

Soon the genetic basis of most human Mendelian diseases will be solved. The next challenge will be to leverage this information to uncover basic mechanisms of disease and develop new therapies. To understand how this transformation is already beginning to unfold, we focus on the ciliopathies, a class of multi-organ diseases caused by disruption of the primary cilium. Through a convergence of data involving mutant gene discovery, proteomics and cell biology, over a dozen phenotypically distinguishable conditions are now united as ciliopathies. Sitting at the interface between simple and complex genetic conditions, these diseases provide clues to the future direction of human genetics.
Until a few years ago, identifying the genetic basis of an inherited human disease was an arduous undertaking, requiring potentially a decade or more of work in ascertainment of families for linkage analysis, followed by endless fine mapping of the locus, and finally sequencing of candidate genes one-by-one until that eureka moment when the likely causative gene was identified. The newly discovered disease gene was often entirely novel, without recognizable domains or a path to understand the disease mechanism. A mouse model was then generated, in which the disease gene was inactivated. In some cases, the mouse faithfully recapitulated the human phenotype, but more often showed no phenotype or phenotypes not clearly related to the human disease. Once established, the model was studied from multiple perspectives to understand the cell biological and biochemical basis of disease, culminating in attempts to test potential therapies. Although successful in a few instances such as losartan treatment for Marfan syndrome (Habashi et al., 2006), this path has not fulfilled the promises of genomic medicine.
This strategy has begun to change over the past ten years due to increased knowledge of human genetic diseases, annotation of the human genome, and an amazing suite of tools to explore disease mechanisms. It is not uncommon now to open up a journal to find that geneticists have solved the molecular basis of a dozen or more conditions. And since we now know a lot more about the function of genes, protein domains, and networks, frequently just the discovery of the molecular cause of a disease can often partially explain its mechanism. For instance, the discovery that the Rett syndrome gene encodes a methyl-CpG-binding protein (Amir et al., 1999) immediately set the stage for a host of important discoveries in epigenetics related to brain function. The types of mutations displayed by patients, known as allelic diversity (Fig. 1), can tell us something about the effect of these disease-causing variants on protein function. By identifying patients with different phenotypes due to specific types of mutations in the same gene (i.e. genocopies) we can understand human disease as a network of related signs and symptoms. For example, specific types of mutations in the gene encoding p53 predispose to very different types of cancers. By comparing the genes mutated in phenotypically related human diseases, we can learn about the disturbed protein networks that underlie them. Finally, by exploring gene-gene and gene-environment interactions, we can begin to characterize genetic and epigenetic modifiers of disease. Perhaps the best example is age-related macular degeneration, in which a substantial part of the risk of disease can be quantified based on gene-environment interactions (Chen et al., 2010).
Figure 1. Human genetic interactions in the ciliopathy spectrum.

Diseased individuals are in color, with severity represented by darker shading. Phenocopies refers to the finding that patients with homozygous or compound heterozygous mutations in two different genes (i.e. AHI1 or INPP5E) can show indistinguishable JBTS phenotypes. Multiple allelism at the same locus indicates that mutations in a single gene (i.e. CEP290) can lead to various distinguishable phenotypes. Modifiers refers to evidence that potentially deleterious sequence changes in a gene like AHI1 can modify in quantifiable ways the phenotype observed in patients with NPHP1 mutations. Black: normal chromosome; Red: mutant chromosome. AHI1: abelson helper integration site 1; INPP5E: inositol polyphosphate-5-phosphatase E; CEP290: centrosomal protein 290kDa; NPHP1: nephrocystin 1. BBS: Bardet-Biedl syndrome; JBTS: Joubert syndrome; LCA: Leber congenital amaurosis; MKS: Meckel syndrome; NPHP: nephronophthisis; RD: retinal dystrophy; SLS: Senior Loken syndrome.
The ciliopathies: one organelle, many disorders
Although they are individually rare conditions, the ciliopathies have emerged as a dynamic new field of biology that exemplifies how genetics can be employed to drive research in basic cell biology and vice versa. The primary cilium is structured with a basal body at its base and a 9-paired microtubule axoneme, surrounded by plasma membrane, but lacking the central pair of microtubules and outer dynein arms that define its cousin, the motile cilium. Primary cilia were first observed more than a century ago and were initially thought to be evolutionary remnants. How is it that biologists missed their importance for so long?
Over a dozen disorders are now considered to be within the ciliopathy spectrum including Joubert syndrome (JBTS), nephronophthisis (NPHP), Senior-Loken syndrome (SLS), orofaciodigital (OFD), Jeune syndrome, autosomal dominant and recessive polycystic kidney disease (ADPKD and ARPKD), Leber congenital amaurosis (LCA), Meckel-Gruber syndrome (MKS), Bardet-Biedl syndrome (BBS), Usher syndrome (US) and some forms of retinal dystrophy (RD). Between them, these conditions involve nearly every major body organ including kidney, brain, limb, retina, liver, and bone (Fig. 2A), highlighting the important role of the primary cilium in development and homeostasis. These conditions were largely defined by clinical geneticists in the middle of the last century, who did their best to ascribe syndromes to unique combinations of clinical features. Individual diseases are known for the most commonly involved or diseased organ: BBS patients display the triad of obesity, polydactyly and retinopathy but can display a host of other pathologies. MKS is a lethal condition at birth with occipital encephalocele, PKD and polydactyly. JBTS is characterized by a very peculiar radiographic finding, known as the “molar tooth sign”, characterized by elongated superior cerebellar peduncles, deepened interpeduncular fossa and cerebellar vermis hypoplasia. For each of these conditions, significant phenotypic variability has been observed even between members of the same family, making clinical diagnosis a challenge.
Figure 2. Phenotypic and interactome diversity of the ciliopathies based upon major organ involvement.
