

Early DNA vaccines were very exciting in small animals, but poorly immunogenic in large animals and humans. Recently, much progress has been made regarding increasing their immune potency. This review summarizes many of these technological advancements and discusses the current status and prospects of DNA vaccines in the clinic targeting specific pathogens.
Abstract
It was discovered almost 20 years ago that plasmid DNA, when injected into the skin or muscle of mice, could induce immune responses to encoded antigens. Since that time, there has since been much progress in understanding the basic biology behind this deceptively simple vaccine platform and much technological advancement to enhance immune potency. Among these advancements are improved formulations and improved physical methods of delivery, which increase the uptake of vaccine plasmids by cells; optimization of vaccine vectors and encoded antigens; and the development of novel formulations and adjuvants to augment and direct the host immune response. The ability of the current, or second-generation, DNA vaccines to induce more-potent cellular and humoral responses opens up this platform to be examined in both preventative and therapeutic arenas. This review focuses on these advances and discusses both preventive and immunotherapeutic clinical applications.
HISTORY OF DNA VACCINES
Current licensed vaccines are predominantly composed of either killed pathogens, pathogen subunits, or live-attenuated viruses. Nonlive vaccines, which confer protection primarily through the induction of CD4+ T- cell and humoral mechanisms, generally do not provide life-long immunity. In contrast, live-attenuated vaccines can mobilize both the cellular and humoral arms of the immune response and generally induce more-prolonged immunity. However, their degree of attenuation can significantly lower the immunogenicity of live vaccines, and the development of live vaccine strategies can be especially challenging when the goal is to target multiple viral subtypes or pathogens. There are also theoretical safety concerns associated with the use of both nonlive and attenuated approaches. These limitations continue to drive the need to develop new vaccine platforms that offer broader immunogenicity.
DNA vaccines first sparked the interested of the scientific community in the early 1990s, when it was reported that plasmid DNA, delivered into the skin or muscle, induced antibody responses to viral and nonviral antigens [1–4]. The simplicity and versatility of this vaccine approach generated a great deal of excitement and inspired additional preclinical studies targeting a plethora of viral and nonviral antigens. In theory, DNA vaccines could generate broad immune responses, similar to the live-attenuated virus platform, without the need for a replicating pathogen.
Owing to the promise of DNA vaccines in small animal studies, clinical trials soon ensued. The first of several of phase I trials, conducted almost 2 decades ago, evaluated the efficacy of a DNA vaccine targeting human immunodeficiency virus type 1 (HIV-1) for therapeutic and prophylactic applications [5]. Other studies shortly followed that targeted cancer or other HIV-1 antigens, influenza, human papillomavirus (HPV), hepatitis, and malaria. However, the results of these early clinical trials were disappointing. The DNA vaccines were safe and well tolerated, but they proved to be poorly immunogenic. The induced antibody titers were very low or nonexistent, CD8+ T-cell responses were sporadic, and CD4+ T-cell responses were of low frequency. However, these studies provided proof of concept that DNA vaccines could safely induce immune responses (albeit low-level responses) in humans.
SECOND-GENERATION DNA VACCINES
Many improvements have been incorporated into the current, or second-generation, DNA vaccines, and these improvements have helped to spark a resurgence of interest in the platform. Second-generation DNA vaccines appear to drive improved cellular and humoral immune responses in both small and large animal models. Importantly, research suggests that newer DNA vaccines can more broadly activate CD8+ cytotoxic T cells (CTL) in larger animal models, compared with earlier DNA approaches [6].
The low immunogenicity of early DNA vaccines is hypothesized to stem, in part, from inefficient uptake of the plasmids by cells due to inefficient delivery. Research has focused on developing novel strategies to enhance transfection efficiency and improve other facets of the DNA platform. These efforts include optimization of the antigens encoded by the plasmids to increase antigen expression on a per cell basis, improved formulation, and inclusion of molecular adjuvants to enhance and direct immune responses [7].
Delivery Approaches
Several physical methods of delivery have been explored to increase the transfection efficiency of DNA vaccines, including needle-free approaches, such as particle bombardment and high-pressure delivery, dermal patches, and electroporation (EP). Particle bombardment approaches use a highly pressurized stream to deliver vaccine plasmids on microscopic heavy metal beads. For example, the PMED device (Pfizer) delivers DNA plasmids, linked to microscopic gold particles, into the skin in a dry powder formulation [8, 9]. High-pressure mediated delivery is conceptually similar to particle bombardment. For example, the Biojector devices (Bioject Medical Technologies) deliver vaccines by forcing liquid through a tiny orifice to create a fine, high-pressure stream that penetrates the skin [10]. One example of noninvasive dermal patch delivery is DermaVir (Genetic Immunity). DermaVir is a self-adhesive patch coated with multiple antigen or adjuvant encoding plasmids and a synthetic polymer that forms pathogen-like nanoparticles [11]. Another promising physical method of delivery is EP, or the application of short electrical pulses to the delivery tissue, which was initially studied over 25 years ago as a method to enhance the efficacy of chemotherapy agents [12]. It was later discovered that EP also increases the uptake of DNA plasmids by cells, resulting in an increase in antigen production [13] and in vaccine immunogenicity [14–16]. Figure 1 demonstrates the magnitude of the increase in immunogenicity that can be achieved when delivering a DNA vaccine by intramuscular injection (IM) with EP (IM+EP), compared with IM alone. EP augmented both antigen-specific production of interferon (IFN) γ (Figure 1A) and seroconversion (Figure 1B). The use of improved delivery has enabled second-generation DNA vaccines to induce cellular immune responses comparable to viral vectors in nonhuman primates (NHPs) [17].
