

Abstract
Accumulation of unfolded proteins in the endoplasmic reticulum (ER) results in ER stress, which subsequently activates the unfolded protein response that induces a transcriptional program to alleviate the stress. Another cellular process that is activated during ER stress is autophagy, a mechanism of enclosing intracellular components in a double-membrane autophagosome, and then delivering it to the lysosome for degradation. Here, we discuss the role of autophagy in cellular response to ER stress, the signaling pathways linking ER stress to autophagy, and the possible implication of modulating autophagy in treatment of diseases such as cancer.
Keywords: Endoplasmic reticulum stress, Autophagy, Apoptosis, Cell survival, Cell death
INTRODUCTION
The endoplasmic reticulum (ER) is an organelle that has essential roles in multiple cellular processes, including intracellular calcium homeostasis, protein secretion and lipid biosynthesis; all of which are required for cell survival and normal cellular functions. Normal ER functions are required for correct folding of newly synthesized proteins and their post-translational modifications, such as glycosylation and disulfide bond formation[1]. ER stress occurs in response to a variety of stimuli, various physiological and pathological conditions that can cause the accumulation of unfolded and misfolded proteins in the ER. Consequently, unfolded protein response (UPR) is triggered to resolve the ensuing stress by activating intracellular signal transduction pathways. In eukaryotic cells, UPR is mediated by three ER membrane-associated proteins, namely, inositol requiring enzyme (IRE)1α, PKR-like eukaryotic initiation factor (eIF)2α kinase (PERK), and activating transcription factor (ATF)6. These ER membrane-associated proteins are inhibited under basal conditions by their association with the chaperone protein Grp78/Bip, but are activated when released from Grp78 during ER stress[2]. These proteins induce signal-transduction events that can alleviate the accumulation of misfolded proteins in the ER by enhancing the protein folding capacity of the ER, by inhibiting new protein synthesis, or by accelerating the degradation of proteins. However, if the function of ER cannot be re-established, extensive or sustained ER stress will eventually induce cell death through activating apoptosis. The phosphorylation of eIF2α at Ser51 by PERK during ER stress downregulates efficient translation of most mRNAs, thereby inhibiting protein synthesis. Under these stressful conditions, only selected mRNAs such as ATF4, are translated. ATF4 induces expression of genes involved in restoring ER homeostasis[1,3]. ATF6 is transported to the Golgi in response to ER stress, where it is cleaved by Golgi-resident proteases S1P (site 1 protease) and S2P (site 2 protease). The cleaved ATF6 N-terminal fragment migrates to the nucleus to activate the transcription of UPR target genes[4]. Under conditions of ER stress, IRE1 processes X-Box binding protein (XBP)1 mRNA to generate mature XBP1 mRNA. Spliced XBP1 mRNA encodes a transcription activator that drives transcription of genes such as ER chaperones, whose products directly participate in ER protein folding. XBP1 also regulates a subset of UPR genes that promotes ER-associated degradation of misfolded proteins and ER biogenesis[5]. In addition to activation of the UPR by the pathways mentioned above, ER-stress leads to a release of Ca2+ from the ER into the cytosol, which, in turn, can activate signaling pathways involved in apoptosis and autophagy.
Autophagy is a lysosomal pathway responsible for the degradation of long-lived proteins, cellular macromolecules and subcellular organelles. Autophagy process involves the formation of double-membrane autophagic vacuoles, known as autophagosomes, which transport cytoplasmic cargo to the lysosome for degradation. Autophagy is induced during starvation in both yeast and higher eukaryotes as a way of breaking down macromolecules to recycle their components[6,7]. Autophagy is also involved in removing damaged or excess organelles. Autophagic activity is controlled by a set of evolutionarily conserved autophagy-related proteins (Atg proteins). The initial nucleation and assembly of the primary autophagosomal membrane requires a kinase complex that consists of class III phosphatidylinositol 3-kinase (PI3K), p150 myristylated protein kinase, and beclin 1. Further elongation of the isolation membrane is mediated by two ubiquitin-like conjugation systems, Atg12-5 and microtubule-associated protein 1A/1B-light chain3 (LC3) systems. Atg12 is activated by Atg7 and transferred to Atg10, and is finally conjugated to Atg5, forming the irreversible Atg12-Atg5 conjugate[8]. The conversion of LC3 results from the free form (LC3-I), which is transformed to a lipid-conjugated membrane-bound form (LC3-II). Accumulation of LC3-II and its localization to vesicular structures are commonly used as markers of autophagy.