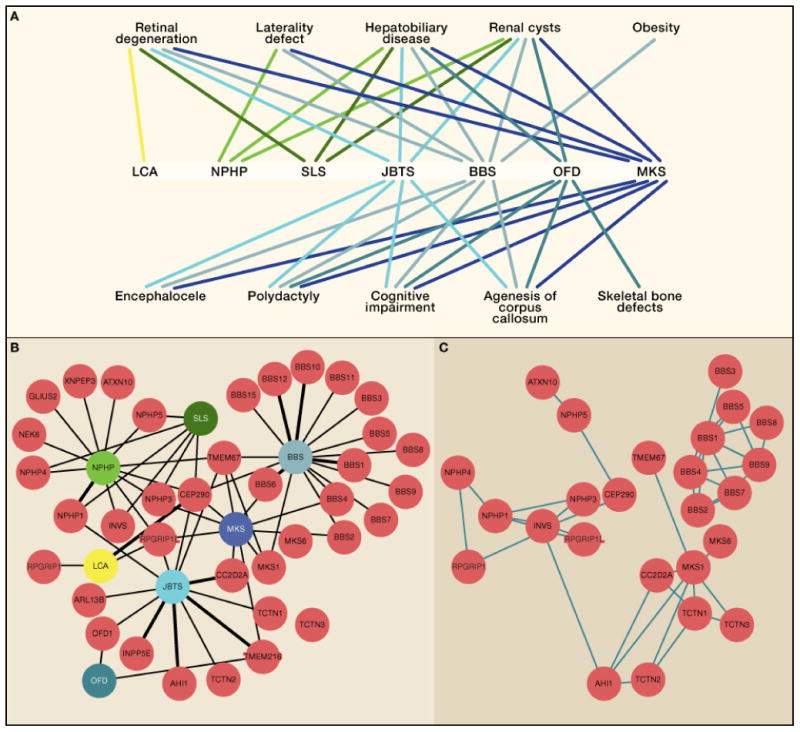
A. Disease is represented below by abbreviation, and involvement of major organ listed above. B. Major ciliopathy diseases (color coded by severity), and gene mutated in each condition (red) linked by black bar with more common causes showing thicker lines. C. The same gene map, now indicating evidence for direct interaction between protein products. Protein interaction networks identified from published data demonstrating major clustering of interactions corresponding to disease networks. Note that genes causing a particular disorder tend to have products that interact, although there are many genes without known connections. Note that some genes like CEP290, which can cause several different diseases, should serve as hubs, but have few demonstrated physical interactions to date. BBS: Bardet-Biedl syndrome; JBTS: Joubert syndrome; LCA: Leber congenital amaurosis; MKS: Meckel syndrome; NPHP: nephronophthisis; OFD: Orofaciodigital syndrome; SLS: Senior Loken syndrome.
Now that these conditions have been united by their underlying cell biology, we can begin to see commonalities between individual syndromes (Fig. 2A). For instance, low muscle tone, cystic kidney disease, agenesis of the brain’s corpus callosum, mental retardation and hyperpnea/apnea are additional clinical features often present in ciliopathy patients. Individuals affected by BBS can have several clinical features common to JBTS such as mental retardation, hypotonia and apnea. However, they also share other symptoms that are usually not present in JBTS, but common to other ciliopathies, such as polydactyly and retinal dystrophy (RD). Moreover, BBS patients are usually obese, a unique trait absent among the other ciliopathies.
The first few genes identified from positional cloning of human or mouse phenotypes for what would eventually become the ciliopathies initially did not point in an obvious way to the cilium as the site of action. It was not until evidence began to accumulate that the encoded proteins localize specifically near the cilium that the field was born (Ansley et al., 2003; Barr and Sternberg, 1999; Kim et al., 2004; Otto et al., 2003; Taulman et al., 2001). Although the localization data is now incontrovertible, the functions of the encoded proteins at the cilium for the most part still remain a mystery, and some of the effects of these proteins do not seem to have direct relevance to ciliary function (Yen et al., 2006). The question that emerges is how a single subcellular organelle can mediate such diverse clinical features.
About 50 genes encoding predominantly ciliary-localized proteins have now been identified that are mutated in these partially overlapping syndromes (Fig. 2B). In a series of positional cloning studies, each of these ciliopathy genes was initially found as causative for a restricted phenotype. Surprisingly, in most instances, this gene identification was followed by reports of mutations in the same gene in a different ciliopathy category (Baala et al., 2007a; Baala et al., 2007b; den Hollander et al., 2006). This occurred with such regularity that the field began to wonder if any genotype-phenotype correlations would stand the test of time. How could mutations in a single gene produce such pleiotropic phenotypic effects in patients? Were these observations exceptions to the rule of strict genotype-phenotype correlation, or were they the exception that proves the rule of widespread phenotypic pleiotropy?
The primary cilium network
The primary cilium is a hair-like, immotile cellular organelle protruding from almost all eukaryotic cells, frequently described as the cell’s “antenna” for transducing extracellular signals. For the purposes of this review, we focus exclusively on diseases involving non-motile cilia, and do not include diseases like primary cilia dyskinesia that involve motile cilia, since there is little phenotypic or genetic overlap. Cilia are generated during interphase from the mother centriole, by coalescence of vesicles at its distal end that fuse with the plasma membrane. After this docking of the mother centriole, the microtubule axoneme protrudes out of the cell, concurrent with recruitment of a host of ciliary-specific proteins. The basal body (i.e. the docked mother centriole) possesses several specialized accessory structures termed transition fibers, basal feet and ciliary rootlets, and is surrounded by the pericentriolar matrix, but for many years the molecular determinants of these structures were unknown.