Figure 1.
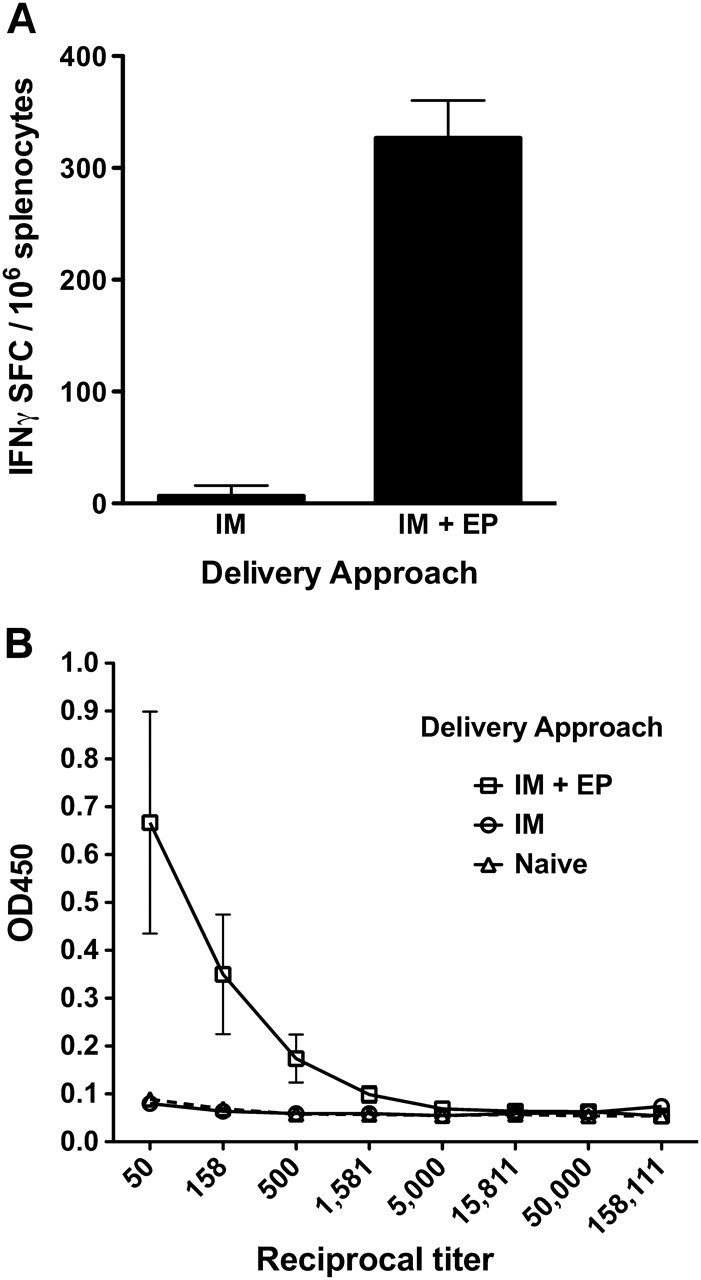
DNA vaccine delivery with electroporation (EP) increases cellular and humoral responses. A DNA vaccine encoding human prostate–specific antigen (PSA) was administered by intramuscular injection (IM) or by IM plus EP (IM+EP). Animals received 2 vaccinations spaced 2 weeks apart. Cellular and humoral responses were determined 1 week after the second immunization. PSA-specific T-cell responses were determined by interferon (IFN) gamma enzyme-linked immunospot (A) and PSA-specific seroconversion by enzyme-linked immunosorbent assay (B). n = 5 per group. OD, optical density; SFC, antigen-specific spot forming cells per 106.
