

Abstract
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is a representative of a large group of polyhalogenated aromatic hydrocarbons that are widespread environmental contaminants. Administration of TCDD to laboratory animals or cultured cells results in a number of adverse effects that are well documented. For example, the effects of TCDD observed in developing organisms indicate that exposure to this class of environmental contaminants significantly alters embryo morphogenesis. However, it is not clear whether tissue regeneration in adult animals may be similarly affected. With this in mind, we examined the impact of TCDD exposure on wound healing using a murine cutaneous wound healing model. Our results indicate that TCDD exposure did not significantly alter the time needed for wound closure. However, in the TCDD-treated mice, a significant decrease in tensile strength in the healed wounds was observed which is indicative of an aberrantly healed wound. Immunostaining revealed that exposure to TCDD increased the population of macrophages detected within the wounded tissue at the latter stages of wound healing. Our findings support the idea that exposure to environmental contaminants such as TCDD is proinflammatory in the wounded tissue, disrupts normal healing and ultimately produces in a poorly healed wound.
Keywords: Skin; wound healing; tensile strength; 2,3,7,8 tetrachlorodibenzo-p-dioxin; extracellular matrix remodeling; macrophages
Introduction
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is considered a prototype and the most toxic of a large group of polyhalogenated aromatic hydrocarbons that are widespread environmental contaminants. Most of the biological effects of TCDD require its activation of the aryl hydrocarbon receptor (AHR), a ligand activated transcription factor (Mandal, 2005). The ability of TCDD to upregulate AHR target genes such as CYP1A1 and CYP1B1 involved in xenobiotic metabolism have been well studied. However, it is not yet known whether modulation of additional gene pathways may also be required to mediate the adverse effects of TCDD and other AHR agonists. The adverse effects associated with TCDD exposure are well documented and include immune dysfunction, reproductive abnormalities, developmental defects and promotion of carcinogenesis (Yoshizawa et al., 2007). In vivo administration of TCDD has also been shown to alter physiological processes such as angiogenesis (Ivnitski-Steele and Walker, 2005), the inflammatory response (Kerkvliet and Oughton, 1993, Pande et al., 2005, Vogel et al., 2007) and remodeling of the extracellular matrix (Aragon et al., 2008, Kung et al., 2009, Mathew et al., 2009). At the cellular level, a number of TCDD-induced effects have been observed including altered cell cycle progression (Marlowe and Puga, 2005), cell migration and adhesion (Peng et al., 2009, Hsu et al., 2007, Kung et al., 2009) and cell fate decisions such as apoptosis, senescence and differentiation (Ray and Swanson, 2009). However, it is not clear whether these cellular effects of TCDD, best characterized in isolated cultured cells, impact cell processes either in the context of healthy multicellular tissues or within tissues under pathological conditions initiated by chronic disease states.
Cutaneous wound healing involves a carefully orchestrated series of events that includes inflammation, cell migration, cell proliferation, angiogenesis, matrix deposition and tissue remodeling (Gurtner et al., 2008, Braiman-Wiksman et al., 2007). Numerous cell types are involved in the wound healing process including immune cells such as neutrophils, monocytes, lymphocytes and dendritic cells, as well as endothelial cells, keratinocytes and fibroblasts. There is little known regarding the effects of TCDD on wound healing. Because TCDD can alter many of the processes required for proper wound healing, we hypothesized that this polyhalogenated aromatic hydrocarbon has an adverse effect on wound healing. We examined this hypothesis using an in vivo model of cutaneous wound healing. Our results indicate that exposure to TCDD leads to healed wounds with low tensile strength, an indicator of an improperly healed wound. Further, we report that exposure to TCDD appears to first, enhance the population of macrophages that are associated with the healing wounds and second, alters the organization of the collagenous extracellular matrix. Taken together, our data indicate that study of the wound healing process may be useful in understanding how environmental contaminants may contribute to the progression of many chronic human diseases.