Recent work has begun to hint at the molecular architecture of these anatomically defined structures. The 9+0 microtubule arrangement of the axoneme emerges from the basal body in a triplet configuration, and shifts to a doublet configuration at the ciliary transition zone (TZ). The Y-shaped microtubule extensions that define the TZ require the Cep290 protein for their formation in Chlamydamonas (Craige et al., 2010), although it is not clear if Cep290 constitutes or otherwise contributes to these structures, or if this function is conserved in vertebrates. The location of the TZ marks the ciliary diffusion barrier; a septin-2 cytoskeleton which separates the ciliary and cytoplasmic compartments (Hu et al., 2010). Attention has shifted to the TZ as the site of action of proteins mutated in several ciliopathies (Garcia-Gonzalo et al., 2011), but clear structure-function relationships are still lacking. Protein synthesis and vesicular transport do not occur inside cilia, and so the assembly of this organelle, its maintenance and function are totally dependent upon an intraflagellar transport system, by which proteins track bidirectionally along the polarized microtubules of the axoneme (Kozminski et al., 1993).
Although the involvement of primary cilia in human diseases is now well established, there are many questions about its function that remain. The current paradigm describes the primary cilium as an organelle for detecting and modulating the response to extracellular signaling molecules and as a location for organizing their cytoplasmic effectors. It is well poised to mediate both effects, as cilia typically protrude in a polarized fashion, which can help the cell interpret the context of extracellular signals. Many cellular receptors are localized to the primary cilium, and the basal body is itself a signaling hub, probably serving as an efficient transit point to transmit signals into the nucleus. In fact, a number of transcription factors undergo processing within or near the cilium prior to nuclear entry. Numerous critical developmental signaling pathways have been directly linked to primary cilia, such as Hedgehog (Hh), canonical and non-canonical Wnt and some forms of PDGF signaling, highlighting its role as a hub (Huangfu et al., 2003; Schneider et al., 2005; Simons et al., 2005). In addition to the modulation of these signaling pathways, primary cilia are essential for mechanical, odor and photoreception. The interpretation of the ciliopathies as a unique group of disorders, associated with defects in a single organelle gave a new direction to the investigation of these human diseases.
Proteomics merges with genomics
The marriage of proteomics with genomics in the area of ciliary biology can be traced to an influential paper showing that the RFX-type transcription factor DAF-19 is essential for assembly of cilia in C. elegans sensory neurons, and regulates several genes encoding intraflagellar transport proteins (Swoboda et al., 2000). The RFX transcription factor family emerged in ciliated proto-eukaryotes but it was only later that the RFX genes were co-opted to regulate expression of cilia-specific genes based upon the presence of an X-box in their promoter (Piasecki et al., 2010). This work set the stage for a comparative genomics approach to search for X-box containing genes that were likely to encode proteins relevant to primary cilia. Two follow-up studies demonstrated the potential of this approach in identifying important components of the primary cilium (Avidor-Reiss et al., 2004; Li et al., 2004) and raised the idea of building a ciliary protein database. The ciliome (Gherman et al., 2006; Inglis et al., 2006), now consists of over 3000 genes encoding proteins either localized to cilia or essential for their assembly or function (Arnaiz et al., 2009).
The ciliary proteome has already proved to be a powerful resource, accelerating the identification of candidate human ciliopathy genes by short-listing positional candidates. For example, the cloning of MKS1, BBS3, BBS5 and the BBS modifier MGC1203 was achieved by sequencing a reduced set of candidate genes (Badano et al., 2006; Chiang et al., 2004; Kyttala et al., 2006; Li et al., 2004). Extending the idea of a ciliary proteome to a ciliary “interactome” was the natural extension of this work, through the identification of proteins that physically interact as part of specific complexes (Eley et al., 2008; Gorden et al., 2008). Through pair-wise testing of potential yeast-two-hybrid interactions (Otto et al., 2005) and identification of binding partners by serial mass spectrometry (Nachury et al., 2007; Sang et al., 2011), a number of discrete, functionally relevant complexes have now emerged as the likely minimal disease-causing modules (Fig. 2B).
An important observation, which has stood the test of time, is that the composition of these specific protein modules could have been predicted based upon the phenotype observed in patients. Specifically, the proteins from seven of the eight most conserved genes mutated in BBS form a core complex termed the BBSome (Nachury et al., 2007), found to play important roles in ciliary protein and vesicular transport. Importantly, this complex does not contain proteins encoded by other ciliopathy diseases genes such as NPHP, JBTS or MKS. This is perhaps surprising considering that mutations in MKS genes can cause BBS (Leitch et al., 2008). Three separate complexes containing many of the proteins mutated in NPHP, JBTS and MKS were recently identified, and although their function is still under investigation, genes for two of the co-purifying proteins, ATXN10 and TCTN2 were found to be mutated in patients matching the same “module” phenotype (Sang et al., 2011). In general, the complexes also display specificity in subcellular localization and function: the MKS/JBTS complex transduces hedgehog signaling and localizes to the ciliary transition zone, whereas the BBS complex forms a coat complex to target vesicles to the cilium, and localizes to ciliary membrane (Jin et al., 2010). Proteins have non-redundant function within a given module and do not associate or function in other modules.
This work has been further corroborated by analyzing a series of C. elegans double mutants, which demonstrate worsened synthetic phenotypes (i.e. functional interactions) only when mutations occur in two different modules (Williams et al., 2011) but not with two different mutations in the same module. For instance, the B9 domain proteins of the MKS module functionally interact with the NPHP module (Williams et al., 2008) but not with most other genes in the MKS module. The conclusion is that each module probably mediates partially separate ciliary functions. Inactivating one component in a module is probably sufficient to fully inactivate the module, so functional interaction is only observed by inactivating a component in a different module. All together, these examples show that the availability of various disease proteomes in combination with the explosion of currently available genomic and transcriptomic datasets are driving forward biological network analysis in human disease.
Genetic complexity of human ciliopathies
Allelic diversity
What can the study of the ciliopathies teach us about the future direction of human genetic disease? Most obviously, that human genetics will be a lot more complex than many of us would have predicted. One obvious example is in the degree of multiple allelism at particular genetic loci. While some of the ciliopathy genes are associated with only a single phenotypic class to date, other gene mutations can result in phenotypes along the entire ciliopathy clinical spectrum. For instance, mutations in CEP290 are reported in MKS, JBTS, NPHP, BBS and LCA, spanning the full breadth of severity (Coppieters et al., 2010b) whereas ARL13B mutations are restricted to patients with JBTS. For ARL13B, it is not clear if this gene is only capable of causing a restricted phenotype or if there are additional mutations to be identified in other ciliopathy class disorders. Current data would suggest the latter, since only hypomorphic mutations in ARL13B gene were identified in humans, whereas comparably more severe phenotypes were observed in mouse and zebrafish (Caspary et al., 2007; Sun et al., 2004), suggesting the full spectrum of disease-causing alleles has not yet been reported.