Formulation and Molecular Adjuvants
Formulation of DNA vaccines in microparticles or liposomes has been reported to increase the uptake of plasmid DNA by cells, thereby increasing the immunogenicity of several different vaccines in animal models and humans [7]. An influenza DNA vaccine formulated in the lipid compound Vaxfectin (Vical) induced protective antibody titers and T-cell responses in many subjects [18]. Another method to improve DNA vaccine immunogenicity is the inclusion of additional plasmids, or additional inserts in the same plasmid, encoding molecular adjuvants. Multiple studies have shown that codelivery of plasmids encoding cytokines, chemokines or costimulatory molecules can augment immune responses. Unlike traditional adjuvants, which stimulate nonspecific inflammation, molecular adjuvants can modulate the adaptive immune response. For example, codelivery of interleukin (IL) 12 or IL-15 was shown to increase the magnitude and functionality of antigen-specific T cells in NHPs [19–21]. Similar to IL-12, IL-28B augments antigen-specific CD8+ T-cell responses, but it also increases CTL-killing ability [22, 23]. Use of granulocyte macrophage colony-stimulating factor (GM-CSF) as a molecular adjuvant has been shown to enhance cellular and humoral responses in NHPs [24, 25]. One study demonstrated codelivery of GM-CSF induced higher avidity in HIV-1–specific antibodies and enhanced neutralizing antibody production, which correlated with a trend towards improved control of a simian-human hybrid virus challenge and re-emergent virus [26].
Antigen Design
Recently, there has also been a focus on designing antigens that successfully target highly variable pathogens. The optimized immunogen sequences are usually designed or selected from a collection of target antigen protein sequences. For example, consensus immunogens are designed to encode the most commonly occurring amino acid at each position in a sequence, whereas mosaic antigens are designed to encode the most immunogenic regions of an antigen [7]. Similarly, center-of-tree immunogens are derived from a native sequence that represents a respective middle of evolutionary diversity, whereas ancestral immunogens are derived from antigen sequences at the root of a phylogenic tree. All of these techniques are an attempt to focus the immune response on a synthetic sequence that is more representative of pathogen diversity. Thus, the host immune response is better educated and responds more effectively to divergent pathogens [27].
SAFETY AND TOLERABILITY OF DNA VACCINES
The DNA platform is conceptually safer and more stable than are conventional vaccine approaches. Plasmids are nonlive and nonreplicating, which leaves little risk for reversion to a disease-causing state or secondary infection. The original concerns associated with the DNA platform were the potential for genomic integration and development of anti-DNA immune responses. Exhaustive research has found little evidence of integration, and the risk for integration appears to be significantly lower than that associated with naturally occurring mutations [28–30]. Induction of anti-DNA immune responses after DNA vaccination has been monitored in multiple NHP studies and clinical trials, but evidence of increased production of such responses or changes in other clinical markers of autoimmunity have not been reported [31]. Overall, multiple studies have reported the DNA platform to be well tolerated and to have an enviable safety record.
SELECTED CLINICAL TARGETS
There are currently 43 clinical trials evaluating DNA vaccines for viral and nonviral diseases listed in the clinicaltrials.gov database (Table 1; Figure 2). The majority (62%) of these trials are investigating vaccines for HIV (33%) or cancers (29%). Almost half (38%) of cancer vaccines currently being investigated are targeting melanoma. The remaining 38% of enrolling or active clinical trials are investigating vaccines for influenza, hepatitis B and C, HPV, and malaria. This review highlights DNA vaccines for influenza, HPV, and HIV-1 as examples of antibody, cellular, and complex immunological targets, respectively. It should be noted, as evidenced by Table 1 and Figure 2, that great strides have also been made in the development of DNA vaccines for many other important clinical targets.
Table 1.
Current DNA Vaccine Clinical Trials
| Phase | No. | Vaccine Targets |
| I | 31 | HIV treatment and prevention, influenza, HPV, cancer (metastatic breast, B cell lymphoma prostate, colorectal), hepatitis B, hepatitis C, malaria |
| I/II | 7 | HIV treatment, cancer (prostate, colorectal), hepatitis B, hepatitis C, HPV, malaria |
| II | 5 | Cancer (prostate, melanoma), HIV treatment, hepatitis B |
NOTE. HIV, human immunodeficiency virus; HPV, human papillomavirus.
Figure 2.

Current DNA vaccine clinical trials. At the time of publication, 43 clinical trials evaluating DNA vaccines were listed as on-going in the clinicaltrials.gov database. The large pie chart shows the percentage of trials by vaccine target. The inset pie chart shows the percentage of trials targeting specific cancers among the 29% of clinical trials that are cancer related. HIV, human immunodeficiency virus; HPV, human papillomavirus.
Influenza
Every year, the scientific and medical communities are charged with the task of determining the appropriate influenza strains to include in the seasonal influenza vaccine. Current vaccine platforms require months to generate sufficient quantities of antigens because of the requirement for the growth of the virus in chicken eggs [32]. This can delay the availability of viral stocks or result in a mismatch between the vaccine strains selected and the actual circulating strains. In 2007, the seasonal influenza vaccine coverage was estimated at only 30% because of mismatches between the strains that were expected to emerge and the strains that actually circulated [33]. In contrast, development of a DNA vaccine for a particular influenza strain could shorten this timeline 2–4-fold and could potentially provide a product in a few months with little chance of mismatch [27].