Baseline levels of autophagy contribute to maintenance of cellular homeostasis through elimination of old or damaged organelles, as well as the turnover of long-lived proteins. Autophagy is frequently activated in response to adverse stress, and has been shown to be involved in many physiological and pathological processes. In starvation conditions, enhanced autophagy provides stressed cells with metabolic intermediates to meet their bioenergetic demands[9]. Moreover, autophagy can be dramatically augmented as a protective and survival mechanism in response to numerous conditions of extracellular or intracellular stress, including hypoxia, radiotherapy and chemotherapy[10]. On the other hand, autophagy does not always promote cell survival; it can be a mechanism of cell death under certain circumstances. For example, under experimental conditions in which apoptotic pathways are blocked, or in response to treatments that specifically trigger caspase-independent autophagy, autophagy can play a pro-death role, causing autophagic cell death[11-13].
Increasing evidence has indicated that ER stress is also a potent trigger of autophagy; another mechanism for removing unfolded proteins that cannot be eliminated by ubiquitin/proteasome system, thus mitigating ER stress and protecting against cell death. It has been reported that ER stress leads to upregulation of the transcription of genes related to autophagy induction, including ATG8, ATG14 and Vacuolar hydrolases aminopeptidase1 (APE1)[14]. In mammalian cells, ER stress has been shown to facilitate the formation of autophagosomes, and induction of autophagy allows removal of toxic misfolded proteins to favor survival of the stressed cells[15-17]. Another function of autophagy during ER stress is degradation of the damaged ER itself. However, autophagy induced by the same chemicals may not confer protection in normal non-transformed cells. For example, the autophagy induced by chemicals, such as A23187, tunicamycin, thapsigargin and brefeldin A protects against cell death in colon and prostate cancer cells, but contributes to cell death in normal cells[18]. ER-stress-induced autophagy is important for clearing polyubiquitinated protein aggregates and for reducing cellular vacuolization in HCT116 colon cancer cells and DU145 prostate cancer cells, thus mitigating ER stress and protecting against cell death. In contrast, autophagy induced by the same chemicals does not confer protection in a normal human colon cell line and in the non-transformed murine embryonic fibroblasts (MEFs) but rather contributes to cell death. Thus, the impact of autophagy on cell survival during ER stress is probably contingent on the status of the cells, which could be explored for tumor-specific therapy.
There is also evidence that autophagy is invoked as a means of killing cells when ER stress is implacable[18,19]. The signaling pathways responsible for autophagy induction and its cellular consequences appear to vary with cell types and the stimuli. A better understanding of the signaling pathways controlling autophagy and cellular fate in response to ER stress will hopefully open new possibilities for the treatment of the numerous diseases associated with ER stress.
CYTOPROTECTIVE AUTOPHAGY INDUCED BY ER STRESS
Initiation of autophagy has been shown to exert protective effects in yeast and mammalian cells in response to ER stress. When the amount of unfolded or misfolded proteins exceeds the capacity of the proteasome-mediated degradation system, autophagy is triggered to remove these proteins. The observations in yeast show that ER-stress-induced autophagy counterbalances ER expansion, removes aggregated proteins from the ER, and plays a cytoprotective role in the case of intense and persistent stress[20,21]. Similarly, autophagy can also act as an ER-associated degradation system in mammalian cells, and it plays a fundamental role in preventing toxic accumulation of disease-associated mutant proteins in the ER. A mutant form of a type-II transmembrane protein dysferlin, a causative agent of human muscle dystrophy, has recently been shown to accumulate and form aggregates in the ER and eventually lead to apoptotic cell death. Inhibition of functional autophagy in Atg5-deficient MEFs further stimulates the aggregation of mutant dysferlin, whereas enhanced autophagy in the rapamycin (mTOR inhibitor)-treated cells reduces accumulation of the mutant protein in the ER[22]. Likewise, ER aggregates of mutant α1-antitrypsin Z, which is associated with the development of chronic liver injury and hepatocellular carcinoma, induce autophagy-mediated removal of the aggregated proteins[16]. These studies did not directly assess the effect of autophagy on cell survival, but as the protein aggregates in the ER are the probable cause of cell death, autophagy capable of degrading them is envisaged to be cytoprotective. Similarly, experimental models for diseases caused by protein aggregates in the cytosol suggest that ER-stress-induced autophagy enhances removal of aggregates and enhances cell survival[23]. In addition, it has been shown that ER itself is the major autophagosomal cargo during ER stress, which suggests that the pro-survival effect of autophagy in this model system could be due to increased removal of unfolded proteins[20].