Materials and Methods
Animals
Female C57BL/6 mice (6–8 weeks of age) (Taconic Laboratories, New York, NY) were used. All procedures were carried out in accordance with the NIH 1996 Guide for the Care and Use of Laboratory Animals and were approved by IACUC at the University of Kentucky.
TCDD treatment and wounding
The mice were treated with TCDD (10 µg/kg dissolved in corn oil) or corn oil by intraperitoneal injection. After 24 hours, the TCDD-treated and control mice were anesthetized by intraperitoneal injection of ketamine (100 mg ketamine/kg) and xylazine (10 mg/kg). The dorsal skin was shaved, depilated and a pair of circular full-thickness wounds on either side of the dorsal midline were made using an 8-mm diameter biopsy punch, as described previously (Frank and Kampfer, 2003). To quantify wound closure, the wounds were photographed at the time of wounding and at the time of euthanization. The wound areas were digitized using Adobe Photoshop CS3 extended version.10.0.1 and the areas of the wounds were then converted from pixels to mm2. Each time point represents 6–8 wounds obtained from 3–4 mice in each group.
Tissue preparation, histology and immunohistochemistry
The wound area including the epithelial margins was excised and the wounds bisected. Wounds from the left side of each animal were fixed overnight in HistoChoice tissue fixative (Amresco); wounds from the right side of each animal were fixed overnight in 4% paraformaldehyde. The fixed samples were stored in 70% ethanol until embedded in paraffin. Five µm sections from the central portion of the wound were deparaffinized and stained with hematoxylin and eosin. The bright field images of the stained sections were acquired on a Nikon Eclipse TE200 equipped with a SPOT Insight color CCD (Model #3.2, Diagnostics Instruments Inc., Sterling Heights, MI). Collagen fibers were stained with picrosirius red and visualized by polarized light microscopy.
For immunohistochemical analysis, the sections were deparaffinized and the samples were treated with DAKO antigen retrieval (Dako Corp., Carpinteria, CA) and then incubated with 0.3% hydrogen peroxide. After incubation with blocking reagent (Perkin Elmer, Waltham MA) the samples were incubated with one of the following antibodies: F4/80 (AbCam, Cambridge UK) for the detection of macrophages; Ki67 (Labvision, Fremont CA) to detect nuclei of dividing cells and MPO (Labvision, Fremont, CA) for the detection of neutrophils. Secondary antibodies were purchased from Jackson Immunoresearch Laboratories (West Grove, PA). The fluorescent signals were amplified using the TSA kit from Perkin Elmer (Waltham MA). The nuclei were counterstained with DAPI (Invitrogen, Carlsbad CA). The stained sections were visualized and photographed using a Nikon inverted TE300 Eclipse microscope. Metamorph software was used for data acquisition and analysis.
The inflammatory cells were quantified as described previously (Furness et al., 1997, McFarland-Mancini et al.). Briefly, the adjacent fields comprising a wound edge were first photographed using a 20× objective. The images were imported into Photoshop and merged into a composite image of each wound edge. Within a square centered on the wedge of staining at the leading edge of the adipose tissue, three 0.5 × 0.5 inch squares were cropped and imported into ImageJ version 1.38×. Image thresholds were set to distinguish between stained macrophages and the unstained background of a representative image; a uniform threshold setting was then used to measure the set of images. The area above the threshold was expressed as a fraction of the total area. Three samples were quantified for each wound, and the mean of three wounds was reported for each experimental condition.
Tensile strength analysis
At the designated time after wounding, the mice (n=6 per group) were euthanized and the skin containing the entire wound was removed and cut at a right angle to the long axis of the wound into 6 mm wide and 12 mm long strip under a stereomicroscope. To measure tensile strength, one end of the strip was connected to a Fort 1000 force transducer (World Precision Instruments, Sarasota, FL) to continuously measure the tension. The other end of the strip was stretched at a constant rate until the wound broke. The tension at which the wound broke was recorded as the tensile strength of the sample.