Influenza presents a particular challenge for the DNA platform because protection is specifically associated with antibodies, and induction of humoral responses was a shortcoming of the original DNA vaccines. New approaches incorporated into the second-generation platform have enabled the induction of humoral responses against a variety of antigens. Thus, the development a DNA vaccine for influenza has become a more reasonable goal. One preclinical study of an H5N1 influenza DNA vaccine showed that protective antibody titers were induced to multiple clades of H5N1 using a single consensus H5 antigen [33]. In further support of this cross-protection approach, it has recently been shown that cross-protective titers can be achieved to viruses that circulated over 90 years apart; namely, the 1918 “Spanish Flu” and the 2009 “Swine Flu” [34]. The concept of cross-neutralization of different influenza strains may be of great significance in future influenza vaccines. Moreover, this concept applies not only to influenza strains with the potential to cause pandemics but also to strains included in seasonal vaccines.
The success of DNA vaccines against multiple strains of influenza in preclinical models has paved the way for their development for the clinic. To that end, there are currently several DNA-based influenza vaccines in various stages of phase I clinical trials, including vaccines against potentially lethal pandemic strains such as H5N1 (Inovio Pharmaceuticals) and H1N1 (National Institutes of Allergy and Infectious Diseases) [35]. A completed phase I clinical trial conducted by Vical demonstrated that formulation of a monovalent H5N1 DNA vaccine in Vaxfectin achieved protective hemagglutination inhibition titers or antibody responses in more than 47% of subjects, and H5-specific T-cell responses were detected in at least 75% of subjects [18]. A phase 1 trial completed by PowderMed demonstrated reductions in disease symptoms and viral shedding in subjects who received a trivalent DNA-based seasonal influenza vaccine, delivered using the PMED device, compared with placebo [36]. The ultimate success of these vaccines could reshape the way physicians and researchers view influenza vaccine development.
HPV
Cervical cancer remains the third leading cause of cancer-related morbidity in women worldwide [37]. Intense research efforts have resulted in US Food and Drug Administration approval of 2 preventive HPV vaccines; Gardasil (Merck) in 2006 and Cervarix (GlaxoSmithKline) in 2009. However, the impact of these vaccines on the global prevalence of HPV infection is slowed because of the high economic burden and logistical issues that hinder widespread vaccination. These preventive HPV vaccines do not induce appreciable levels of cellular immune responses and, thus, cannot clear established HPV infections or HPV-associated lesions. Thus, the DNA platform, which can drive strong cellular responses, is a logical approach for this task.
Some candidate HPV therapeutic vaccines utilize the E6 and E7 oncoproteins as antigens to target HPV-16 and HPV-18, which are present in HPV-associated cervical cancer and cervical intraepithelial neoplasia (CIN). E6 and E7 are ideal therapeutic targets, because they play an integral role in the generation and maintenance of HPV-associated disease and are constituently expressed in HPV-associated cancer and precursor lesions [38]. One interesting DNA vaccine strategy is the use of fusion consensus antigens that encode multiple antigens in the same vector. For example, HPV-16 and HPV-18 E6/E7 fusion consensus vaccines, delivered by EP, demonstrated encouraging results in NHPs and are currently being evaluated in a phase I clinical trial (Inovio Pharmaceuticals) [39, 40].
Several other therapeutic HPV DNA vaccine clinical trials have been recently completed or are currently ongoing. ZYC101 (Eisai Pharmaceuticals), a microencapsulated DNA vaccine encoding multiple HPV-16 E7-specific CTL epitopes, was well tolerated in 2 different phase I trials [41, 42]. An alternative version of this vaccine, ZYC101a, which includes HPV-16 and HPV-18 E6- and E7-derived CTL epitopes, was moved into a phase II study in women with CIN2/3. In this study, the proportion of subjects with resolved lesions was higher in the treatment groups, but this result did not reach statistical significance [43]. A phase II/III trial of ZYC101a is currently underway. A different phase I study investigated a HPV-16 E7-specific vaccine, pNGVL4a-Sig/E7detox/HSP70 (NCI), administered by IM at escalating doses. The vaccine was well tolerated, but it failed to induce significant antibody or T-cell responses [44] and is currently undergoing reevaluation as a component of a DNA and viral-vector heterologous prime-boost strategy.
HIV
The development of a vaccine to prevent or control HIV-1 infection has been an elusive goal since the virus was first described in 1981. Unlike conventional vaccine targets, inducing broadly neutralizing antibodies against HIV-1 has proven to be exceedingly challenging [45]. Also, because of the complexity of HIV-1, it is likely that an effective vaccine will be required to modulate broad cellular and humoral responses. Neither the recombinant protein gp120 nor the Ad5-vaccine used in the STEP trial was effective at preventing HIV infection [45, 46].