Autophagy can be protective against ER stress in several circumstances including cancer progression. Autophagy protects colon and prostate cells from ER stress and cell death induced by A23187, tunicamycin, thapsigargin and brefeldin A[18]. Treatment of neuroblastoma SK-N-SH cells with ER stressors, tunicamycin and thapsigargin, induces formation of autophagosomes, and Atg5-deficient cells and 3-methyladenine (3-MA)-treated cells demonstrate increased vulnerability to ER stress, as well as more rapid activation of cascape-3, as compared with the non-transfected and non-treated cells when subjected to ER stress[24]. Upon exposure of HeLa cells to HIV-1 Tat-induced autophagy, suppression of autophagy by 3-MA or knockdown of Atg5 significantly increases cell death, indicating that autophagy protects against cell death during ER stress[25]. These results indicate that autophagy plays pivotal roles in protecting against cell death induced by ER stress.
AUTOPHAGY AS A CELL DEATH MECHANISM IN APOPTOSIS-DEFICIENT CELLS DURING ER STRESS
ER stress can cause necrotic cell death in bak-/-bax-/- cells that are defective in apoptosis, and ER-stress-induced necrosis has been known to be associated with autophagy[19,26]. Autophagy can be induced to similar levels in the wild-type and bak-/-bax-/- cells in response to ER stress, but the resulting outcome of this response appears to be different. Inhibition of autophagy by 3-MA or by silencing of Atg5 using shRNA significantly enhances the viability of bak-/-bax-/- cells when ER stress is present, indicating that autophagy can enhance cell death in the bak-/-bax-/- cells. In contrast, 3-MA enhances ER-stress-induced cell death in apoptosis-competent wild-type cells[19]. These findings suggest that autophagy may have opposite effects in determining cell fate in response to ER stress in apoptosis-competent cells in which autophagy serves as a survival mechanism, and in apoptosis-deficient cells that utilize autophagy as a means to promote non-apoptotic cell death. This may be because, in the ER-stressed cells in which excessive stress fails to induce apoptosis, the stress status keeps escalating to a point where autophagy is massively induced, leading to subsequent cellular damage and necrosis. Therefore, autophagy may act as a death mechanism that substitutes for deficient apoptosis under ER stress via crosstalk with necrosis.
Autophagy can play differential role in cancer and non-transformed cells. For instance, disturbing ER homeostasis and/or functions by certain chemicals can elicit autophagy in primary colon cells, and suppression of autophagy by 3-MA reduces cell death. Nevertheless, 3-MA or depletion of beclin1 in colon carcinoma cells sensitizes tumor cells to the same treatments. In addition, suppression of autophagy induced by the same chemicals in the immortalized but non-transformed MEFs by deletion of Atg5 also reduces cell death, indicating that non-transformed cells may be especially sensitive to ER-stress-induced autophagy[18]. These observations suggest that autophagy can contribute to ER-stress-induced cell death in different scenarios, which may be dependent on cellular status under given stimulation. The differential role of autophagy in promoting survival of cancer cells or death of non-transformed cells might be related to the level at which ER stress is compensated.
