

Abstract
Autophagy, the pathway whereby cell components are degraded by lysosomes, is involved in the cell response to environmental stresses, such as nutrient deprivation, hypoxia or exposition to chemotherapeutic agents. Under these conditions, which are reminiscent of certain phases of tumor development, autophagy either promotes cell survival or induces cell death. This strengthens the possibility that autophagy could be an important target in cancer therapy, as has been proposed. Here, we describe the regulation of survival and death by autophagy and apoptosis, especially in cultured breast cancer cells. In particular, we discuss whether autophagy represents an apoptosis-independent process and/or if they share common pathways. We believe that understanding in detail the molecular mechanisms that underlie the relationships between autophagy and apoptosis in breast cancer cells could improve the available treatments for this disease.
Keywords: Autophagy, Apoptosis, Survival, Breast cancer cells, Signaling pathways
AUTOPHAGY
Autophagy is the process whereby organelles and other cell components are degraded by lysosomes. There are various types of autophagy, including macroautophagy, microautophagy and chaperone-mediated autophagy[1]. Macroautophagy, hereafter called autophagy, is the most important form of autophagy and involves the formation of double-membrane vacuoles, named autophagosomes, containing cytosol and organelles. Autophagosomes then fuse with endosomes and lysosomes to form autolysosomes (Figure 1), which undergo a gradual acidification, by a proton pump, and degradation, by hydrolytic enzymes, of their content[2]. Autophagosome formation is a complex mechanism in which different autophagy-related (Atg) proteins participate, including Beclin 1 and LC3 (Atg6 and Atg8 in yeast, respectively), and which also requires the cell cytoskeleton[1,3,4]. Autophagy occurs at basal levels in almost all cells, and its main function is the degradation of cell components, including long-lived proteins, protein aggregates and organelles produced in excess, aged, damaged and potentially dangerous or no longer needed[5,6]. Under starvation conditions, autophagy provides the cells with molecules (amino acids, fatty acids, monosaccharides and nucleotides) that can be used for biosynthetic purposes. Some of these molecules can also be utilized as energy sources and the ensuing biosyntheses require energy. Therefore, it appears logical that part of them can be used to produce this energy, as has been postulated by many authors[7-10]. However, direct experimental proof for a role of autophagy in restoring the energy levels in the cell is still missing, probably because of the difficulties derived from the fact that this energy would be immediately used by the cells recovering from stress. Autophagy has also an important role in normal development, differentiation, and tissue remodeling in multicellular organisms, as well as in their adaptation to several stresses[5,11].
Figure 1.

Morphology of autophagic vacuoles. Typical autophagic vacuoles from 3T3 mouse fibroblasts incubated in a nutrient-poor medium containing cytoplasmic material at early (Avi) and late (Avd) degradation stages. Mit: Mitochondria; N: Nucleus.
Regarding cancer, which is the general subject of this Topic Highlight, a tumor suppressor role for autophagy has been also proposed, removing injured mitochondria that could increase the production of reactive oxygen species (ROS) and the number of mutations in cancer cells[11].
Role of autophagy in survival and death of tumor cells in response to environmental stress
In the previous decade, several reports have suggested a role for autophagy in cell survival at different stages of tumor development and in the tumor cell response to anticancer therapy[4,11,12], and this role of autophagy has become a major research topic. Under stress conditions, like deprivation of growth factors or nutrients, hypoxia or exposition to chemotherapeutic agents, cells induce autophagy to provide biosynthetic precursors and, perhaps also (but see above), energy, or to eliminate injured cell components, thus preventing cell death[7,13,14]. Therefore, autophagy may allow cancer cells to survive under nutrient and oxygen-poor conditions, reminiscent of certain microenvironments in poorly vascularized tumors[15]. Autophagy can also contribute to cell survival by removing injured targets of ROS and proteins carrying mutations that could lead to an irreversible stage conducive to cell death[16]. Under the aggressive stress conditions experienced by tumor cells, their autophagy levels are higher than normal and, therefore, disruption of this increased autophagy by therapeutic manipulations will make difficult the adaptation of these cells to extreme environments, and contribute to cancer therapy. However, chemical inhibitors of autophagy also prevent the death of cancer cells induced by a variety of agents[17]. This opposite role of autophagy as an executioner of cell death[18-20] and, thus, playing a role as a tumor suppressor[11], could probably be explained by a persistent degradation of components essential for cell survival[4,21]. Therefore, it appears that, in addition to its conventional role in cell survival, autophagy can be also a death-promoter, in particular when the stimulus is too intense, when autophagy is extensive, or under conditions of inhibition of apoptosis. The level of autophagy that represents the point of no return leading to cell death has not been clearly defined and should be determined experimentally in each specific system. However, some authors have considered that a situation in which the total area of autophagic vacuoles is equal or greater than that of the remaining cytoplasm would irreversibly lead to cell death[20,22].