In the case of CEP290, despite the identification of over 100 unique disease-causing mutations, the ability to predict phenotype based upon genotype is extremely limited. The mechanism underlying the different clinical outcomes of distinct mutations is not always clear. One possibility might relate to the type of mutations and their locations as predictors of phenotypic severity. While there are some types of mutations associated with particular phenotypes, such as the c.2991+1655A-->G variant present in 21% of all LCA patients, (den Hollander et al., 2006), the exact same mutation can be seen in two different ciliopathy classes (Coppieters et al., 2010b).
There are other examples of pleiotropy in the ciliopathies. NPHP1 mutations are found in pure NPHP and NPHP with RD (a combination known as SLS); INPP5E mutations are found in JBTS as well as BBS-like conditions. The case of TMEM67 deserves special attention, in that mutations cause a broad range of phenotypic combinations that comprise renal cyst, liver fibrosis, central nervous system malformations, retinal manifestations and postaxial polydactyly. The gene was first identified as both a cause of MKS3 and the origin of the multiple phenotypes of the Wpk rat, which include PKD, agenesis of the corpus callosum and hydrocephalus (Smith et al., 2006). Thereafter more than 80 TMEM67 mutations were identified, not only associated with MKS but also with a peculiar form of JBTS involving liver fibrosis and several renal and liver ciliopathy syndromes in which missense mutations predominate (Brancati et al., 2009; Iannicelli et al., 2010). Interestingly, the presence of two truncating alleles or a missense mutation within exons 8–15 associates with the lethal MKS phenotypes, suggesting an essential function of this region of the encoded protein that is yet to be identified.
Modifiers
While allelism offers one perspective with which to view phenotypic pleiotropy, epistatic interactions and mutation load offer another important perspective that deserves consideration. Such reports are starting to emerge across the full spectrum of human disease (Gu et al., 2009; Oprea et al., 2008) but are still limited in number due to underpowered studies. Within the ciliopathies, recent studies of families with more than one affected child and larger population screenings have highlighted an important role of epistatic interactions, whereby the effect of one gene modifies the phenotypic attributes of a different gene (Leitch et al., 2008; Wiszniewski et al., 2011) (Fig. 1c). For this review, we distinguish this from mutational load, whereby the total genetic burden from accumulated deleterious variants sums to produce the phenotype. It is now clear that the precise disease manifestations and probably the timing of the appearance of symptoms are subject to modification, which could be in the form of stochastic, environmental or genetic inputs. The identification of genetically encoded modifiers offers the potential to understand gene networks, improve prognostic information and identify targetable biochemical processes for the development of therapeutic treatments.
Evidence that heritable factors in humans can alter the course of ciliopathies came initially from an elegant series of experiments involving the role of the MGC1203 gene (Badano et al., 2006). The encoded protein, CCDC28B, contains a coiled-coil domain, and was identified in a yeast-two hybrid screen as a BBS4-interacting protein. After demonstrating CCDC28B interaction with several BBS proteins, the authors screened a BBS cohort for potential MGC1203 mutations. While none of the patients carried a mutation known to cause BBS, a C to T transition (C430T) in MGC1203 generated a splice defect in about 10% of gene products. To test the involvement of C430T as a potential modifier, they screened BBS and control cohorts for this variant, finding that 6.2% of patients vs. 1.4% of controls carried the variant, representing an over-transmission of the variant in transmission disequilibrium testing. The authors reported three families with affected siblings carrying a homozygous mutation on BBS1 in which the RD severity was associated with the presence of the transition, suggesting that this variant increases the likelihood of developing more severe BBS symptoms when associated with a known BBS mutation.
Since then, other examples of epistatic effects on RD associated with ciliopathies have emerged. Mutations in either AHI1 or RPGRIP1L, which both encode ciliary-localized proteins, were initially reported to cause JBTS. The AHI1 gene product physically interacts with nephrocystin-1 (Eley et al., 2008), the product of the most commonly mutated NPHP gene, NPHP1. Because over 50% of patients with AHI1 mutations display RD/LCA in addition to JBTS, investigators considered that AHI1 might be mutated in isolated RD/LCA. However, screening a cohort of 176 mixed ancestry patients with LCA failed to demonstrate any causative mutations, indicating that AHI1 mutations do not lead to isolated LCA in the absence diagnostic JBTS features (Louie et al., 2010). However, Ahi1 mutant mice predominantly displayed an LCA/RD phenotype, which was more severe when synthetically combined with an Nphp1 mutant allele, prompting investigation of epistatic interactions. Among a cohort of 153 NPHP1-mutant patients, there was a significant enrichment for a heterozygous c.C2488T change leading to a functional p.R830W substitution in those with RD. This translated into a 7.5-fold increase in RD in this population, which represents one of the highest known risk alterations so far described for any human disease. The c.C2488T allele therefore significantly increases the risk of developing RD in the presence of an NPHP1 mutation.
This association is not restricted to those with RD, as the same c.C2488T allele was more commonly found in individuals with NPHP1 mutations displaying a more severe neurological phenotype (Tory et al., 2007). Nor are AHI1 variants solely restricted to modifying NPHP1 phenotypes, as three unrelated patients with the exact same CEP290 genotype (p.R1465X) presenting with different clinical phenotypes showed variants in AHI1 that might explain this discordance. AHI1 heterozygous transversions (p.N811K and p.H758P) were found associated with greater severity of nephrological and neurological phenotypes (Coppieters et al., 2010a). Intriguingly the AHI1 variants in this case had no effect on the presence of RD. Thus, these epistatic interactions are both genotype-specific and phenotype-specific.