In an effort to increase HIV-specific immune responses, several clinical trials have investigated heterologous prime-boost approaches that combine DNA-based and viral-based vaccines with recombinant protein vaccines. The concept of combining a vaccine platform that induces T-cell responses (DNA or viral-vector vaccines) with one that induces antibody responses (recombinant protein vaccines) to induce broad HIV-1–specific immunity has shown promise in a recently completed efficacy trial (RV144). This trial incorporated a multiple-antigen viral-vector prime (ALVAC) to induce HIV-1–specific T cells, followed by a recombinant gp120 protein boost (AIDSVAX) to generate HIV-1–specific antibodies. In a modified intent-to-treat analysis, this heterologous prime-boost approach demonstrated 31% efficacy for prevention of HIV-1 acquisition, but it did not affect viral load in subjects who were not protected [47]. Although post hoc analysis of the RV144 trial is ongoing, the success of 2 platforms that are ineffective individually suggests that a preventive HIV vaccine will most likely require induction of cellular and humoral responses. Other studies are investigating conceptually similar heterologous prime-boost strategies by combining a DNA prime with a recombinant protein boost. For example, a phase I clinical trial (DP6-001) demonstrated priming with a multiple-antigen polyvalent DNA vaccine, and boosting a recombinant HIV-1 envelope protein induced cross-subtype antibody and cellular responses [48].
Combining a DNA prime and viral boost creates a synergistic enhancement in the magnitude of antigen-specific CD8+ T-cell responses. A phase I trial that combined a multi-clade DNA vaccine prime with an Ad5 boost demonstrated that this strategy was capable of eliciting humoral responses in addition to cellular responses [49]. Preclinical studies also suggested that this approach increases not only the magnitude but also the quality of the humoral response [50]. This combination is now being explored in a larger efficacy trial. The National Institutes of Health Vaccine Research Center, in collaboration with the HIV Vaccine Trial Network, is evaluating the efficacy of this approach to reduce viral loads in patients who become infected after vaccination (HVTN 505) [51]. Other viral vectors, such as modified vaccinia ankara (MVA), are also being investigated for use in HIV-1 vaccine strategies. A phase IIa trial (HVTN 205) (GeoVax) is currently evaluating a multiple-antigen DNA prime followed by an MVA boost encoding the same antigens.
First-generation DNA vaccines were shown to stimulate T cell responses and antibodies, although at levels insufficient to prevent HIV-1 infection. The advent of improved methods of physical delivery and other new technologies has spurred a second wave of clinical trials investigating DNA as a stand-alone platform. A phase I clinical trial (HVTN-080) is currently underway to determine the safety of Pennvax-B, a DNA vaccine encoding HIV-1 gag, pol and env, and molecular adjuvant IL-12 delivered by EP [52]. The use of molecular adjuvants is of particular interest for HIV-1 vaccine development. In addition to increasing the magnitude of the immune response, some molecular adjuvants can also alter the homing of antigen-specific cells to specific target tissues. For example, an NHP study demonstrated codelivery of muscosal chemokines induced trafficking of antigen-specific T cells to the gut mucosa, which could position immune effector cells in a more advantageous location to dampen initial HIV-1 viral replication [53].
FUTURE DIRECTIONS
A great deal of progress has been made since the disappointment of the original DNA vaccine clinical trials almost 16 years ago. Advancements in antigen design, improved formulations, inclusion of molecular adjuvants, and physical methods of delivery have greatly enhanced the immunogenicity of second-generation DNA vaccines. The improved performance has spurred a renewed interest in the platform, which is reflected by the numerous ongoing clinical trials investigating DNA vaccines for preventative and therapeutic applications. There are several gene-based vaccines approved for use in veterinary practice for targeting canine melanoma (Merial), West Nile virus (Wyeth), fish hematopoietic necrosis virus (Novartis), and swine growth hormone–releasing hormone (Inovio). Research is still continuing to explore combining other vaccine platforms with DNA, enhanced methods for delivery, and new molecular adjuvants. The results of on-going clinical trials will be pivotal for providing insight into the progress of this platform and determining the impact of the technological advances integrated into the second-generation DNA platform.
Acknowledgments
Financial support. This work was supported by a grant from the National Institutes of Health (R01AI092843) and National Institute of Allergy and Infectious Diseases (PO1-AI071739).
Potential conflicts of interest. D. B. W. has grant funding and collaborations or funding by consulting including serving or chairing scientific advisory committees for commercial entities. Compensation from this work can include consulting fees or stock payments. In the interest of disclosure DBW therefore notes potential conflicts associated with this work with Pfizer, Bristol Myers Squibb, VIRxSYS, Ichor, Inovio, Merck, Althea, Aldevron, and possibly others. All other authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed in the Acknowledgments section.