In all these cases, the conventional inhibitors of autophagy and the concentrations used by most authors to block or promote survival of cancer cells under in vitro conditions[13,14,18,23,24] were the following: 3-methyladenine (5-10 mmol/L), chloroquine (10 μmol/L) and bafilomycin A1 (0.1 μmol/L). To the best of our knowledge, these chemicals have not yet been used for clinical treatment of cancer, except for chloroquine, which has been used in patients with glioblastoma multiforme. Thus, in these antitumoral clinical trials, chloroquine, or its lower toxicity analog hydroxychloroquine, have been used (150 mg/d, for 12 mo) as autophagy inhibitors in combination with proapoptotic drugs, increasing, in this way, twofold the median survival of these patients[25-28].
In summary, autophagy may either promote or inhibit survival in tumor cells, and the threshold to decide between both opposite processes will depend on the extent of the cell degradation produced[29], as well as on many other factors, such as the genetic context of the cell and the nature and intensity of the stimulus needed to reduce cell survival[30].
Autophagy in the context of cell death
In recent decades, studies in the field of cell death have focused on understanding the molecular mechanisms of apoptosis (often called programmed cell death, and now also referred to as cell death type I). Apoptosis is the form of cell death in which a group of cysteinyl aspartate-specific proteases, called caspases, become activated to cleave different proteins (and the caspases themselves) that ultimately produce loss of cell function, and cell death. In apoptosis, initiator caspases (2, 8, 9 and 10) activate executioner caspases (3, 6 and 7, of which, caspase-3 is the major and most widespread effector of the process)[31,32]. The essential feature of apoptosis, which makes it different from classical necrosis, is that it is a self-directed cell destruction process through caspase activation. Hundreds of caspase substrates have been described[33] and different biochemical and morphological changes in the nucleus and cytoplasm (e.g. cell contraction, membrane blebbing, externalization of phosphatidylserine, chromatin condensation into one or more masses, DNA fragmentation, limited proteolysis of certain substrates, and heterophagic elimination of apoptotic bodies by neighboring cells) have been used to identify apoptotic cells[17,34,35]. Two well-established molecular pathways (extrinsic and intrinsic) activate caspases and trigger apoptosis. The first is the death-receptor-mediated pathway, which is activated by ligands that bind to specific receptors on the plasma membrane, such as the tumor necrosis factor receptor 1 and Fas. The other is the mitochondrial pathway, which takes place through permeabilization of these organelles, followed by the release of apoptotic molecules such as cytochrome c (which triggers the formation of larger complexes called apoptosomes), apoptosis-inducing factor (AIF), or endonuclease G[17,31-33].
In addition to canonical apoptosis and necrosis, diverse experimental evidence has shown that cells can die through alternative pathways[36]. Thus, there is a form of cell death, whose main feature is the appearance of abundant autophagic vacuoles in the cytoplasm of dying cells, known as autophagic or type II cell death, and several of its characteristics, based mainly on morphological criteria, have been described in recent years[20]. Type II cell death would occur because of persistent autophagy with excessive degradation of cell components essential for survival[4,21], and it is usually accompanied by inhibition of the phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3kinase/Akt/mTOR) signaling pathway[37,38], which is the main regulator of autophagy, and by increased levels of LC3-II[1], a protein that is recruited to autophagosomes and that, under certain conditions, can be used as a reliable marker for autophagy[39,40]. However, different studies have found that some of the apoptotic cell death features cited above are also associated with an increased autophagy[18,29]. Therefore, the question is raised as to whether or not apoptosis and autophagy represent two independent processes.
