

Abstract
Chirality can be used as a design tool to control the mechanical rigidity of hydrogels formed from self-assembling peptides. Hydrogels prepared from enantiomeric mixtures of self-assembling β-hairpins show non-additive, synergistic, enhancement in material rigidity compared to gels prepared from either pure enantiomer, with the racemic hydrogel showing the greatest effect. CD spectroscopy, TEM, and AFM indicate that this enhancement is defined by nanoscale interactions between enantiomers in the self-assembled state.
Keywords: Hydrogel, Peptide, Self-Assembly, Chirality, Enantiomers
As an emerging class of biomaterials, hydrogels prepared from self-assembling peptides1-8 have gained special interest in the drug delivery and tissue engineering fields, in part, due to their ability to be naturally degraded by proteolytic enzymes9-11. One logical way to control the rate of protease-mediated degradation is to simply dope the naturally occurring L-isomeric peptide with its D-enantiomer, which is incapable of being proteolyzed 12. In the course of applying this methodology, we discovered a large, non-additive, synergistic enhancement of the mechanical rigidity of gels prepared from enantiomeric mixtures of self-assembling peptides, with the racemate displaying the greatest degree of enhancement. To our knowledge, this phenomenon is unprecedented in the literature and may represent a new design modality for the preparation of self-assembling materials.
MAX1 is a twenty amino acid peptide that undergoes triggered β-hairpin folding and self-assembles into a structurally well-defined fibril network that leads to the formation of a mechanically rigid hydrogel 13, 14, Figure 1. MAX1 is composed of N- and C-terminal β-stands containing alternating hydrophobic (valine) and hydrophilic (lysine) residues. A central four residue sequence (-VDPPT-) connects the two stands and is designed to adopt a type II’ β-turn when folding is triggered. Hydrogel formation is initiated with temporal resolution by controlling the folded state of the peptide. At neutral pH and low ionic strength, electrostatic repulsion between protonated lysine side chains keeps the peptide unfolded. Increasing the ionic strength with NaCl to 150 mM screens the positive charge, allowing the peptide to fold into a facially amphiphilic β-hairpin. Once folded, these peptides are designed to self assemble into a β-sheet rich network of fibrils where each fibril is composed of a bilayer of folded hairpins that have hydrogen-bonded along the fibril long axis. The resulting network of fibrils constitutes a self-supporting hydrogel 15-17.
Figure 1.
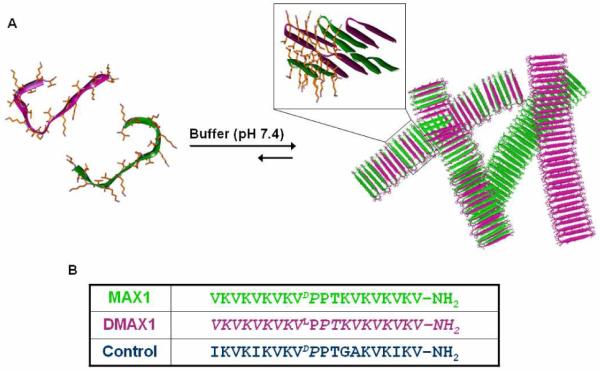
(A) Assembly mechanisms for enantiomeric peptides leading to the formation of a fibrillar network that defines hydrogelation. Enantiomers can either self-sort to form fibrils that are homogeneous with respect to enantiomer (solid colored fibrils) or enantiomers can co-assemble to form a network of fibrils that are heterogeneous with respect to enantiomer (multi-colored fibrils). (B) Sequences of enantiomers MAX1, DMAX1 and the non-isomeric Control Peptide. D-amino acid residues are italicized.
The enantiomer of MAX1, namely DMAX1 (Figure 1B) was initially prepared18 for doping experiments intended to control the enzymatic susceptibility of the fibril network. DMAX1 alone undergoes triggered β-hairpin folding and self-assembly affording hydrogels rich in β-sheet structure in an identical manner to MAX1. For example, Figure 2A shows a circular dichroism spectrum for a 1 wt % gel formed by MAX1 (open squares) that displays a minimum in mean residue ellipticity at 216 nm ([θ]216), indicative of β-sheet struc ture19. Also shown is the spectrum for a gel prepared from pure enantiomer DMAX1 (filled diamonds). As expected, the spectrum is the perfect mirror image of its enantiomeric MAX1 gel. Hydrogels prepared from 3:1 and 1:3 MAX1:DMAX1 molar ratios show symmetrical spectra with reduced negative and positive values of [θ]216 values, respectively. For the racemic hydrogel formed from equal amounts of MAX1 and DMAX1, each enantiomer absorbs equal amounts of circularly polarized light resulting in the baseline spectrum evident in Figure 2A. Since the CD spectrum of the racemic gel is uninformative, FTIR spectroscopy was used to confirm the β-sheet structure within the fibrils that define this hydrogel. The spectrum shown in the inset of Figure 2A shows characteristic absorptions due to anti-parallel β-sheet secondary structure at 1615 and 1680 cm−1 20, thus verifying the β-sheet nature of the network.