Western blot analysis
Protein lysates were prepared from the excised wounds following homogenization in RIPA buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP40 and 1% sodium deoxycholate) containing protease inhibitor cocktail and phosphatase inhibitor cocktails I and II (Sigma, St. Louis MO). The tissue homogenates were centrifuged for 15 min at 14,000 × g, the supernatants were removed and aliquots (~100 µg of total protein) were separated by sodium dodecyl sulfate gel electrophoresis. After transfer to nitrocellulose, the proteins were detected using antibodies that recognized either CYP1A1 (Xenotech LLC, Lexena KS) or β-actin (Sigma, St Louis, MO).
Statistical Analyses
All data are presented as mean ± standard error of mean. Statistical analysis was performed using GraphPad Prism (GraphPad Software Inc., CA). For the wound closure and tensile strength analyses, significance was determined using the unpaired T test. For all other analyses, significance was determined by one way ANOVA analysis followed by Tukey’s Multiple Comparison Test at a p < 0.05 level of significance.
Results
Impact of TCDD exposure on wound closure
To determine whether exposure to TCDD alters the rate of wound closure, mice were pretreated with either TCDD or the corn oil control. Twenty four hours later, the mice were subjected to excisional wounding. The size of the wounded areas was then quantitated daily. As shown in Figure 1A, wound closure was complete in the corn oil control group at 10 days post-wounding. The wound areas of the TCDD-treated versus the control groups, were significantly different at 4 days post wounding (P<0.0005 level), at 7 days post-wounding (P<0.01 level) and at 2, 5, 8 days post-wounding (P<0.05 level). The area under the curve was 206.1 and 170.2 for the corn oil and TCDD-treated groups, respectively. To confirm that the 10 µg/kg dose of TCDD was sufficient for activating AHR-mediated events within the skin, the wounded areas were removed at the indicated post-wounding time points and analyzed for expression of the prototypical AHR target gene, CYP1A1. As shown in Figure 1B, CYP1A1 expression was increased in the samples obtained from the TCDD- treated mice, as compared to that of the control, at all time points examined.
Figure 1. Effect of TCDD on cutaneous wound closure and skin CYP1A1 protein expression.


The mice (n=3–4) were treated with either TCDD or corn oil 24 hours prior to wounding and the wounded tissue (two wounds/mice) was evaluated at the indicated time points, post-wounding. (A) The wounded tissue was photographed and quantitated. (B) The wounded tissue was excised and the protein was extracted from the tissue and was analyzed for expression of CYP1A1 and β-actin. *P<0.05; **P<0.01, P<0.0005. The data are shown as mean ± s.e.m.
Exposure to TCDD reduces tensile strength of the healed wounds
At the completion of healing, the wound is marked by dermal reorganization leading to a gain in strength and elasticity (Brainman-Wiksman et. al., 2007). Appropriate tissue remodeling indicative of a properly healed wound is often measured by analyzing skin strength. We determined the tensile strength of the skin obtained from the corn oil- and TCDD-treated mice at 15 and 20 days post wounding. A time dependent increase in tensile strength typical of normal wound healing was observed in the control group (Figure 2). This trend, however, is not recapitulated in the TCDD-treated samples. Further, the tensile strength of the skin obtained from the TCDD- treated mice was significantly lower than that of the corn oil control at both time points examined.
Figure 2. Exposure to TCDD reduces tensile strength of the healed wound.

The mice were treated with either TCDD or corn oil (n=6). After 24 hours, the mice were wounded and 2 × 0.5 cm skin strips containing the healed wounded area was dissected under microscope. The tensile strength of the strips was then measured by isometric tension method using a Ford 1000 transducer (WPI- World Precision Instrument). The data are shown as mean ± s.e.m.
Exposure to TCDD does not appear to significantly alter cell proliferation in the wounded tissue
We performed H & E staining on sections of the wounded tissues and found that no gross differences in the histological appearance of the samples obtained from the corn oil versus TCDD-treated mice were readily apparent (data not shown). Cell proliferation was analyzed by staining the wound sections for expression of Ki67. As shown in Figure 3, Ki67 staining could be detected after day 1 post-wounding. TCDD did not significantly alter the fraction of Ki67-positive nuclei within the epidermis at any time point examined. However, at some time points (i.e., days 2 and 5 post wounding), decreases in the TCDD-treated samples as compared to the control were observed that were not statistically significant.