In this regard, different reports indicate that autophagy can act independently of the apoptotic signaling pathways. Thus, because preservation of most cytoplasmic organelles is among the classic hallmarks of apoptosis, autophagic cell death, which comprises an extensive sequestration and degradation of mitochondria, endoplasmic reticulum (ER) and other cell components, has been considered by some authors as a different category of cell death on its own[41,42]. In addition, other evidence supports that extensive autophagy may be a caspase-independent form of cell death. For example, blockage of caspase activity prevents Bax-induced poly (ADP ribose) polymerase and DNA cleavage, but not cytosolic vacuolation and non-apoptotic cell death[43]. In the same lines of evidence, it has been shown that death-associated protein kinase proteins positively regulate membrane blebbing and autophagy, but apparently not nuclear fragmentation, and that these events occur in a caspase-independent manner[44].
However, it is also quite clear that autophagy can also coexist and crosstalk with apoptosis. Indeed, several molecules that regulate apoptosis are among the different targets of the PI3-kinase/Akt/mTOR signaling pathway[45,46] and proteins, such as Beclin 1, phosphatase and tensin homolog, apoptosis-specific protein, and the product of the steroid-inducible gene E93 can establish interconnections between autophagy and apoptosis[29,42]. Therefore, different evidence appears to indicate that apoptosis and extensive autophagy represent two forms of cell death with independent, but also with common pathways (Figure 2). However, the molecular details of these latter relationships remain poorly known.
Figure 2.
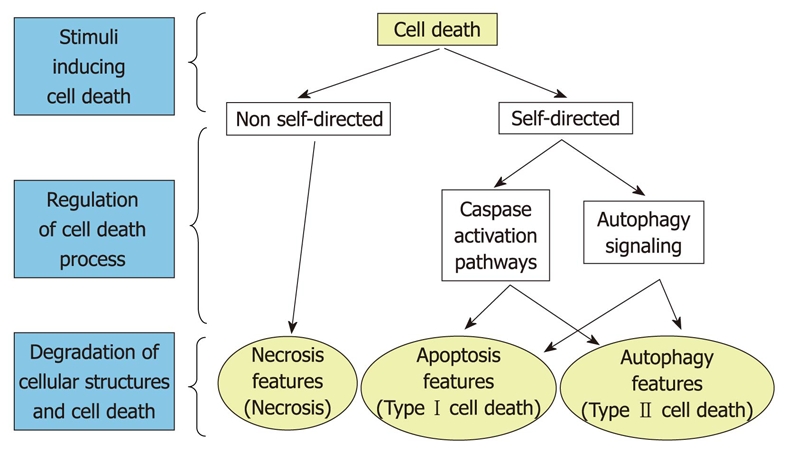
Main forms of cell death. Autophagy is located in the proper context in relation to classical necrosis and apoptosis. Crossing arrows indicate the existence of common links for apoptosis and autophagy. The entire process of cell death has been divided into three phases: stimulation, regulation and degradation. Note the absence of a regulation phase in necrosis.
MOLECULAR MECHANISMS OF AUTOPHAGY AND APOPTOSIS IN BREAST CANCER CELLS
As mentioned above, autophagy can promote or inhibit tumor survival depending on many factors, such as the specific cell type with the set of mutations that it carries, the stage of tumor development, and the stimulus that induces autophagy plus the extent of the resulting autophagy. Therefore, being aware of the heterogeneity in the survival/death response, which makes it difficult to generalize the different observations, and to limit the problem, we update the data on this topic in breast cancer cells. We have chosen these cells because of the growing number of recent studies on the role of autophagy in survival and death, compared to other experimental models.
Role of autophagy in survival and death of breast cancer cells in response to environmental stress
Studies on autophagy in breast cancer cells, mainly in MCF-7 cells, indicate that, in chemotherapeutic treatments, induction of autophagy plays a protective role in the resistance to apoptosis induced by anticancer drugs, such as the inhibitor of DNA topoisomerase I camptothecin[29], epirubicin[47], which intercalates DNA strands, different ligands that stimulate the antiestrogen binding site (AEBS), including tamoxifen[48,49], or 4-hydroxytamoxifen, an active metabolite of tamoxifen that binds to the estrogen receptor α[50,51]. Consistent with this idea, treatment of estrogen-receptor-positive breast cancer cells with the antiestrogen tamoxifen, combined with histone deacetylase inhibition, maintains a subpopulation of cells with an elevated autophagy and a remarkable resistance to apoptosis. These apoptosis-resistant cells only become apoptotic after inhibition of autophagy[52]. Also, and in the same line of evidence, the anticancer properties of lucanthone have been recently related to its ability to induce apoptosis and inhibit autophagy in breast cancer cell lines[53]. Further indications for a promoting effect on breast malignant cell development by autophagy are provided by recent reports showing that the tumor suppressor BRCA1 (breast cancer type 1 susceptibility) negatively regulates autophagy in MDA-MB-231[54] and in MCF-7[55] breast cancer cells. Thus, it could be that mutations in the BRCA1 gene or reduced expression of the encoded protein facilitate tumor development by preventing apoptosis through autophagy activation. Nevertheless, a death-promoting effect has also been reported for autophagy; for example, in MCF-7 cells subjected to oxidative damage by photodynamic therapy[56] or in MCF-7 cells overexpressing Bcl-2 in the presence of the antineoplastic factor brevinin-2R[57]. Table 1 shows the specific anticancer effects on apoptosis and/or autophagy of various agents tested under in vitro conditions in breast cancer cells.