Figure 2.

(A) CD wavelength spectra of 1 wt % hydrogels containing pure MAX1 (□), 3:1 MAX1:DMAX1 (▲), 1:1 MAX1:DMAX1 (●), 1:3 MAX1:DMAX1 (▼), and pure DMAX1 (◆) (inset) IR spectrum of 1:1 MAX1:DMAX1 gel (B) Dynamic time sweep rheological data measuring the storage moduli of 1 wt % hydrogels containing pure MAX1 (□), 3:1 MAX1:DMAX1 (▲), 1:1 MAX1:DMAX1 (●), 1:3 MAX1:DMAX1 (▼), and pure DMAX1 (◆). (C) Storage moduli of 1 wt % hydrogels composed of varying mole fractions of peptide enantiomers (N=3).
As a consequence of their enantiomeric relationship, both MAX1 and DMAX1 independently form hydrogels with nearly identical mechanical properties. Figure 2B shows oscillatory rheology data that reports on the mechanical rigidity of pure MAX1 and DMAX1 gels as well as composite gels formed by mixing different molar ratios of the two enantiomers. Here, time sweep experiments were performed that monitor the evolution of the storage modulus (G’, a measure of the material’s mechanical rigidity) as a function of time after folding, self-assembly and ultimate gelation is initiated by the addition of saline buffer to a solution of unfolded peptide. 1 wt % solutions of pure MAX1 or DMAX1 form gels within minutes that cure over a time period of two hours with ultimate storage moduli that are nearly equivalent (200 Pa). Surprisingly, when a 1 wt % solution of an equal molar mixture of enantiomers is triggered, gelation occurs more rapidly resulting in a network whose rigidity is over four-fold greater (800 Pa) than gels prepared from either pure enantiomer. Keeping in mind that the total amount of peptide in the racemic gel is equal to that of either pure enantiomeric gel, this result was truly unexpected. This observation suggests that biomolecular chiralty may represent a new tool for materials design and can be used to influence the mechanical properties of self-assembled materials. This effect can be controllably modulated by adjusting the molar ratio of β-hairpin enantiomers. For example, hydrogels prepared from 3:1 and 1:3 MAX1:DMAX1 molar ratios have nearly identical values of G’ that are intermediate between the racemic hydrogel and the gels formed by either pure enantiomer. The titratable character of this material enhancement effect is exemplified in Figure 2C where the storage modulus is plotted with respect to the mole fraction of DMAX1 relative to MAX1 for five independent gels. The Job plot shows a clear maximum at 0.5 mole fraction with excellent symmetry.
A control peptide was designed to investigate whether the cause of the enhancement in material rigidity is truly dictated by the specific chiral relationship between MAX1 and DMAX1. It may be possible that any two peptides of opposite chirality but differing sequence, and thus not strictly related enantiomerically, can produce the same effect. The control peptide was carefully designed to undergo triggered folding and self-assembly at a rate similar to both pure MAX1 and DMAX1 enantiomers and to form a gel that provides a rigidity nearly identical to that of both pure enantiomeric gels. This was accomplished by altering the identity of residues on the hydrophobic face of MAX1 while keeping the sequence length and gross overall chirality the same (Figure 1B). If the exact sequence of MAX1 and DMAX1 is unimportant and only the gross chiralty of the peptides is at play, then a gel formed from equimolar mixtures of the Control and DMAX1 should also display an enhancement in material rigidity. Oscillatory rheology reveals that a 1 wt % hydrogel composed of a 1:1 molar ratio of Control:DMAX1 shows no large enhancement in material rigidity, Figure S7. This simple experiment clearly illustrates that the specific chiral relationship between MAX1 and DMAX1 as enantiomers is important to the observed enhancement in material rigidity.