SIGNALING PATHWAYS INVOLVED IN AUTOPHAGY DURING ER STRESS
Although autophagy is known to be associated with ER stress, the precise molecular mechanisms by which autophagy is activated under ER stress is not yet fully elucidated. PERK, IRE1 and increased [Ca2+]i have been implicated as mediators of ER-stress-induced autophagy in mammalian cells, as depicted in Figure 1.
Figure 1.
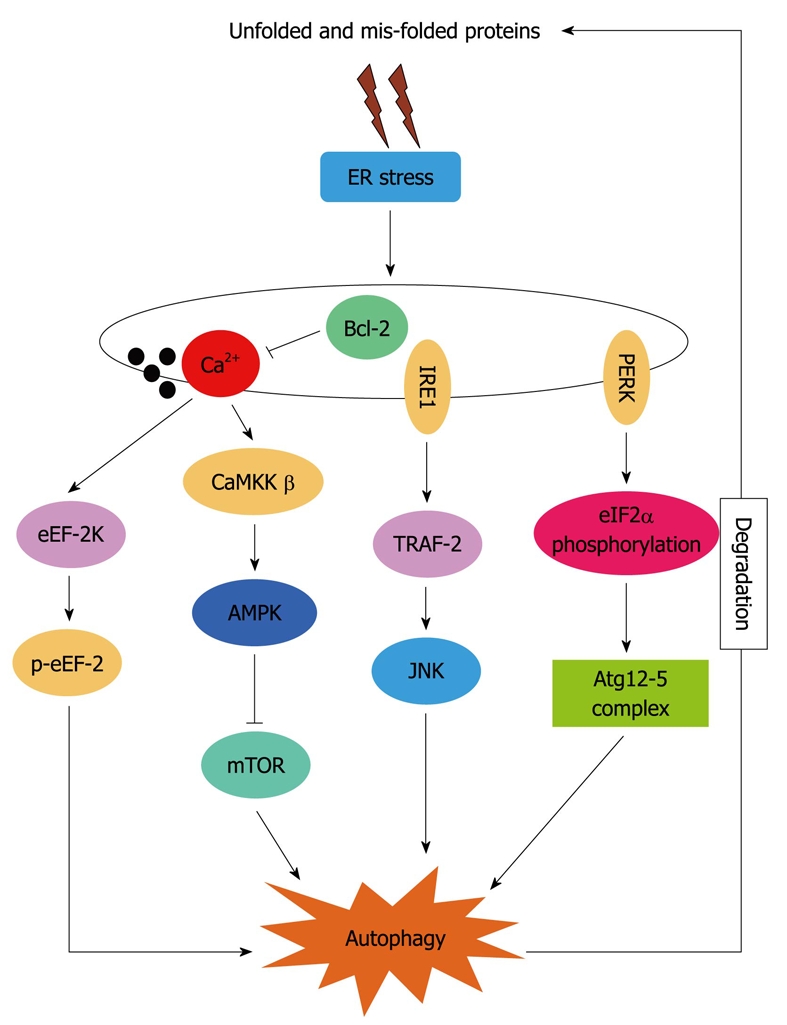
Hypothetical signaling pathways involved in endoplasmic reticulum stress-induced autophagy. ER: Endoplasmic reticulum; Bcl-2: B-cell lymphoma/leukemia 2; IRE1: Inositol requiring enzyme 1; eEF: Eukaryotic elongation factor; eIF: Eukaryotic initiation factor; JNK: c-Jun N-terminal kinase.