Table 1.
Agents inducing anticancer mechanisms in cultured breast cancer cells
| Agent | Model | Anticancer mechanism | Citation |
| Camptothecin | MCF-7 | Apoptosis↑ | [29] |
| Epirubicin | MCF-7 | Apoptosis↑ | [47] |
| Tamoxifen | MCF-7 | Apoptosis↑ | [48,49] |
| 4-hydroxytamoxifen | MCF-7, T-47D | Apoptosis↑ | [50,51] |
| Lucanthone | MDA-MB-231 | Apoptosis↑, autophagy↓ | [25,53] |
| Chloroquine | Breast cancer carcinoma1 | Apoptosis↑, autophagy↓ | [69] |
| Photodynamic therapy | MCF-7 | Autophagy↑ | [56] |
| Tunicamycin | MCF-7 | Autophagy↑ | [59] |
Ex vivo model.
In conclusion, in breast cancer cell lines, autophagy mainly facilitates their survival and adaptation to adverse environments, whereas apoptosis has the opposite effect, and the final outcome, in terms of survival or death of the cells, will depend on many factors. Therefore, it appears that, at least in breast cancer cells, both apoptosis induction and autophagy inhibition have positive therapeutic implications depending on context.
Functional links of autophagy and apoptosis in cultured breast cancer cells
In breast cancer MCF-7 cells, camptothecin induces both apoptosis, demonstrated by deficient (sub-G1) DNA content and by chromatin condensation, and autophagy, demonstrated by increased levels of Beclin 1 and autophagosomes[58]. Also, in various breast cancer cells, sterol accumulation promoted by binding of various ligands, such as tamoxifen, to microsomal AEBS, induces both apoptosis and autophagy[48,49]. However, other treatments have opposite effects in both processes (Table 1). For example, in MDA-MB-231 breast cancer cells, lucanthone induces apoptosis and inhibits autophagy[25]. This experimental evidence suggests the existence of common links between apoptosis and autophagy in breast cancer cells. However, the door to the molecular mechanisms that link apoptosis and autophagy in breast cancer cells has only recently begun to open, and current knowledge is discussed below.
Thus, different proteins that belong to the mitochondrial pathway of apoptosis have also been shown to crosstalk with Atg proteins and to regulate autophagy in cultured breast cancer cells. For example, in MCF-7 cells, which lack caspase-3, expression of an ectopic caspase-3 reduces the enhanced autophagy produced by tunicamycin (an inductor of ER stress) or/and by radiation[59]. This effect is accompanied by a decrease in the levels of phosphorylated eukaryotic initiation factor 2α, which at the same time increases protein synthesis[59]. Therefore, caspase-3 may be a switch between type I and II cell death[17,60]. In these same cells, activation of another apoptosis promoter, protein Bid, also affects apoptosis and autophagy in opposite directions, because it not only stimulates apoptosis but also reduces autophagy by inhibition of Beclin 1[58]. In contrast, and also in MCF-7 cells, the antiapoptotic protein Bcl-2 regulates both processes in the same direction, because it negatively regulates the levels of three Atg proteins (Beclin 1, Atg5 and LC3-II), thus inhibiting autophagy[61]. Recently, a gene network signaling model has also indicated a central role for Bcl-2 and Beclin 1 in the apoptotic and autophagic responses to endocrine therapies in breast cancer cells, and has identified nuclear factor κB, interferon regulatory factor-1, and the X-box binding protein-1 as new key proteins that regulate Bcl-2 and Beclin 1 in these responses[62].