In studies that use model systems, it is possible to manipulate gene structure or expression through a variety of techniques, but investigation of genetic modifiers in humans is limited to naturally occurring variations. This might seem like a huge drawback initially, but it has tremendous benefits in the long run because large numbers of patients can be analyzed for the more common variants. Several heterozygous variants have been reported in RPGRIP1L in ciliopathy cohorts, and while their individual rarity precluded further investigation, one remarkable exception, the heterozygous p.A229T, is common enough to apply statistical analysis. The RPGRIP1L protein interacts with RPGR, a ciliary protein frequently mutated in RD, and this interaction is disrupted in the presence of the p.A229T transversion (Khanna et al., 2009). Individuals with this variant alone have normal vision, suggesting that this heterozygous variant is silent in isolation. However, it is enriched in ciliopathy patients with RD compared to those without RD. There are probably many such gene-gene interactions, but defining those of highest effect size, and the mechanisms by which these epistatic effects are manifest will require new experimental strategies.
Mutational load
Is it possible that such second site mutations might modify not only the expressivity but also the penetrance of disease? BBS has long provided the classic example of oligogenicity (Katsanis et al., 2001), representing the idea that second site mutations are necessary to produce an observable phenotype. The demonstration that three mutant alleles at two different loci were required for pathogenicity was among the first example of oligogenic inheritance. In this example, investigators observed that both affected and healthy children of an outbred family carried two different nonsense mutations in BBS2 (p.Y24X and p.Q59X). In an effort to identify the differential genetic trait that caused the disease in the affected sibling, they identified a heterozygous nonsense potentially deleterious sequence variant (PDSV) in BBS6 (Q147X) that was absent in the healthy sibling, suggesting that the three different mutant alleles were necessary to produce this phenotype. Subsequently, additional examples were described in which BBS patients carried three mutant alleles in two different loci (Eichers et al., 2004). However, in such examples the segregation does not exclude the possibility that two mutations in the same gene are sufficient to cause disease in a different genetic background (Mykytyn et al., 2003). Moreover, the lack of recurrence of such combinations in BBS cohorts studied complicates the validation of the oligogenic hypothesis.
Although oligogenic inheritance in BBS is still hotly contested, several reports of possible oligogenic inheritance in other ciliopathies have emerged more recently (Hoefele et al., 2007; Hopp et al., 2011), where only single heterozygous PDSVs are detected, or where patients with two deleterious mutations in one gene also carry a heterozygous PDSV in a second gene. The evidence for oligogenicity is somewhat tempered by the natural limitations of the technology used to support the findings, and whether such variants are functionally relevant and whether they are overrepresented in patients compared with similarly investigated controls is still a matter of debate. Nevertheless, the observed phenotypic and genetic variability, together with an apparent overabundance of PDSVs in ciliopathy genes have led to the general acceptance of a “mutational load” hypothesis. Defining the mutational load required for the manifestation of a given phenotypic combination or expressivity is now the main challenging issue. One potential reason for optimism is the emergence of next-generation sequencing, which will allow genome-wide unbiased exploration of the mutational load hypothesis. For instance, it would fascinating to test whether patients with ciliopathy phenotypes carry greater burdens or particular patterns of mutations in cilia-specific genes compared with housekeeping genes on a genome-wide scale.
IPSCs, cell based screens and treatments for ciliopathies
What does the future of human genetics hold and how can disorders like the ciliopathies benefit from new technological advances? Whole-genome (WGS) and whole-exome sequencing (WES) is clearly the breakthrough most likely to impact the study of Mendelian disorders like ciliopathies. The exons, accounting for ~1% of the genome, harbor most disease-causing mutations of high-effect size, and therefore WES is a reasonable approach for finding novel disease-causing genes. This is evident by the abundance of novel disease-causing genes identified in recent years. The limitations of WES can be overcome with WGS, with its ability to identify not just variants in coding regions of the genes, but also variants in introns, untranslated regions, and non-coding RNAs, such as the recent discovery of mutations in the U4atac shRNA in microcephalic osteodysplastic primordial dwarfism type I (Edery et al., 2011; He et al., 2011). Additionally, with genome-wide approaches, these datasets offer the possibility to directly test the mutational load hypothesis and identify a host of epistatic modifiers. At this point, we still require better bioinformatic tools to predict resultant human phenotypes, but this technology is already greatly benefiting the field of human genetics with an explosion in the number of newly discovered human disease genes. The ciliopathies are no exception (Gilissen et al., 2010; Hopp et al., 2011).
Combining these genetic discoveries with proteomic research is another huge area for the future, as molecular geneticists take advantage of the explosion of new human disease genes. Identifying functional complexes, modules and genetic pathways through the identification of protein binding partners and animal modeling will yield more robust platforms from which to consider therapeutics. An example is in PKD, one of the most common Mendelian diseases. Based upon the knowledge of similarities in phenotype between PKD and tuberous sclerosis complex (TSC), investigators wondered if there was a possible functional connection. The two genes mutated in TSC are involved in the mTOR pathway, and some symptoms are treatable with mTOR inhibitors like rapamycin. Investigators hypothesized that PKD1 and tuberin (one of the genes mutated in TSC) might interact. After confirming this hypothesis, they tested whether, like TSC, PKD might be abrogated by mTOR inhibitors. Strikingly, rapamycin results in a significant reversal of renal cystogenesis in two different mouse models (Shillingford et al., 2006), and these findings led to a clinical trial in humans. More recent work hints at the possibility that mTOR inhibitors may benefit other ciliopathy conditions (Tobin and Beales, 2008). Although the human trials were not successful and will need to be repeated with different endpoints, the story exemplifies the streamlining of approaches as investigators move from human disease genes to new treatments.
