis of alternative splicing in medulloblastoma identifies splicing patterns characteristic of normal cerebellar development. Cancer Research. 2011;71(6):2045–2055. doi: 10.1158/0008-5472.CAN-10-2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo X, Chen QR, Song YK, Wei JS, Khan J. Exon array analysis reveals neuroblastoma tumors have distinct alternative splicing patterns according to stage and MYCN amplification status. BMC Medical Genomics. 2011;4(35) doi: 10.1186/1755-8794-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheung HC, Baggerly KA, Tsavachidis S, et al. Global analysis of aberrant pre-mRNA splicing in glioblastoma using exon expression arrays. BMC Genomics. 2008;9(216) doi: 10.1186/1471-2164-9-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.French PJ, Peeters J, Horsman S, et al. Identification of differentially regulated splice variants and novel exons in glial brain tumors using exon expression arrays. Cancer Research. 2007;67(12):5635–5642. doi: 10.1158/0008-5472.CAN-06-2869. [DOI] [PubMed] [Google Scholar]
- 50.Zhang C, Zhang Z, Castle J, et al. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes and Development. 2008;22(18):2550–2563. doi: 10.1101/gad.1703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nature Structural and Molecular Biology. 2009;16(2):130–137. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ule J, Ule A, Spencer J, et al. Nova regulates brain-specific splicing to shape the synapse. Nature Genetics. 2005;37(8):844–852. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- 53.Zhang C, Frias MA, Mele A, et al. Integrative modeling defines the nova splicing-regulatory network and its combinatorial controls. Science. 2010;329(5990):439–443. doi: 10.1126/science.1191150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warzecha CC, Jiang P, Amirikian K, et al. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO Journal. 2010;29(19):3286–3300. doi: 10.1038/emboj.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Molecular Cell. 2009;33(5):591–601. doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warzecha CC, Shen S, Xing Y, Carstens RP. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biology. 2009;6(5):546–562. doi: 10.4161/rna.6.5.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 58.Schwerk C, Schulze-Osthoff K. Regulation of apoptosis by alternative pre-mRNA splicing. Molecular Cell. 2005;19(1):1–13. doi: 10.1016/j.molcel.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 59.Moore MJ, Wang Q, Kennedy CJ, Silver PA. An alternative splicing network links cell-cycle control to apoptosis. Cell. 2010;142(4):625–636. doi: 10.1016/j.cell.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boger-Nadjar E, Vaisman N, Ben-Yehuda S, Kassir Y, Kupiec M. Efficient initiation of S-phase in yeast requires Cdc40p, a protein involved in pre-mRNA splicing. Molecular and General Genetics. 1998;260(2-3):232–241. doi: 10.1007/s004380050891. [DOI] [PubMed] [Google Scholar]
- 61.Li X, Wang J, Manley JL. Loss of splicing factor ASF/SF2 induces G2 cell cycle arrest and apoptosis, but inhibits internucleosomal DNA fragmentation. Genes and Development. 2005;19(22):2705–2714. doi: 10.1101/gad.1359305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang N, Kaur R, Akhter S, Legerski RJ. Cdc5L interacts with ATR and is required for the S-phase cell-cycle checkpoint. EMBO Reports. 2009;10(9):1029–1035. doi: 10.1038/embor.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Merdzhanova G, Edmond V, De Seranno S, et al. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death and Differentiation. 2008;15(12):1815–1823. doi: 10.1038/cdd.2008.135. [DOI] [PubMed] [Google Scholar]
- 64.Merdzhanova G, Gout S, Keramidas M, et al. The transcription factor E2F1 and the SR protein SC35 control the ratio of pro-angiogenic versus antiangiogenic isoforms of vascular endothelial growth factor-A to inhibit neovascularization in vivo. Oncogene. 2010;29(39):5392–5403. doi: 10.1038/onc.2010.281. [DOI] [PubMed] [Google Scholar]
- 65.Edmond V, Moysan E, Khochbin S, et al. Acetylation and phosphorylation of SRSF2 control cell fate decision in response to cisplatin. EMBO Journal. 2011;30(3):510–523. doi: 10.1038/emboj.2010.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nature Structural and Molecular Biology. 2007;14(3):185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Piekielko-Witkowska A, Wiszomirska H, Wojcicka A, et al. Disturbed expression of splicing factors in renal cancer affects alternative splicing of apoptosis regulators, oncogenes, and tumor suppressors. PLoS ONE. 2010;5(10) doi: 10.1371/journal.pone.0013690. Article ID e13690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ghigna C, Giordano S, Shen H, et al. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Molecular Cell. 2005;20(6):881–890. doi: 10.1016/j.molcel.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 69.Babic I, Jakymiw A, Fujita DJ. The RNA binding protein Sam68 is acetylated in tumor cell lines, and its acetylation correlates with enhanced RNA binding activity. Oncogene. 2004;23(21):3781–3789. doi: 10.1038/sj.onc.1207484. [DOI] [PubMed] [Google Scholar]
- 70.Busà R, Paronetto MP, Farini D, et al. The RNA-binding protein Sam68 contributes to proliferation and survival of human prostate cancer cells. Oncogene. 2007;26(30):4372–4382. doi: 10.1038/sj.onc.1210224. [DOI] [PubMed] [Google Scholar]
- 71.Lukong KE, Larocque D, Tyner AL, Richard S. Tyrosine phosphorylation of Sam68 by breast tumor kinase regulates intranuclear localization and cell cycle progression. Journal of Biological Chemistry. 2005;280(46):38639–38647. doi: 10.1074/jbc.M505802200. [DOI] [PubMed] [Google Scholar]