eIF2α is phosphorylated in response to various stresses, including starvation, viral infection and ER stress. The relationship between autophagy and eIF2α phosphorylation has been shown during starvation-induced autophagy in Saccharomyces cerevisiae and during starvation- and virus-infection-induced autophagy in mammalian cells[27]. Thus, it is possible that various stressful conditions that activate eIF2α kinases, including ER stress, may have an ability to induce autophagy in mammalian cells. Consistent with this hypothesis, the PERK-eIF2α signaling pathway has been reported to link ER stress to autophagy. A novel mutant form of a type-II transmembrane protein dysferlin aggregates and accumulates in the ER and induces eIF2α phosphorylation and LC3 conversion. Inhibition of autophagy by depletion of Atg5 inhibits degradation of mutant dysferlin. Furthermore, dephosphorylation of eIF2α also stimulates aggregation of mutant dysferlin in the ER, suggesting that ER-stress-induced eIF2α phosphorylation may regulate autolysosome formation. Rapamycin, which induces eIF2α phosphorylation-mediated LC3 conversion, inhibits mutant dysferlin aggregation in the ER[22]. These results indicate that mutant dysferlin aggregated on the ER membrane stimulates autophagosome formation via activating ER-stress-induced eIF2α phosphorylation.
Kouroku et al[23] have reported that ER stress caused by ectopic expression of polyQ72 upregulates Atg12 expression and induces autophagy, as demonstrated by an increase in conversion of LC3-I to LC3-II and an increase in LC3-postive vesicles in mouse embryonic carcinoma cells and MEFs. The polyQ72-induced LC3 conversion is inhibited in cells containing the eIF2α A/A mutation and dominant negative-PERK, strongly suggesting that the PERK/eIF2α pathway, an ER stress response signal, plays an essential role in polyQ72-induced Atg12 upregulation and LC3 conversion. However, the molecular mechanism by which eIF2α phosphorylation regulates LC3 conversion remains unclear. Atg12, a component of Atg5-Atg12-Atg16 complex, as well as CHOP mRNA, are selectively upregulated by polyQ72 via eIF2α phosphorylation. Thus, one possible explanation is that the eIF2α phosphorylation-dependent selective translation of transcription factors increases the expression of Atg12, resulting in the formation of Atg5-Atg12-Atg16 complex, followed by conversion of LC3-I to LC3-II[23].
Contradictory to the above, some studies have shown that IRE1 is crucial for autophagosome formation and LC3-II conversion after treatment with ER stressors. Imaizumi and co-workers have suggested that IRE1, rather than PERK, links UPR to autophagy[24]. Using MEFs deficient in IRE1α or ATF6 and embryonic stem cells deficient in PERK, they have demonstrated that accumulation of LC3-positive vesicles triggered by tunicamycin or thapsigargin fully depends on IRE1, but not PERK or ATF6. Thapsigargin-induced accumulation of LC3-positive vesicles is also completely inhibited in MEFs deficient in tumor necrosis factor receptor-associated factor (TRAF)-2, a cytosolic adaptor molecule that links active IRE1 to the activation of c-Jun N-terminal kinase (JNK). Additionally, a pharmacological inhibitor of JNK, SP600125, effectively inhibits the LC3 translocation in this model system, suggesting that IRE1-TRAF2-JNK pathway is essential for induction of autophagy in MEFs challenged with ER stressors. Yorimitsu et al[28] have reported that the Ire1-Hac1 signaling pathway is required for induction of autophagy. They have examined autophagy under ER stress conditions in the absence of Ire1 or Hac1, and have found that, in both ire1D(-/-) and hac1D(-/-) cells, ER-stress-induced autophagy was blocked. Starvation-induced autophagy was not affected in these cells. These observations suggest that under ER stress, the Ire-Hac1 signaling pathway is involved in autophagy induction; probably through the UPR.
The release of Ca2+ can activate various kinases and proteases that are possibly involved in the autophagy pathway. Thapsigargin increases [Ca2+]C and induces autophagy, as measured by LC3 translocation, electron microscopy and degradation rate of long-lived proteins, and this is effectively inhibited by Ca2+ chelators[29]. The same study has further demonstrated that Ca2+-mediated autophagy is dependent on the calmodulin-dependent protein kinase kinase-β/AMP-activated protein kinase pathway that ultimately leads to the inhibition of mTORC1, as demonstrated by decreased phosphorylation of the mTORC1 substrate p70S6K1.