Because animal models can frequently be significantly different in terms of organ specificity and severity compared with their human counterparts, they have been of limited benefit in directly exploring disease pathogenesis. For instance, both published mouse models for the most commonly mutated gene in human NPHP, NPHP1, shows no detectable kidney phenotype. This could be a result of functional differences in the kidney in humans versus mice, differences in the types of mutations, or in the genetic background. Although NPHP1 mutant mice have some evidence of ciliopathy-like features, including aberrant sperm maturation and a background-dependent genetic interaction with AHI1 in RD (Jiang et al., 2009; Jiang et al., 2008; Louie et al., 2010), in general the use of animal models of human disease often requires careful assessment of relevance.
As in many other diseases, investigators that continue to return to patients and patient-derived samples for clues to pathogenesis are more apt to make disease-relevant discoveries. Induced pluripotent stem cells (IPSCs) offer such an opportunity, especially for diseases like ciliopathies, in which the genetic background is probably critical for phenotypic expressivity. For this reason, it might be preferable to work with a patient sample carrying a known disease-causing mutation in the disease-causing genetic background than a sample from a knockout mouse where the species or the species’ genetic background may not be appropriate for full expressivity. IPSCs and other cellular reprogramming strategies offer an opportunity to make discoveries in disease-relevant cells. For example, a recent application of the spheroid cellular assay to probe mechanistic insights of kidney cysts (Otto et al., 2010) might greatly benefit from the use of patient-derived cells.
Functional genomic cell-based screens also offer a complementary approach to the study of human disease, by leap-frogging past animal models in the identification of potential treatment-relevant targets. In the past year, a number of such cell-based high-throughput or high-content screening systems to identify genes required for cilium assembly and maintenance or signaling pathways known to depend upon the cilium have emerged. By combining Stable Isotope Labeling with Amino acids in Cell culture (SILAC) with BAC transgenesis in human cells, a list of 135 new centriolar components that are specific to either the mother or daughter centriole were identified (Jakobsen et al., 2011). Given the important coordination of centriole function with ciliogenesis, this list is likely to lead to many new functional discoveries in the future. Genome-wide RNAi screening in cells has identified the multitasking kinase Stk11 (a.k.a Lkb1), as a key factor in cilium stability and an integrator of Shh and Wnt signals in cells, two key pathways modulated by the cilium (Jacob et al., 2011). Finally, two recent papers established the genetic requirements of cilium assembly and length in mammalian cells, highlighting the important contribution of the actin cytoskeleton and showing it is possible to uncouple ciliary cargo transport from cilia formation in vertebrates (Kim et al., 2010; Lai et al., 2011).
Rapidly evolving sequencing methods combined with the underlying growth of informatic algorithms can provide the power to uncover both new causes as well as new potential treatments in human disease. Using the relatively simple example of the ciliopathies, we can see how the paradigm is shifting in experimental biology to one less reliant on the pure study of animal models in favor of using humans as a model to study human disease. Further integration of these new approaches has the potential to yield improved diagnosis and opens the window for new therapies across the field of genetics.
Figure 3. Discovery pipeline for mechanism of disease: past and future paradigms.
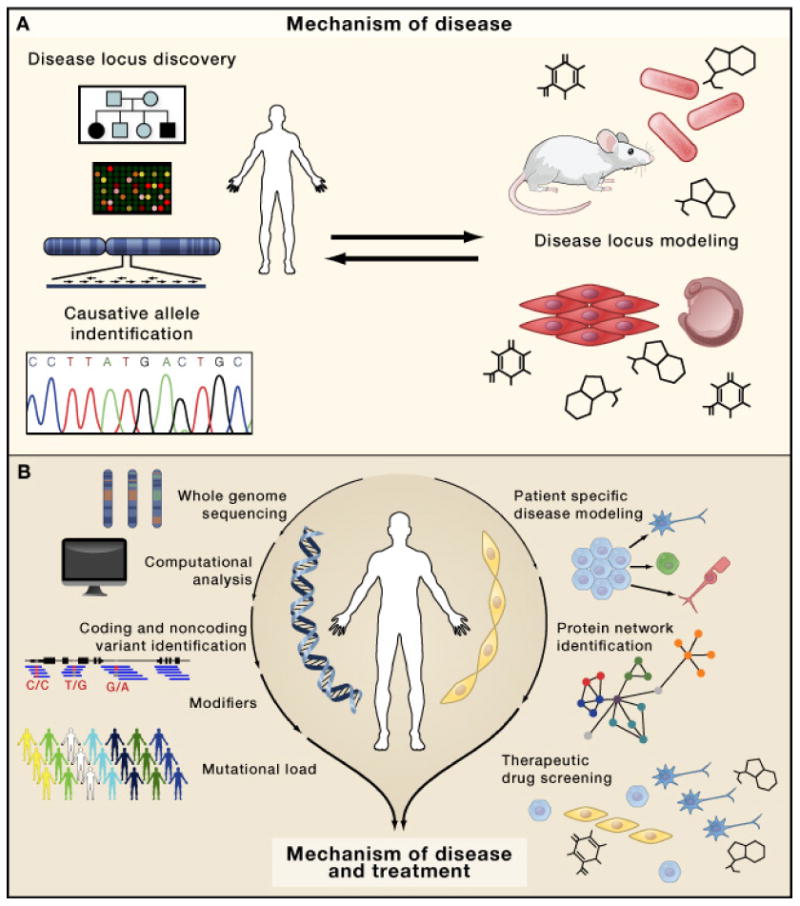
A. Former strategy to identify disease mechanisms, starting with ascertainment of pedigrees for linkage, disease mapping to a particular chromosomal locus, and candidate gene Sanger sequencing. Once a mutant gene was identified, animal models were created to understand the mechanism. B. The future paradigm bypasses the need for informative pedigrees and disease mapping, instead going directly from patient ascertainment to genome sequencing, then to variant identification and expanded to identification of modifiers and mutational load. In parallel, patient-specific disease modeling using human cells coupled with protein interaction networks and therapeutic drug screening can further uncover disease mechanisms and help develop better treatments.