- 72.Paronetto MP, Farini D, Sammarco I, et al. Expression of a truncated form of the c-kit tyrosine kinase receptor and activation of Src kinase in human prostatic cancer. American Journal of Pathology. 2004;164(4):1243–1251. doi: 10.1016/S0002-9440(10)63212-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rajan P, Gaughan L, Dalgliesh C, et al. Regulation of gene expression by the RNA-binding protein Sam68 in cancer. Biochemical Society Transactions. 2008;36(3):505–507. doi: 10.1042/BST0360505. [DOI] [PubMed] [Google Scholar]
- 74.Liu K, Li L, Nisson PE, Gruber C, Jessee J, Cohen SN. Neoplastic transformation and tumorigenesis associated with Sam68 protein deficiency in cultured murine fibroblasts. Journal of Biological Chemistry. 2000;275(51):40195–40201. doi: 10.1074/jbc.M006194200. [DOI] [PubMed] [Google Scholar]
- 75.Taylor SJ, Resnick RJ, Shalloway D. Sam68 exerts separable effects on cell cycle progression and apoptosis. BMC Cell Biology. 2004;5(5) doi: 10.1186/1471-2121-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bielli P, Busa R, Paronetto MP, Sette C. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocrine-Related Cancer. 2011;18(4):R91–R102. doi: 10.1530/ERC-11-0041. [DOI] [PubMed] [Google Scholar]
- 77.Matter N, Herrlich P, König H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature. 2002;420(6916):691–695. doi: 10.1038/nature01153. [DOI] [PubMed] [Google Scholar]
- 78.Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. Journal of Cell Biology. 2007;176(7):929–939. doi: 10.1083/jcb.200701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Valacca C, Bonomi S, Buratti E, et al. Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. Journal of Cell Biology. 2010;191(1):87–99. doi: 10.1083/jcb.201001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paronetto MP, Cappellari M, Busà R, et al. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Research. 2010;70(1):229–239. doi: 10.1158/0008-5472.CAN-09-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Llorian M, Schwartz S, Clark TA, et al. Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nature Structural and Molecular Biology. 2010;17(9):1114–1123. doi: 10.1038/nsmb.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xue Y, Zhou Y, Wu T, et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Molecular Cell. 2009;36(6):996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cheung HC, Hai T, Zhu W, et al. Splicing factors PTBP1 and PTBP2 promote proliferation and migration of glioma cell lines. Brain. 2009;132(8):2277–2288. doi: 10.1093/brain/awp153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463(7279):364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.He X, Pool M, Darcy KM, et al. Knockdown of polypyrimidine tract-binding protein suppresses ovarian tumor cell growth and invasiveness in vitro. Oncogene. 2007;26(34):4961–4968. doi: 10.1038/sj.onc.1210307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jin W, Bruno IG, Xie TX, Sanger LJ, Cote GJ. Polypyrimidine tract-binding protein down-regulates fibroblast growth factor receptor 1 α-exon inclusion. Cancer Research. 2003;63(19):6154–6157. [PubMed] [Google Scholar]
- 87.Boutz PL, Stoilov P, Li Q, et al. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes and Development. 2007;21(13):1636–1652. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Clower CV, Chatterjee D, Wang Z, Cantley LC, Heidena MGV, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(5):1894–1899. doi: 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hatakeyama K, Ohshima K, Fukuda Y, et al. Identification of a novel protein isoform derived from cancer-related splicing variants using combined analysis of transcriptome and proteome. Proteomics. 2011;11(11):2275–2282. doi: 10.1002/pmic.201100016. [DOI] [PubMed] [Google Scholar]
- 90.Bates DO, Cui TG, Doughty JM, et al. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Research. 2002;62(14):4123–4131. [PubMed] [Google Scholar]
- 91.Nowak DG, Woolard J, Amin EM, et al. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. Journal of Cell Science. 2008;121(20):3487–3495. doi: 10.1242/jcs.016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 93.Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. International Journal of Biochemistry and Cell Biology. 2011;43(7):969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 94.Medina MW, Gao F, Naidoo D, et al. Coordinately regulated alternative splicing of genes involved in cholesterol biosynthesis and uptake. PLoS ONE. 2011;6(4, article e19420) doi: 10.1371/journal.pone.0019420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Prinos P, Garneau D, Lucier JF, et al. Alternative splicing of SYK regulates mitosis and cell survival. Nature Structural and Molecular Biology. 2011;18(6):673–679. doi: 10.1038/nsmb.2040. [DOI] [PubMed] [Google Scholar]
- 96.Pajares MJ, Ezponda T, Catena R, Calvo A, Pio R, Montuenga LM. Alternative splicing: an emerging topic in molecular and clinical oncology. The Lancet Oncology. 2007;8(4):349–357. doi: 10.1016/S1470-2045(07)70104-3. [DOI] [PubMed] [Google Scholar]
- 97.Ward AJ, Cooper TA. The pathobiology of splicing. Journal of Pathology. 2010;220(2):152–163. doi: 10.1002/path.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ghigna C, De Toledo M, Bonomi S, et al. Pro-metastatic splicing of Ron proto-oncogene mRNA can be reversed: therapeutic potential of bifunctional oligonucleotides and indole derivatives. RNA Biology. 2010;7(4):495–503. doi: 10.4161/rna.7.4.12744. [DOI] [PubMed] [Google Scholar]
- 99.Tazi J, Bakkour N, Stamm S. Alternative splicing and disease. Biochimica et Biophysica Acta. 2009;1792(1):14–26. doi: 10.1016/j.bbadis.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]