Eukaryotic elongation factor (eEF)-2 is a 93-kDa monomeric guanine nucleotide-binding protein and is an essential mediator of the ribosomal elongation step during mRNA translation. eEF-2 promotes the GTP-dependent translocation of the nascent protein chain from the A-site to the P-site of the ribosome, and is an essential regulatory factor for protein synthesis. The phosphorylation of eEF-2 on Thr56 by eEF-2 kinase is known to inhibit its translational function, by reducing its affinity for ribosomes[30]. It is known that eIF2α phosphorylation is required for phosphorylation of eEF-2 during nutrient starvation. eEF-2K is also required for activation of autophagy caused by various stresses, including ER stress[31], nutrient depletion[32], and Akt inhibition[33], suggesting that phosphorylation of eEF-2 serves as an integrator of various cell stresses for autophagy signaling. However, PERK and the phosphorylation of eIF2α are dispensable for eEF-2 phosphorylation during ER stress, indicating that eEF-2 phosphorylation can be triggered by multiple signaling pathways, including the PERK/eIF2α pathway. The phosphorylation of eEF-2 by tunicamycin or thapsigargin treatment is significantly inhibited in the presence of the Ca2+ chelator BAPTA-AM, indicating that activation of eEF-2 kinase relies on Ca2+ flux during ER stress. Thus, phosphorylation of eEF-2 may be a common mediator of autophagy during starvation or ER stress. These results suggest that eEF-2 kinase plays an important regulatory role in mediating autophagy in response to multiple stress stimuli, and can be activated in an eIF2α-dependent or -independent manner.
B-cell lymphoma/leukemia 2 (Bcl-2) is an anti-apoptotic protein located at mitochondrial, ER and nuclear membranes, and to a lesser extent in the cytoplasm. Accumulating evidence suggests that Bcl-2 can inhibit or activate autophagy, depending on different model systems. The opposite effects of Bcl-2 on autophagy may be attributed to its post-translational modifications or different subcellular localizations. Inhibition of autophagy by Bcl-2 is shown by the fact that it blocks autophagosome accumulation induced by starvation, vitamin D analog EB1089, ATP and Xestospogin B[18,29,34]. At least two mechanisms have been proposed for Bcl-2-mediated inhibition of autophagy: a direct interaction with beclin 1; and regulation of ER Ca2+ stores, possibly via its binding to IP3R[29,34]. Beclin 1 is a Bcl-2-interacting protein that promotes autophagosome formation when in complex with class III PI3K and p150 myristylated kinase. Bcl-2 has been suggested to function as an autophagy brake by inhibiting the formation of this autophagy-promoting protein complex. ER-localized Bcl-2 lowers the steady-state level of Ca2+ in the ER and thereby reduces stimulus-induced Ca2+ fluxes from the ER. Thus, it may inhibit Ca2+-dependent autophagy by reducing the increase in [Ca2+]C. This hypothesis is supported by data showing that ER-localized Bcl-2 effectively inhibits autophagy induced by Ca2+ mobilizing agents that depend on ER Ca2+ stores (EB1089and ATP)[35]. Bcl-2 at the ER may depend on beclin1 binding to decrease the amount of Ca2+ released from the ER following agonist stimulation. Alternatively, ER-targeted Bcl-2 may be able to inhibit autophagy by other means, depending on the signaling pathway involved in autophagy induction.
CONCLUSION
Autophagy is important for the clearance of unfolded/misfolded proteins and for relief of ER stress induced by various stresses. The current studies, as discussed above, encourage the development of autophagy-promoting therapies for diseases associated with protein aggregates in the ER or cytosol. It is known that activation of ER stress and autophagy is associated with dealing with amyloid β-peptide accumulation in the brain; the major cause of Alzheimer’s disease. In cancer cells, autophagy helps to alleviate ER stress, and inhibits cell death. If this proves to be the case, combination therapies with ER stressors and autophagy inhibitors may also be useful in cancer therapy. The direct link between ER stress and autophagy has been reported for less than 1 year. Thus, many burning questions concerning the signaling pathways linking ER stress to autophagy, the mechanisms by which ER is selected as autophagic cargo, the crosstalk between ER-stress-induced autophagy and cell death pathways (apoptosis and necrosis), and the impact of autophagy in diseases associated with ER stress, remain largely unanswered. Future research will hopefully clarify these issues and pave the way for pharmacological exploitation of the signaling pathways involved in crosstalk between autophagy and apoptosis or necrosis. As the roles of autophagy can either be pro-survival or pro-death depending on context, it is conceivable that manipulating autophagy would have an impact on therapeutic outcome of various diseases. For instance, if autophagy activation contributes to resistance of cancer cells to certain therapies, use of autophagy inhibitors could be beneficial; in contrast, if induction of autophagy facilitates cell killing by cancer therapeutics, co-treatment with autophagy activators could reinforce the therapy. How to exploit autophagy as a therapeutic intervention remains an area of extensive investigation.