Acknowledgments
NA is supported by a training grant from the California Institute for Regenerative Medicine, GN is supported by a grant from the Deutsche Forschungsgemeinschaft (DFG). Work in the Gleeson lab is supported by grants from the NIH (R01NS041537, P01HD070494, R01NS052455, P30NS047101, R01NS048453). JGG is an Investigator of the Simons Foundation Autism Research Initiative (SFARI) and the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, Kim JC, Ross AJ, Eichers ER, Teslovich TM, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425:628–633. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- Arnaiz O, Malinowska A, Klotz C, Sperling L, Dadlez M, Koll F, Cohen J. Cildb: a knowledgebase for centrosomes and cilia. Database (Oxford) 2009;2009:bap022. doi: 10.1093/database/bap022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, Zuker CS. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117:527–539. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, Rattenberry E, et al. Pleiotropic Effects of CEP290 (NPHP6) Mutations Extend to Meckel Syndrome. Am J Hum Genet. 2007a;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, et al. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007b;80:186–194. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badano JL, Leitch CC, Ansley SJ, May-Simera H, Lawson S, Lewis RA, Beales PL, Dietz HC, Fisher S, Katsanis N. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006;439:326–330. doi: 10.1038/nature04370. [DOI] [PubMed] [Google Scholar]
- Barr MM, Sternberg PW. A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature. 1999;401:386–389. doi: 10.1038/43913. [DOI] [PubMed] [Google Scholar]
- Brancati F, Iannicelli M, Travaglini L, Mazzotta A, Bertini E, Boltshauser E, D’Arrigo S, Emma F, Fazzi E, Gallizzi R, et al. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum Mutat. 2009;30:E432–442. doi: 10.1002/humu.20924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary T, Larkins CE, Anderson KV. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Chen Y, Bedell M, Zhang K. Age-related macular degeneration: genetic and environmental factors of disease. Mol Interv. 2010;10:271–281. doi: 10.1124/mi.10.5.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang AP, Nishimura D, Searby C, Elbedour K, Carmi R, Ferguson AL, Secrist J, Braun T, Casavant T, Stone EM, et al. Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3) Am J Hum Genet. 2004;75:475–484. doi: 10.1086/423903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters F, Casteels I, Meire F, De Jaegere S, Hooghe S, van Regemorter N, Van Esch H, Matuleviciene A, Nunes L, Meersschaut V, et al. Genetic screening of LCA in Belgium: predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes. Hum Mutat. 2010a;31:E1709–1766. doi: 10.1002/humu.21336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters F, Lefever S, Leroy BP, De Baere E. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat. 2010b;31:1097–1108. doi: 10.1002/humu.21337. [DOI] [PubMed] [Google Scholar]
- Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010;190:927–940. doi: 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edery P, Marcaillou C, Sahbatou M, Labalme A, Chastang J, Touraine R, Tubacher E, Senni F, Bober MB, Nampoothiri S, et al. Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science. 2011;332:240–243. doi: 10.1126/science.1202205. [DOI] [PubMed] [Google Scholar]
- Eichers ER, Lewis RA, Katsanis N, Lupski JR. Triallelic inheritance: a bridge between Mendelian and multifactorial traits. Ann Med. 2004;36:262–272. doi: 10.1080/07853890410026214. [DOI] [PubMed] [Google Scholar]
- Eley L, Gabrielides C, Adams M, Johnson CA, Hildebrandt F, Sayer JA. Jouberin localizes to collecting ducts and interacts with nephrocystin–1. Kidney Int. 2008 doi: 10.1038/ki.2008.377. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011 doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherman A, Davis EE, Katsanis N. The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia. Nat Genet. 2006;38:961–962. doi: 10.1038/ng0906-961. [DOI] [PubMed] [Google Scholar]
- Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, van Lier B, Steehouwer M, van Reeuwijk J, Kant SG, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, van Beersum SE, Mans DA, Hikida A, Eckert M, Knutzen D, et al. CC2D2A Is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet. 2008;83:559–571. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Harley IT, Henderson LB, Aronow BJ, Vietor I, Huber LA, Harley JB, Kilpatrick JR, Langefeld CD, Williams AH, et al. Identification of IFRD1 as a modifier gene for cystic fibrosis lung disease. Nature. 2009;458:1039–1042. doi: 10.1038/nature07811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Liyanarachchi S, Akagi K, Nagy R, Li J, Dietrich RC, Li W, Sebastian N, Wen B, Xin B, et al. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science. 2011;332:238–240. doi: 10.1126/science.1200587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefele J, Wolf MT, O’Toole JF, Otto EA, Schultheiss U, Deschenes G, Attanasio M, Utsch B, Antignac C, Hildebrandt F. Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol. 2007;18:2789–2795. doi: 10.1681/ASN.2007020243. [DOI] [PubMed] [Google Scholar]
- Hopp K, Heyer CM, Hommerding CJ, Henke SA, Sundsbak JL, Patel S, Patel P, Consugar MB, Czarnecki PG, Gliem TJ, et al. B9D1 is revealed as a novel Meckel syndrome (MKS) gene by targeted exon-enriched next-generation sequencing and deletion analysis. Hum Mol Genet. 2011;20:2524–2534. doi: 10.1093/hmg/ddr151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 2010;329:436–439. doi: 10.1126/science.1191054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Iannicelli M, Brancati F, Mougou-Zerelli S, Mazzotta A, Thomas S, Elkhartoufi N, Travaglini L, Gomes C, Ardissino GL, Bertini E, et al. Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum Mutat. 2010;31:E1319–1331. doi: 10.1002/humu.21239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis PN, Boroevich KA, Leroux MR. Piecing together a ciliome. Trends Genet. 2006;22:491–500. doi: 10.1016/j.tig.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Jacob LS, Wu X, Dodge ME, Fan CW, Kulak O, Chen B, Tang W, Wang B, Amatruda JF, Lum L. Genome-wide RNAi screen reveals disease-associated genes that are common to Hedgehog and Wnt signaling. Sci Signal. 2011;4:ra4. doi: 10.1126/scisignal.2001225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen L, Vanselow K, Skogs M, Toyoda Y, Lundberg E, Poser I, Falkenby LG, Bennetzen M, Westendorf J, Nigg EA, et al. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J. 2011;30:1520–1535. doi: 10.1038/emboj.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang ST, Chiou YY, Wang E, Chien YL, Ho HH, Tsai FJ, Lin CY, Tsai SP, Li H. Essential role of nephrocystin in photoreceptor intraflagellar transport in mouse. Hum Mol Genet. 2009;18:1566–1577. doi: 10.1093/hmg/ddp068. [DOI] [PubMed] [Google Scholar]