Unlike the apoptotic regulation of autophagy in breast cancer cells, a possible control of apoptosis by autophagy remains to be investigated in detail. However, it is known in other cell types that the PI3-kinase/Akt/mTOR signaling pathway, which has an inhibitory effect on autophagy, can interact with proteins that regulate apoptosis[45,46]. Moreover, it has been speculated that the selective removal of damaged mitochondria generating ROS by autophagy (mitophagy) could inhibit the mitochondrial pathway of apoptosis[6,63,64]. Furthermore, lysosomal cathepsins can establish a link between apoptosis and autophagy, because they are released from lysosomes into the cytosol in response to death stimuli, and induce apoptosis[65]. More specifically, it has been described in other cell lines that cathepsin D activates the proapoptotic protein Bax, which triggers the release of AIF from mitochondria[66], and that papain-like lysosomal cathepsins are able to cleave the proapoptotic protein Bid[67]. Also in MCF-7 breast cancer cells, papain-like cysteine cathepsins, probably including cathepsin B[58], activate Bid, which promotes apoptosis and reduces autophagy. A further example of a lysosomal cathepsin regulating apoptosis is provided by MDA-MB-231 breast cancer cells, in which lucanthone inhibits autophagy, probably by affecting lysosomal acidification, and induces a cathepsin-D-mediated apoptosis. This apoptosis probably occurs by lysosomal membrane permeabilization, subsequently releasing cathepsin D into the cytosol, which cleaves caspases[25,53].
In addition to mitochondria and lysosomes, the ER has also been shown to be involved in the regulation of autophagy and apoptosis. Thus, in MCF-7 cells, the ER transmembrane protein kinase-like ER kinase (PERK) increases autophagy and reduces the fraction of cells that survive radiation and/or a treatment with tunicamycin, and this PERK-controlled autophagy can be inhibited by caspase-3[59].
Thus, the above-mentioned examples support a molecular link between autophagy and apoptosis. In contrast, in breast adenocarcinoma MCF-7 cells overexpressing Bcl-2, the antineoplastic factor brevinin-2R leads to mitochondrial dysfunction (demonstrated by a reduction in mitochondrial membrane potential and in cellular ATP levels, and by an increase of ROS levels), autophagosome formation and cell death. These effects occur without involving apoptotic effectors (such as caspase activation and the mitochondrial release of the AIF or of endonuclease G)[57]. Thus, it appears that autophagic cell death can also occur independently of apoptosis. All these molecular mechanisms are summarized in Figure 3.
Figure 3.

Model of regulation of autophagy and apoptosis in breast cancer cells. Different molecules described to act as a link between apoptosis and autophagy are shown (see text for details). Ellipses indicate classical regulators of apoptosis. Moreover, organelles, such as mitochondria, endoplasmic reticulum and lysosomes, appear to be involved in this regulation. Arrow-headed lines and bar-headed lines indicate activation and inhibition, respectively, of their corresponding targets.
Although this Topic Highlight is focused on breast cancer cells in vitro, and limited information is available in vivo, we briefly summarize the most relevant information available under these last conditions. In a breast tumor xenograft model, Bcl-2 reduces autophagy by inhibition of Beclin 1, as it also occurs in vitro[68]. Moreover, samples from patients with breast ductal carcinoma and their corresponding mouse xenografts, show an increase in many autophagic markers, and this autophagy is necessary for the ex vivo survival of all these samples, as shown with 50 μmol/L chloroquine[69]. This observation is again in agreement with the survival function for autophagy observed in vitro. Interestingly, as we discussed above, the use of chloroquine in clinical trials has increased the survival of glioblastoma patients[25-28]. Therefore, all these data support that inhibition of autophagy offers a potential therapy in breast cancer.
In summary, several lines of evidence under in vitro conditions indicate that, in breast cancer cells, although apoptosis and autophagy can coexist as independent pathways, they are also interconnected processes. Molecular links are represented by classic apoptosis-regulator proteins (caspase-3, Bid and Bcl-2), which inhibit autophagy by acting on Atg proteins. Upstream of these regulators of apoptosis are cytosol-released lysosomal cathepsins, which induce apoptosis by activating proapoptotic proteins. In addition, new candidates to interact with these proteins that link apoptosis and autophagy are now emerging, as illustrated by the above-mentioned studies with a gene network signaling model, and elucidation of their specific function could contribute to understand further this complex mechanism.