Footnotes
Supported by Grants from the US Public Health Service R01CA135038 (Yang JM), and from the Department of Defense BC103654 (Cheng Y)
Peer reviewer: Yannick Goumon, PhD, INSERM Unit 575, Physiopathology of the Nervous System, 5 rue Blaise Pascal, 67200 Strasbourg, France
S- Editor Cheng JX L- Editor Kerr C E- Editor Zheng XM
References
- 1.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 2.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 3.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 4.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 6.Klionsky DJ, Ohsumi Y. Vacuolar import of proteins and organelles from the cytoplasm. Annu Rev Cell Dev Biol. 1999;15:1–32. doi: 10.1146/annurev.cellbio.15.1.1. [DOI] [PubMed] [Google Scholar]
- 7.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 9.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 10.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 13.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 14.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 15.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, Yoshimori T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem. 2006;281:4467–4476. doi: 10.1074/jbc.M509409200. [DOI] [PubMed] [Google Scholar]
- 17.Kruse KB, Brodsky JL, McCracken AA. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell. 2006;17:203–212. doi: 10.1091/mbc.E04-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–4710. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 19.Ullman E, Fan Y, Stawowczyk M, Chen HM, Yue Z, Zong WX. Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ. 2008;15:422–425. doi: 10.1038/sj.cdd.4402234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kruse KB, Dear A, Kaltenbrun ER, Crum BE, George PM, Brennan SO, McCracken AA. Mutant fibrinogen cleared from the endoplasmic reticulum via endoplasmic reticulum-associated protein degradation and autophagy: an explanation for liver disease. Am J Pathol. 2006;168:1299–1308; quiz 1404-1405. doi: 10.2353/ajpath.2006.051097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Hum Mol Genet. 2007;16:618–629. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 23.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 24.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu RF, Ma Z, Liu Z, Terada LS. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol Cell Biol. 2010;30:3553–3568. doi: 10.1128/MCB.01445-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janssen K, Horn S, Niemann MT, Daniel PT, Schulze-Osthoff K, Fischer U. Inhibition of the ER Ca2+ pump forces multidrug-resistant cells deficient in Bak and Bax into necrosis. J Cell Sci. 2009;122:4481–4491. doi: 10.1242/jcs.055772. [DOI] [PubMed] [Google Scholar]
- 27.Tallóczy Z, Jiang W, Virgin HW, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299–30304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 30.Ryazanov AG, Shestakova EA, Natapov PG. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature. 1988;334:170–173. doi: 10.1038/334170a0. [DOI] [PubMed] [Google Scholar]
- 31.Py BF, Boyce M, Yuan J. A critical role of eEF-2K in mediating autophagy in response to multiple cellular stresses. Autophagy. 2009;5:393–396. doi: 10.4161/auto.5.3.7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu H, Yang JM, Jin S, Zhang H, Hait WN. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res. 2006;66:3015–3023. doi: 10.1158/0008-5472.CAN-05-1554. [DOI] [PubMed] [Google Scholar]
- 33.Cheng Y, Ren X, Zhang Y, Patel R, Sharma A, Wu H, Robertson GP, Yan L, Rubin E, Yang JM. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt Inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res. 2011;71:2654–2663. doi: 10.1158/0008-5472.CAN-10-2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 35.Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, Molgó J, Díaz J, Lavandero S, Harper F, et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14:1029–1039. doi: 10.1038/sj.cdd.4402099. [DOI] [PubMed] [Google Scholar]