References
- 1.Tang DC, DeVit M, Johnston SA. Genetic immunization is a simple method for eliciting an immune response. Nature. 1992;356:152–154. doi: 10.1038/356152a0. [DOI] [PubMed] [Google Scholar]
- 2.Ulmer JB, Donnelly JJ, Parker SE, et al. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science. 1993;259:1745–1749. doi: 10.1126/science.8456302. [DOI] [PubMed] [Google Scholar]
- 3.Wang B, Agadjanyan MG, Srikantan V, et al. Molecular cloning, expression, and biological characterization of an HTLV-II envelope glycoprotein: HIV-1 expression is permissive for HTLV-II-induced cell fusion. AIDS Res Hum Retroviruses. 1993;9:849–860. doi: 10.1089/aid.1993.9.849. [DOI] [PubMed] [Google Scholar]
- 4.Fynan EF, Webster RG, Fuller DH, Haynes JR, Santoro JC, Robinson HL. DNA vaccines: protective immunizations by parenteral, mucosal, and gene-gun inoculations. Proc Natl Acad Sci U S A. 1993;90:11478–11482. doi: 10.1073/pnas.90.24.11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacGregor RR, Boyer JD, Ugen KE, et al. First human trial of a DNA-based vaccine for treatment of human immunodeficiency virus type 1 infection: safety and host response. J Infect Dis. 1998;178:92–100. doi: 10.1086/515613. [DOI] [PubMed] [Google Scholar]
- 6.Wang R, Doolan DL, Le TP, et al. Induction of antigen-specific cytotoxic T lymphocytes in humans by a malaria DNA vaccine. Science. 1998;282:476–480. doi: 10.1126/science.282.5388.476. [DOI] [PubMed] [Google Scholar]
- 7.Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet. 2008;9:776–788. doi: 10.1038/nrg2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yager EJ, Dean HJ, Fuller DH. Prospects for developing an effective particle-mediated DNA vaccine against influenza. Expert Rev Vaccines. 2009;8:1205–1220. doi: 10.1586/erv.09.82. [DOI] [PubMed] [Google Scholar]
- 9.Roy MJ, Wu MS, Barr LJ, et al. Induction of antigen-specific CD8+ T cells, T helper cells, and protective levels of antibody in humans by particle-mediated administration of a hepatitis B virus DNA vaccine. Vaccine. 2000;19:764–778. doi: 10.1016/s0264-410x(00)00302-9. [DOI] [PubMed] [Google Scholar]
- 10.Mumper RJ, Cui Z. Genetic immunization by jet injection of targeted pDNA-coated nanoparticles. Methods. 2006;31:255–262. doi: 10.1016/s1046-2023(03)00138-5. [DOI] [PubMed] [Google Scholar]
- 11.Lisziewicz J, Calarota SA, Lori F. The potential of topical DNA vaccines adjuvanted by cytokines. Expert Opin Biol Ther. 2007;7:1563–1574. doi: 10.1517/14712598.7.10.1563. [DOI] [PubMed] [Google Scholar]
- 12.Okino M, Mohri H. Effects of a high-voltage electrical impulse and an anticancer drug on in vivo growing tumors. Jpn J Cancer Res. 1987;78:1319–1321. [PubMed] [Google Scholar]
- 13.Titomirov AV, Sukharev S, Kistanova E. In vivo electroporation and stable transformation of skin cells of newborn mice by plasmid DNA. Biochim Biophys Acta. 1991;1088:131–134. doi: 10.1016/0167-4781(91)90162-f. [DOI] [PubMed] [Google Scholar]
- 14.Rosati M, Valentin A, Jalah R, et al. Increased immune responses in rhesus macaques by DNA vaccination combined with electroporation. Vaccine. 2008;26:5223–5229. doi: 10.1016/j.vaccine.2008.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirao LA, Wu L, Khan AS, et al. Combined effects of IL-12 and electroporation enhances the potency of DNA vaccination in macaques. Vaccine. 2008;26:3112–3120. doi: 10.1016/j.vaccine.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otten G, Schaefer M, Doe B, et al. Enhancement of DNA vaccine potency in rhesus macaques by electroporation. Vaccine. 2004;22:2489–2493. doi: 10.1016/j.vaccine.2003.11.073. [DOI] [PubMed] [Google Scholar]
- 17.Hirao LA, Wu L, Satishchandran A, et al. Comparative analysis of immune responses induced by vaccination with SIV antigens by recombinant Ad5 vector or plasmid DNA in rhesus macaques. Mol Ther. 2010;18:1568–1576. doi: 10.1038/mt.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith LR, Wloch MK, Ye M, et al. Phase 1 clinical trials of the safety and immunogenicity of adjuvanted plasmid DNA vaccines encoding influenza A virus H5 hemagglutinin. Vaccine. 2010;28:2565–2572. doi: 10.1016/j.vaccine.2010.01.029. [DOI] [PubMed] [Google Scholar]