Figure 3. Impact of TCDD exposure on keratinocyte proliferation within the wounded tissue.


(A) Sections of the excised wounds obtained from either the corn oil or TCDD- treated mice were stained with either Ki67 (left panels) or DAPI (right panels) to identify proliferating keratinocytes within the epidermal layer. Samples obtained at one, two and three days post-wounding were analyzed. Small arrows indicate the apical and basal surfaces of the epidermis whereas the arrowheads indicate the wound edge. (B) The percent of DAPI-stained epidermal nuclei that stained positive for the Ki67 antigen were quantitated. The data are shown as mean ± s.e.m.
Impact of TCDD exposure on the presence of polymorphonuclear neutrophils and macrophages within the wounded tissue
An important aspect of proper wound healing is the recruitment and retention of inflammatory cells. The early phase of wound healing (i.e., 1–3 days post wounding) is characterized by an infiltration of neutrophils into the wounded tissue (Broughton et al., 2006, Eming et al., 2007a). As an indirect measure of neutrophil content, we subjected the wounded tissue sections to myeloperoxidase staining. As show in Figure 4, the abundance of neutrophils was statistically indistinguishable in wounded tissues obtained from the TCDD-treated versus corn oil-treated mice at any time point examined. As the wound healing process progresses (i.e., approximately 3–4 days post wounding), a marked infiltration of macrophages is typically observed (Broughton et al., 2006, Eming et al., 2007a). The timing of appearance and relative levels of macrophage invasion were visualized by positive staining for the F4/80 antigen. In the corn oil-treated mice, the infiltration of macrophages reached a peak level at days 3–6 post-wounding (Figure 5) which is consistent with previous reports (Eming et al., 2007b). In the TCDD-treated samples, the population of macrophages was increased, relative to that of the corn oil samples, at days 6, 8 and 15 post wounding. Interestingly, the increase in F4/80 staining in the TCDD-treated samples observed at the latter times points (i.e., 15 days post wounding) coincided with the appearance of the adipose tissue (data not shown).
Figure 4. Impact of TCDD exposure on neutrophil cell numbers within the wound.

Sections of the excised wounds were obtained from either the corn oil- or TCDD- treated mice and stained with the anti-MPO antibody to visualize the wound neutrophil numbers. The samples analyzed represented the left edges of the wounds obtained at one, two, three and four days post-wounding. The MPO reactive cells were quantitated from the wound areas using Image J software. Data are shown as mean ± s.e.m.
Figure 5. Impact of TCDD exposure on macrophage cell numbers within the wound.

Sections of the excised wounds were obtained from either the corn oil- or TCDD- treated mice and were stained with the anti-F4/80 antibody to visualize wound macrophage cell numbers. The samples analyzed represented the left edges of the wounds obtained at the indicated days post-wounding. The F4/80 reactive cells were quantitated from the wound areas using Image J software. Data are shown as mean ± s.e.m.
Impact of TCDD exposure on collagen organization
Low tensile strength of tissues is often associated with aberrant structure and composition of the collagenous extracellular matrix network (Wenstrup et al., 2006). With this in mind, we hen examined the collagen organization in excised wounded tissues. Representative histology of some of the differences that were observed at the latter time points is shown in Figure 6. Here, it appears that in the TCDD-treated wound tissue, a less organized and more randomly orientated collagen matrix is formed as compared to the lamellar pattern seen in the corn oil control.
Figure 6. Analysis of collagen organization in the corn oil- and TCDDtreated mice.