CONCLUSION
Autophagy is a physiological process of lysosomal degradation that, in response to environmental stresses, may either promote cell survival or death depending on many factors. In addition to canonical apoptosis (type I cell death) and necrosis, extensive autophagy represents an alternative form of cell death (type II). In breast cancer cells, autophagy and apoptosis share some common proteins from their signaling routes. Thus, classical regulators of apoptosis, such as Bid, Bcl-2 and caspases, appear to crosstalk with Atg proteins and, in consequence, regulate autophagy. Moreover, lysosomal cathepsins provide an important link between both processes, by acting on target proteins of the apoptotic signaling pathways. However, autophagy in breast cancer cells can also be an apoptosis-independent process. Therefore, the relationships between autophagy and apoptosis are quite complex, but we predict that a better understanding of the underlying molecular mechanisms could contribute in the near future to anticancer therapy.
Footnotes
Supported by Ministerio de Ciencia e Innovación, Grant No. BFU 2008-00186 and Generalitat Valenciana, No. ACOMP07-187
Peer reviewers: Rong Shao, PhD, Assistant Professor, University of Massachusetts Amherst, Pioneer Valley Science Institute, 3601 Main St, Springfield, MA 01107, United States; Beric Henderson, PhD, NHMRC Senior Research Fellow, University of Sydney, Westmead Millennium Institute, Darcy Road, PO Box 412, Westmead NSW 2145, Australia
S- Editor Cheng JX L- Editor Kerr C E- Editor Zheng XM
References
- 1.Knecht E, Aguado C, Cárcel J, Esteban I, Esteve JM, Ghislat G, Moruno JF, Vidal JM, Sáez R. Intracellular protein degradation in mammalian cells: recent developments. Cell Mol Life Sci. 2009;66:2427–2443. doi: 10.1007/s00018-009-0030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- 3.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12 Suppl 2:1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hippert MM, O’Toole PS, Thorburn A. Autophagy in cancer: good, bad, or both? Cancer Res. 2006;66:9349–9351. doi: 10.1158/0008-5472.CAN-06-1597. [DOI] [PubMed] [Google Scholar]
- 5.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 6.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–103. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 8.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 9.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 10.Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007;14:548–558. doi: 10.1038/sj.cdd.4402030. [DOI] [PubMed] [Google Scholar]
- 11.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 13.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 14.Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 16.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 17.Bursch W, Karwan A, Mayer M, Dornetshuber J, Fröhwein U, Schulte-Hermann R, Fazi B, Di Sano F, Piredda L, Piacentini M, et al. Cell death and autophagy: cytokines, drugs, and nutritional factors. Toxicology. 2008;254:147–157. doi: 10.1016/j.tox.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 18.Jia L, Dourmashkin RR, Allen PD, Gray AB, Newland AC, Kelsey SM. Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br J Haematol. 1997;98:673–685. doi: 10.1046/j.1365-2141.1997.2623081.x. [DOI] [PubMed] [Google Scholar]
- 19.Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta. 2003;1603:113–128. doi: 10.1016/s0304-419x(03)00004-0. [DOI] [PubMed] [Google Scholar]
- 20.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 21.Abreu MM, Sealy L. The C/EBPbeta isoform, liver-inhibitory protein (LIP), induces autophagy in breast cancer cell lines. Exp Cell Res. 2010;316:3227–3238. doi: 10.1016/j.yexcr.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 23.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 24.Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- 25.Carew JS, Nawrocki ST, Cleveland JL. Modulating autophagy for therapeutic benefit. Autophagy. 2007;3:464–467. doi: 10.4161/auto.4311. [DOI] [PubMed] [Google Scholar]
- 26.Savarino A, Lucia MB, Giordano F, Cauda R. Risks and benefits of chloroquine use in anticancer strategies. Lancet Oncol. 2006;7:792–793. doi: 10.1016/S1470-2045(06)70875-0. [DOI] [PubMed] [Google Scholar]