- 19.Boyer JD, Robinson TM, Kutzler MA, et al. SIV DNA vaccine co-administered with IL-12 expression plasmid enhances CD8 SIV cellular immune responses in cynomolgus macaques. J Med Primatol. 2005;34:262–270. doi: 10.1111/j.1600-0684.2005.00124.x. [DOI] [PubMed] [Google Scholar]
- 20.Xin KQ, Hamajima K, Sasaki S, et al. IL-15 expression plasmid enhances cell-mediated immunity induced by an HIV-1 DNA vaccine. Vaccine. 1999;17:858–866. doi: 10.1016/s0264-410x(98)00271-0. [DOI] [PubMed] [Google Scholar]
- 21.Egan MA, Chong SY, Megati S, et al. Priming with plasmid DNAs expressing interleukin-12 and simian immunodeficiency virus gag enhances the immunogenicity and efficacy of an experimental AIDS vaccine based on recombinant vesicular stomatitis virus. AIDS Res Hum Retroviruses. 2005;21:629–643. doi: 10.1089/aid.2005.21.629. [DOI] [PubMed] [Google Scholar]
- 22.Morrow MP, Yan J, Pankhong P, et al. Unique Th1/Th2 phenotypes induced during priming and Memory Phases using IL-12 or IL-28B vaccine adjuvants in rhesus macaques. Clin Vaccine Immunol. 2010;17:1493–1499. doi: 10.1128/CVI.00181-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrow MP, Yan J, Pankhong P, et al. IL-28B/IFN-lambda3 drives granzyme B loading and significantly increases CTL killing activity in macaques. Mol Ther. 2010;18:1714–1723. doi: 10.1038/mt.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai L, Vodros D, Kozlowski PA, et al. GM-CSF DNA: an adjuvant for higher avidity IgG, rectal IgA, and increased protection against the acute phase of a SHIV-89.6P challenge by a DNA/MVA immunodeficiency virus vaccine. Virology. 2007;369:153–167. doi: 10.1016/j.virol.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loudon PT, Yager EJ, Lynch DT, et al. GM-CSF increases mucosal and Systemic immunogenicity of an H1N1 influenza DNA vaccine administered into the Epidermis of non-human primates. PLoS One. 2010;5:e11021. doi: 10.1371/journal.pone.0011021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson HL, Montefiori DC, Villinger F, et al. Studies on GM-CSF DNA as an adjuvant for neutralizing Ab elicited by a DNA/MVA immunodeficiency virus vaccine. Vaccine. 2006;352:285–294. doi: 10.1016/j.virol.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Lambert LC, Fauci AS. Influenza vaccines for the future. N Engl J Med. 2010;363:2036–2044. doi: 10.1056/NEJMra1002842. [DOI] [PubMed] [Google Scholar]
- 28.Sheets RL, Stein J, Manetz TS, et al. Biodistribution of DNA plasmid vaccines against HIV-1, Ebola, severe acute respiratory syndrome, or West Nile virus is similar, without integration, despite differing plasmid backbones or gene inserts. Toxicol Sci. 2006;91:610–619. doi: 10.1093/toxsci/kfj169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ledwith BJ, Manam S, Troilo PJ, et al. Plasmid DNA vaccines: assay for integration into host genomic DNA. Dev Biol (Basel) 2000;104:33–43. [PubMed] [Google Scholar]
- 30.Wang Z, Troilo PJ, Wang X, et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004;11:711–721. doi: 10.1038/sj.gt.3302213. [DOI] [PubMed] [Google Scholar]
- 31.Tavel JA, Martin JE, Kelly GG, et al. Safety and immunogenicity of a Gag-Pol candidate HIV-1 DNA vaccine administered by a needle-free device in HIV-1-seronegative subjects. J Acquir Immune Defic Syndr. 2007;44:601–605. doi: 10.1097/QAI.0b013e3180417cb6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minor P. Vaccines against seasonal and pandemic influenza and the implications of changes in substrates for virus production. Clin Infect Dis. 2010;50:560–565. doi: 10.1086/650171. [DOI] [PubMed] [Google Scholar]
- 33.Laddy DJ, Yan J, Kutzler M, et al. Heterosubtypic protection against pathogenic human and avian influenza viruses via in vivo electroporation of synthetic consensus DNA antigens. PLoS One. 2008;3:e2517. doi: 10.1371/journal.pone.0002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei CJ, Boyington JC, Dai K, et al. Cross -neutralization of 1918 and 2009 influenza viruses: role of glycans in viral evolution and vaccine design. Sci Transl Med. 2010;24ra21 doi: 10.1126/scitranslmed.3000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. NIH Clinical trials database. http://clinicaltrials.gov/ct2/results?term=influenza+dna. Accessed April 2011. [Google Scholar]