Although a molecular level understanding of this enhancement is not yet known, nano-scale interrogation of local fibril morphology via Transmission Electron Microscopy (TEM) and Atomic Force Microscopy (AFM) show that the racemic fibrils are mophologically similar to the fibrils formed by either pure enantiomer. Figure 3A shows a representative TEM image of fibrils from a racemic network. The local fibril morphology is well-defined, having ~3 nm widths that are in agreement with the model of self-assembly presented in Figure 1 where the fibril width is defined by the length of a folded hairpin in the self-assembled state. AFM was used to measure a fibril height of ~2 nm, which is consistent with the height of a hairpin bilayer formed by the hydrophobic collapse of two hairpins during self-assembly, Figures 1, 3B. TEM and AFM images of fibrils obtained from gels of pure enantiomers show similar morphologies (Supporting Information). The microscopy data of fibrils from pure and racemic gels indicates that the enhancement in material rigidity is not due to any gross differences in local fibril morphology.
Figure 3.
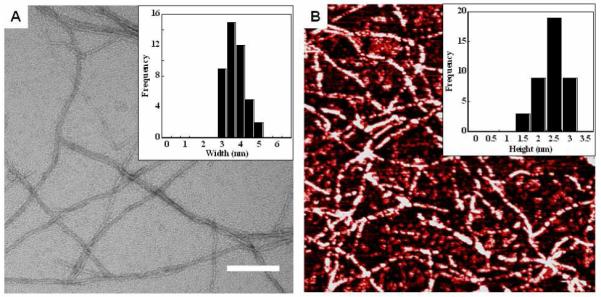
(A) TEM of fibrils obtained from a 1 wt % racemic gel. Scale bar = 100 nm (inset) Multiple fibril width measurements (N=43) reveal average widths of 2.5-3 nm. (B) AFM revealing an average fiber height of 2.5 nm from multiple measurements (N=40). A 1 μm × 1 μm square is shown.
Taken together, the spectroscopy, rheology and microscopy data suggests that the cause of the enhancement in mechanical properties is due to energetically favorable interactions between enantiomers 21,22. Interactions between enantiomers can occur at the molecular level and/or at the hydrogel network level, both scenarios are depicted in Figure 1. For example, if favorable molecular level interactions between enantiomers guides self-assembly during fibril formation, then a hydrogel network composed of heterogeneous fibrils will be obtained; in this scenario, each fibril contains both enantiomers, Figure 1 (multicolored fibrils). However if the interactions between enantiomers are at the hydrogel network level, then a system could be envisioned where the enantiomers self-segregate23,24, forming homogenous fibrils composed of only one enantiomer. Inter-enantiomer contact could then be made across distinct fibrils in the network, Figure 1 (solid colored fibril). This scenario is much like that observed for classical interpenetrating networks formed by synthetic polymers25,26. Interestingly, IR spectra of pure enantiomeric gels and the racemic hydrogel are nearly identical suggesting that the extended secondary structure formed in all these formulations is similar irrespective of the exact nature of the network, Figure S8. We are currently working towards a molecular and network level understanding of the structure and the interactions responsible for the enhancement in material rigidity, which will be reported in due course.
Stereocomplexation can be used to drive gel formation of synthetic polymers27,28. The use of chiralty in designing self-assembled biomaterials has largely centered on controlling the proteolytic susceptibility of implantable scaffolds. The work presented in this communication suggests that the impact of chirality may reach far beyond this immediate use. One can envision using enantiomers, diastereomers, meso compounds and racemates of self-assemblers to control the mechanistic pathways by which molecules assemble to produce novel shapes, network morphologies and material properties.
Supplementary Material
ACKNOWLEDGMENT
The authors would like to acknowledge Ulrich Baxa for his work with TEM imaging and for intellectual conversations.
Funding Sources This work was supported by the Intramural Research Program of the National Cancer Institute of the National Institutes of Heath.