- 27.Sotelo J, Briceño E, López-González MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006;144:337–343. doi: 10.7326/0003-4819-144-5-200603070-00008. [DOI] [PubMed] [Google Scholar]
- 28.Garber K. Inducing indigestion: companies embrace autophagy inhibitors. J Natl Cancer Inst. 2011;103:708–710. doi: 10.1093/jnci/djr168. [DOI] [PubMed] [Google Scholar]
- 29.Motyl T, Gajkowska B, Zarzyńska J, Gajewska M, Lamparska-Przybysz M. Apoptosis and autophagy in mammary gland remodeling and breast cancer chemotherapy. J Physiol Pharmacol. 2006;57 Suppl 7:17–32. [PubMed] [Google Scholar]
- 30.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–6206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- 32.Kitazumi I, Tsukahara M. Regulation of DNA fragmentation: the role of caspases and phosphorylation. FEBS J. 2011;278:427–441. doi: 10.1111/j.1742-4658.2010.07975.x. [DOI] [PubMed] [Google Scholar]
- 33.Logue SE, Martin SJ. Caspase activation cascades in apoptosis. Biochem Soc Trans. 2008;36:1–9. doi: 10.1042/BST0360001. [DOI] [PubMed] [Google Scholar]
- 34.Zhivotosky B, Orrenius S. Assessment of apoptosis and necrosis by DNA fragmentation and morphological criteria. Curr Protoc Cell Biol. 2001;Chapter 18:Unit 18.3. doi: 10.1002/0471143030.cb1803s12. [DOI] [PubMed] [Google Scholar]
- 35.Schutters K, Reutelingsperger C. Phosphatidylserine targeting for diagnosis and treatment of human diseases. Apoptosis. 2010;15:1072–1082. doi: 10.1007/s10495-010-0503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lockshin RA, Zakeri Z. Caspase-independent cell death? Oncogene. 2004;23:2766–2773. doi: 10.1038/sj.onc.1207514. [DOI] [PubMed] [Google Scholar]
- 37.Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–2462. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 38.Yang YP, Liang ZQ, Gu ZL, Qin ZH. Molecular mechanism and regulation of autophagy. Acta Pharmacol Sin. 2005;26:1421–1434. doi: 10.1111/j.1745-7254.2005.00235.x. [DOI] [PubMed] [Google Scholar]
- 39.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 40.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bursch W, Hochegger K, Torok L, Marian B, Ellinger A, Hermann RS. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J Cell Sci. 2000;113(Pt 7):1189–1198. doi: 10.1242/jcs.113.7.1189. [DOI] [PubMed] [Google Scholar]
- 42.Lefranc F, Facchini V, Kiss R. Proautophagic drugs: a novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist. 2007;12:1395–1403. doi: 10.1634/theoncologist.12-12-1395. [DOI] [PubMed] [Google Scholar]
- 43.Xiang J, Chao DT, Korsmeyer SJ. BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc Natl Acad Sci USA. 1996;93:14559–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157:455–468. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castedo M, Ferri KF, Kroemer G. Mammalian target of rapamycin (mTOR): pro- and anti-apoptotic. Cell Death Differ. 2002;9:99–100. doi: 10.1038/sj.cdd.4400978. [DOI] [PubMed] [Google Scholar]
- 46.Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, Eshraghi M, Manda KD, Wiechec E, Los M. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat. 2007;10:13–29. doi: 10.1016/j.drup.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 47.Sun WL, Chen J, Wang YP, Zheng H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy. 2011;7:1035–1044. doi: 10.4161/auto.7.9.16521. [DOI] [PubMed] [Google Scholar]
- 48.de Medina P, Payré B, Boubekeur N, Bertrand-Michel J, Tercé F, Silvente-Poirot S, Poirot M. Ligands of the antiestrogen-binding site induce active cell death and autophagy in human breast cancer cells through the modulation of cholesterol metabolism. Cell Death Differ. 2009;16:1372–1384. doi: 10.1038/cdd.2009.62. [DOI] [PubMed] [Google Scholar]
- 49.de Medina P, Silvente-Poirot S, Poirot M. Tamoxifen and AEBS ligands induced apoptosis and autophagy in breast cancer cells through the stimulation of sterol accumulation. Autophagy. 2009;5:1066–1067. doi: 10.4161/auto.5.7.9820. [DOI] [PubMed] [Google Scholar]
- 50.Samaddar JS, Gaddy VT, Duplantier J, Thandavan SP, Shah M, Smith MJ, Browning D, Rawson J, Smith SB, Barrett JT, et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol Cancer Ther. 2008;7:2977–2987. doi: 10.1158/1535-7163.MCT-08-0447. [DOI] [PubMed] [Google Scholar]
- 51.Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, Jackson WH, Barrett JT. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy. 2009;5:400–403. doi: 10.4161/auto.5.3.7784. [DOI] [PubMed] [Google Scholar]