- 36.Evans K, McElwaine-Johnn H, Sharpe M, et al. DNA vaccination protects against an influenza challenge in a double-blind randomised placebo-controlled phase 1b clinical trial. Vaccine. 2009;27:2506–12. doi: 10.1016/j.vaccine.2009.02.061. [DOI] [PubMed] [Google Scholar]
- 37.World Health Organization. GLOBOCAN. 2008. Cervical cancer incidence and mortality worldwide in 2008. http://globocan.iarc.fr/factsheets/cancers/cervix.asp. Accessed 10 May 2010. [Google Scholar]
- 38.Lin K, Roosinovich E, Ma B, Hung CF, Wu TC. Therapeutic HPV DNA vaccines. Immunol Res. 2010;47:86–112. doi: 10.1007/s12026-009-8141-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan J, Harris K, Khan AS, Draghia-Akli R, Sewell D, Weiner DB. Cellular immunity induced by a novel HPV18 DNA vaccine encoding an E6/E7 fusion consensus protein in mice and rhesus macaques. Vaccine. 2008;26:5210–5215. doi: 10.1016/j.vaccine.2008.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan J, Reichenbach DK, Corbitt N, et al. Induction of antitumor immunity in vivo following delivery of a novel HPV-16 DNA vaccine encoding an E6/E7 fusion antigen. Vaccine. 2009;27:431–440. doi: 10.1016/j.vaccine.2008.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheets EE, Urban RG, Crum CP, et al. Immunotherapy of human cervical high-grade cervical intraepithelial neoplasia with microparticle-delivered human papillomavirus 16 E7 plasmid DNA. Am J Obstet Gynecol. 2003;188:916–926. doi: 10.1067/mob.2003.256. [DOI] [PubMed] [Google Scholar]
- 42.Klencke B, Matijevic M, Urban RG, et al. Encapsulated plasmid DNA treatment for human papillomavirus 16-associated anal dysplasia: a phase I study of ZYC101. Clin Cancer Res. 2002;8:1028–1037. [PubMed] [Google Scholar]
- 43.Garcia F, Petry KU, Muderspach L, et al. ZYC101a for treatment of high-grade cervical intraepithelial neoplasia: a randomized controlled trial. Obstet Gynecol. 2004;103:317–326. doi: 10.1097/01.AOG.0000110246.93627.17. [DOI] [PubMed] [Google Scholar]
- 44.Trimble CL, Peng S, Kos F, et al. A phase I trial of a human papillomavirus DNA vaccine for HPV16+ cervical intraepithelial neoplasia 2/3. Clin Cancer Res. 2009;15:361–367. doi: 10.1158/1078-0432.CCR-08-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pitisuttithum P, Gilbert P, Gurwith M, et al. Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J Infect Dis. 2006;194:1661–1671. doi: 10.1086/508748. [DOI] [PubMed] [Google Scholar]
- 46.Buchbinder SP, Mehrotra DV, Duerr A, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372:1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–2210. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 48.Wang S, Kennedy JS, West K, et al. Cross-subtype antibody and cellular immune responses induced by a polyvalent DNA prime-protein boost HIV-1 vaccine in healthy human volunteers. Vaccine. 2008;26:3947–3957. doi: 10.1016/j.vaccine.2007.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Catanzaro AT, Koup RA, Roederer M, et al. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 candidate vaccine delivered by a replication-defective recombinant adenovirus vector. J Infect Dis. 2006;194:1638–1649. doi: 10.1086/509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vaine M, Wang S, Hackett A, Arthos J, Lu S. Antibody responses elicited through homologous or heterologous prime-boost DNA and protein vaccinations differ in functional activity and avidity. Vaccine. 2010;28:2999–3007. doi: 10.1016/j.vaccine.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Safety and effectiveness of HIV-1 DNA plasmid vaccine and HIV-1 recombinant adenoviral vector vaccine in HIV-uninfected, circumcised men and male-to-female (MTF) transgender persons who have sex with men. 2010 http://www.clinicaltrials.gov/ct2/show/NCT00865566?term=Ad5+DNA&rank=3. Accessed 12 June 2010. [Google Scholar]
- 52.Safety and effectiveness of PENNVAX-B vaccine alone, with Il-12, or IL15 cell, 15 in healthy adults. 2010 NIH clinicaltrials.gov. http://clinicaltrials.gov/ct2/show/NCT00528489. Accessed 10 May 2010. [Google Scholar]
- 53.Kraynyak KA, Kutzler MA, Cisper NJ, et al. Systemic immunization with CCL27/CTACK modulates immune responses at mucosal sites in mice and macaques. Vaccine. 28:1942–1951. doi: 10.1016/j.vaccine.2009.10.095. [DOI] [PMC free article] [PubMed] [Google Scholar]