Footnotes
ASSOCIATED CONTENT
Supporting Information. HPLC, mass and IR spectra, rheology, TEM and AFM images of peptides used in this study. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- (1).Branco MC, Sigano DM, Schneider JP. Curr. Opin. Chem. Biol. 2011;15:427–434. doi: 10.1016/j.cbpa.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bowerman CJ, Nilsson BL. J. Am. Chem. Soc. 2010;132:9526–9527. doi: 10.1021/ja1025535. [DOI] [PubMed] [Google Scholar]
- (3).Collier JH, Hu BH, Ruberti JW, Zhang J, Shum P, Thompson DH, Messersmith PB. J. Am. Chem. Soc. 2001;123:9463–9464. doi: 10.1021/ja011535a. [DOI] [PubMed] [Google Scholar]
- (4).Kopecek J, Yang JY. Acta Biomater. 2009;5:805–816. doi: 10.1016/j.actbio.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ryadnov MG, Bella A, Timson S, Woolfson DN. J. Am. Chem. Soc. 2009;131:13240. doi: 10.1021/ja905539h. + [DOI] [PubMed] [Google Scholar]
- (6).Pires MM, Przybyla DE, Chmielewski J. Angew. Chem.-Int. Edit. 2009;48:7813–7817. doi: 10.1002/anie.200902375. [DOI] [PubMed] [Google Scholar]
- (7).Kyle S, Aggeli A, Ingham E, McPherson MJ. Biomaterials. 2010;31:9395–9405. doi: 10.1016/j.biomaterials.2010.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gao Y, Kuang Y, Guo ZF, Guo ZH, Krauss IJ, Xu BJ. Am. Chem. Soc. 2009;131:13576–13577. doi: 10.1021/ja904411z. [DOI] [PubMed] [Google Scholar]
- (9).Giano MC, Pochan DJ, Schneider JP. Biomaterials. 2011;32:6471–6477. doi: 10.1016/j.biomaterials.2011.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Galler KM, Aulisa L, Regan KR, D’Souza RN, Hartgerink JD. J. Am. Chem. Soc. 132:3217–3223. doi: 10.1021/ja910481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chau Y, Luo Y, Cheung ACY, Nagai Y, Zhang SG, Kobler JB, Zeitels SM, Langer R. Biomaterials. 2008;29:1713–1719. doi: 10.1016/j.biomaterials.2007.11.046. [DOI] [PubMed] [Google Scholar]
- (12).Luo ZL, Zhao XJ, Zhang SG. Macromol. Biosci. 2008;8:785–791. doi: 10.1002/mabi.200800003. [DOI] [PubMed] [Google Scholar]
- (13).Schneider JP, Pochan DJ, Ozbas B, Rajagopal K, Pakstis L, Kretsinger J. J. Am. Chem. Soc. 2002;124:15030–15037. doi: 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- (14).Ozbas B, Kretsinger J, Rajagopal K, Schneider JP, Pochan DJ. Macromolecules. 2004;37:7331–7337. [Google Scholar]
- (15).Branco MC, Nettesheim F, Pochan DJ, Schneider JP, Wagner NJ. Biomacromolecules. 2009;10:1374–1380. doi: 10.1021/bm801396e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Yucel T, Micklitsch CM, Schneider JP, Pochan DJ. Macromolecules. 2008;41:5763–5772. doi: 10.1021/ma702840q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hule RA, Nagarkar RP, Hammouda B, Schneider JP, Pochan DJ. Macromolecules. 2009;42:7137–7145. doi: 10.1021/ma9003242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nagarkar RP, Schneider JP. Methods Mol. Biol. 2008;474:61–77. doi: 10.1007/978-1-59745-480-3_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Johnson WC. Proteins. 1990;7:205–214. doi: 10.1002/prot.340070302. [DOI] [PubMed] [Google Scholar]
- (20).Kubelka J, Keiderling TA. J. Am. Chem. Soc. 2001;123:12048–12058. doi: 10.1021/ja0116627. [DOI] [PubMed] [Google Scholar]
- (21).Brizard A, Oda R, Huc I. Low Molecular Mass Gelators: Design, Self-Assembly, Function. Vol. 256. Springer-Verlag Berlin; Berlin: 2005. Chirality effects in self-assembled fibrillar networks; pp. 167–218. [DOI] [PubMed] [Google Scholar]
- (22).Smith DK. Chem. Soc. Rev. 2009;38:684–694. doi: 10.1039/b800409a. [DOI] [PubMed] [Google Scholar]
- (23).Messmore BW, Sukerkar PA, Stupp SI. J. Am. Chem. Soc. 2005;127:7992–7993. doi: 10.1021/ja051183y. [DOI] [PubMed] [Google Scholar]
- (24).Makarevic J, Jokic M, Raza Z, Stefanic Z, Kojic-Prodic B, Zinic M. Chem.-Eur. J. 2003;9:5567–5580. doi: 10.1002/chem.200304573. [DOI] [PubMed] [Google Scholar]
- (25).Sperling LH. Interpenetrating Polymer Networks - an Overview. In: Klempner D, Sperling LH, Utracki LA, editors. Interpenetrating Polymer Networks. Vol. 239. Amer Chemical Soc; Washington: 1994. pp. 3–38. [Google Scholar]
- (26).Kostanski LK, Huang RX, Filipe CDM, Ghosh RJ. Biomater. Sci.-Polym. Ed. 2009;20:271–297. doi: 10.1163/156856208X3999107. [DOI] [PubMed] [Google Scholar]
- (27).Hiemstra C, Zhong Z, Li L, Dijkstra PJ, Feijen J. Biomacromolecules. 2006;7:2790–2795. doi: 10.1021/bm060630e. [DOI] [PubMed] [Google Scholar]
- (28).Yu JM, Jerome R. Macromolecules. 1996;29:8371–8378. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.