- Jiang ST, Chiou YY, Wang E, Lin HK, Lee SP, Lu HY, Wang CK, Tang MJ, Li H. Targeted disruption of Nphp1 causes male infertility due to defects in the later steps of sperm morphogenesis in mice. Hum Mol Genet. 2008;17:3368–3379. doi: 10.1093/hmg/ddn231. [DOI] [PubMed] [Google Scholar]
- Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, Nachury MV. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell. 2010;141:1208–1219. doi: 10.1016/j.cell.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, Hoskins BE, Scambler PJ, Davidson WS, Beales PL, Lupski JR. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–2259. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman MI, Waseem N, Chakarova CF, et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009;41:739–745. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee JE, Heynen-Genel S, Suyama E, Ono K, Lee K, Ideker T, Aza-Blanc P, Gleeson JG. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464:1048–1051. doi: 10.1038/nature08895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JC, Badano JL, Sibold S, Esmail MA, Hill J, Hoskins BE, Leitch CC, Venner K, Ansley SJ, Ross AJ, et al. The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet. 2004;36:462–470. doi: 10.1038/ng1352. [DOI] [PubMed] [Google Scholar]
- Kozminski KG, Johnson KA, Forscher P, Rosenbaum JL. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci U S A. 1993;90:5519–5523. doi: 10.1073/pnas.90.12.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyttala M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestila M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet. 2006;38:155–157. doi: 10.1038/ng1714. [DOI] [PubMed] [Google Scholar]
- Lai CK, Gupta N, Wen X, Rangell L, Chih B, Peterson AS, Bazan JF, Li L, Scales SJ. Functional characterization of putative cilia genes by high-content analysis. Mol Biol Cell. 2011;22:1104–1119. doi: 10.1091/mbc.E10-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- Li JB, Gerdes JM, Haycraft CJ, Fan Y, Teslovich TM, May-Simera H, Li H, Blacque OE, Li L, Leitch CC, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117:541–552. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- Louie CM, Caridi G, Lopes VS, Brancati F, Kispert A, Lancaster MA, Schlossman AM, Otto EA, Leitges M, Grone HJ, et al. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat Genet. 2010;42:175–180. doi: 10.1038/ng.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mykytyn K, Nishimura DY, Searby CC, Beck G, Bugge K, Haines HL, Cornier AS, Cox GF, Fulton AB, Carmi R, et al. Evaluation of complex inheritance involving the most common Bardet-Biedl syndrome locus (BBS1) Am J Hum Genet. 2003;72:429–437. doi: 10.1086/346172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- Oprea GE, Krober S, McWhorter ML, Rossoll W, Muller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320:524–527. doi: 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto EA, Hurd TW, Airik R, Chaki M, Zhou W, Stoetzel C, Patil SB, Levy S, Ghosh AK, Murga-Zamalloa CA, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet. 2010;42:840–850. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, Muerb U, O’Toole JF, Helou J, Attanasio M, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005;37:282–288. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- Otto EA, Schermer B, Obara T, O’Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piasecki BP, Burghoorn J, Swoboda P. Regulatory Factor X (RFX)-mediated transcriptional rewiring of ciliary genes in animals. Proc Natl Acad Sci U S A. 2010;107:12969–12974. doi: 10.1073/pnas.0914241107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006;103:5466–5471. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, Morgan NV, Goranson E, Gissen P, Lilliquist S, et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet. 2006;38:191–196. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- Sun Z, Amsterdam A, Pazour GJ, Cole DG, Miller MS, Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131:4085–4093. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- Swoboda P, Adler HT, Thomas JH. The RFX-type transcription factor DAF-19 regulates sensory neuron cilium formation in C. elegans. Mol Cell. 2000;5:411–421. doi: 10.1016/s1097-2765(00)80436-0. [DOI] [PubMed] [Google Scholar]
- Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK. Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol Biol Cell. 2001;12:589–599. doi: 10.1091/mbc.12.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin JL, Beales PL. Restoration of renal function in zebrafish models of ciliopathies. Pediatr Nephrol. 2008;23:2095–2099. doi: 10.1007/s00467-008-0898-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tory K, Lacoste T, Burglen L, Moriniere V, Boddaert N, Macher MA, Llanas B, Nivet H, Bensman A, Niaudet P, et al. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol. 2007;18:1566–1575. doi: 10.1681/ASN.2006101164. [DOI] [PubMed] [Google Scholar]
- Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol. 2011;192:1023–1041. doi: 10.1083/jcb.201012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CL, Winkelbauer ME, Schafer JC, Michaud EJ, Yoder BK. Functional redundancy of the B9 proteins and nephrocystins in Caenorhabditis elegans ciliogenesis. Mol Biol Cell. 2008;19:2154–2168. doi: 10.1091/mbc.E07-10-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiszniewski W, Lewis RA, Stockton DW, Peng J, Mardon G, Chen R, Lupski JR. Potential involvement of more than one locus in trait manifestation for individuals with Leber congenital amaurosis. Hum Genet. 2011;129:319–327. doi: 10.1007/s00439-010-0928-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HJ, Tayeh MK, Mullins RF, Stone EM, Sheffield VC, Slusarski DC. Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer’s vesicle cilia function. Hum Mol Genet. 2006;15:667–677. doi: 10.1093/hmg/ddi468. [DOI] [PubMed] [Google Scholar]