- 52.Thomas S, Thurn KT, Biçaku E, Marchion DC, Münster PN. Addition of a histone deacetylase inhibitor redirects tamoxifen-treated breast cancer cells into apoptosis, which is opposed by the induction of autophagy. Breast Cancer Res Treat. 2011;130:437–447. doi: 10.1007/s10549-011-1364-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carew JS, Espitia CM, Esquivel JA, Mahalingam D, Kelly KR, Reddy G, Giles FJ, Nawrocki ST. Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J Biol Chem. 2011;286:6602–6613. doi: 10.1074/jbc.M110.151324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fan S, Meng Q, Saha T, Sarkar FH, Rosen EM. Low concentrations of diindolylmethane, a metabolite of indole-3-carbinol, protect against oxidative stress in a BRCA1-dependent manner. Cancer Res. 2009;69:6083–6091. doi: 10.1158/0008-5472.CAN-08-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Esteve JM, Armengod ME, Knecht E. BRCA1 negatively regulates formation of autophagic vacuoles in MCF-7 breast cancer cells. Exp Cell Res. 2010;316:2618–2629. doi: 10.1016/j.yexcr.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 56.Xue LY, Chiu SM, Oleinick NL. Atg7 deficiency increases resistance of MCF-7 human breast cancer cells to photodynamic therapy. Autophagy. 2010;6:248–255. doi: 10.4161/auto.6.2.11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghavami S, Asoodeh A, Klonisch T, Halayko AJ, Kadkhoda K, Kroczak TJ, Gibson SB, Booy EP, Naderi-Manesh H, Los M. Brevinin-2R(1) semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway. J Cell Mol Med. 2008;12:1005–1022. doi: 10.1111/j.1582-4934.2008.00129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lamparska-Przybysz M, Gajkowska B, Motyl T. Cathepsins and BID are involved in the molecular switch between apoptosis and autophagy in breast cancer MCF-7 cells exposed to camptothecin. J Physiol Pharmacol. 2005;56 Suppl 3:159–179. [PubMed] [Google Scholar]
- 59.Kim KW, Moretti L, Mitchell LR, Jung DK, Lu B. Endoplasmic reticulum stress mediates radiation-induced autophagy by perk-eIF2alpha in caspase-3/7-deficient cells. Oncogene. 2010;29:3241–3251. doi: 10.1038/onc.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fazi B, Bursch W, Fimia GM, Nardacci R, Piacentini M, Di Sano F, Piredda L. Fenretinide induces autophagic cell death in caspase-defective breast cancer cells. Autophagy. 2008;4:435–441. doi: 10.4161/auto.5669. [DOI] [PubMed] [Google Scholar]
- 61.Akar U, Chaves-Reyez A, Barria M, Tari A, Sanguino A, Kondo Y, Kondo S, Arun B, Lopez-Berestein G, Ozpolat B. Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy. 2008;4:669–679. doi: 10.4161/auto.6083. [DOI] [PubMed] [Google Scholar]
- 62.Clarke R, Shajahan AN, Riggins RB, Cho Y, Crawford A, Xuan J, Wang Y, Zwart A, Nehra R, Liu MC. Gene network signaling in hormone responsiveness modifies apoptosis and autophagy in breast cancer cells. J Steroid Biochem Mol Biol. 2009;114:8–20. doi: 10.1016/j.jsbmb.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14:500–510. doi: 10.1038/sj.cdd.4402039. [DOI] [PubMed] [Google Scholar]
- 64.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Minarowska A, Minarowski L, Karwowska A, Gacko M. Regulatory role of cathepsin D in apoptosis. Folia Histochem Cytobiol. 2007;45:159–163. [PubMed] [Google Scholar]
- 66.Bidère N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C, Senik A. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem. 2003;278:31401–31411. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- 67.Cirman T, Oresić K, Mazovec GD, Turk V, Reed JC, Myers RM, Salvesen GS, Turk B. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem. 2004;279:3578–3587. doi: 10.1074/jbc.M308347200. [DOI] [PubMed] [Google Scholar]
- 68.Oh S, Xiaofei E, Ni D, Pirooz SD, Lee JY, Lee D, Zhao Z, Lee S, Lee H, Ku B, et al. Downregulation of autophagy by Bcl-2 promotes MCF7 breast cancer cell growth independent of its inhibition of apoptosis. Cell Death Differ. 2011;18:452–464. doi: 10.1038/cdd.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Espina V, Mariani BD, Gallagher RI, Tran K, Banks S, Wiedemann J, Huryk H, Mueller C, Adamo L, Deng J, et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PLoS One. 2010;5:e10240. doi: 10.1371/journal.pone.0010240. [DOI] [PMC free article] [PubMed] [Google Scholar]