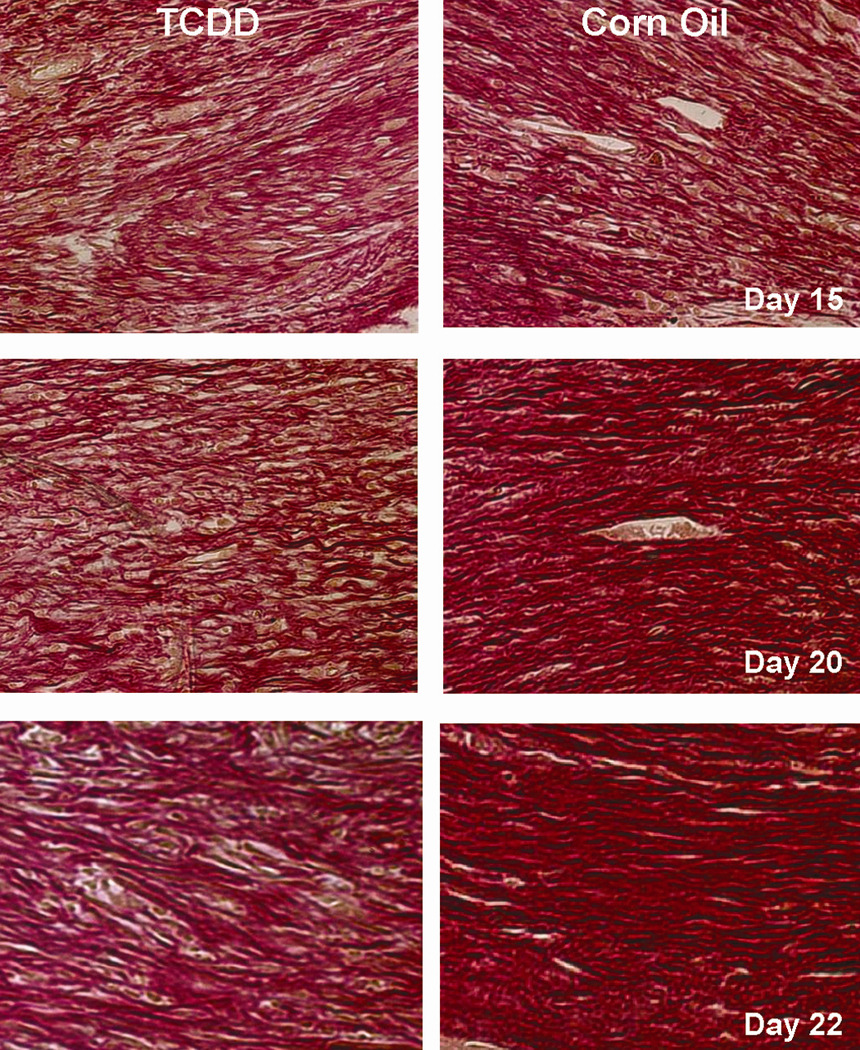
Histological sections of the wounded tissues obtained either the corn oil- (left panels) or TCDD-treated (right panels) mice at the indicated post-wounding time points were prepared and were subjected to picro-sirius red staining. The sections were photographed with a 40X objective.
Discussion
In this study, we used the cutaneous wound healing model to assess the extent to which exposure to TCDD interferes with the complex process of wound repair. We chose an in vivo model of wound healing to increase the relevance and ability to extrapolate our results to human health. We show that exposure to TCDD is capable of significantly decreasing the tensile strength of the healed wound which is indicative of a TCDD-mediated event that affects the late stage maturation and remodeling phase of wound healing. A role for the AHR in mediating the effects of TCDD in this study is implied by the observation that a single administration of TCDD induced the expression of the prototypical AHR target gene, CYP1A1, an effect which was sustained for at least 8 days post-wounding (Figure 1B).
In this study, we have found that TCDD exposure decreases the tensile strength of the healed wound (Figure 2) and contributes to disorganized collagen formation (Figure 6). During the maturation and remodeling phase of normal wound healing, the collagen is deposited in an organized and well-mannered network (Gurtner et al., 2008, Broughton et al., 2006). The elasticity and mechanical properties (i.e., the tensile strength) of the healed tissue (Cavalcante et al., 2005) are a consequence of the amounts, as well as the organization of collagen. For example, high levels of collagen correlate with an increase in tissue stiffness, but excess amounts of collagen lead to an alteration in matrix organization and a subsequent decrease in tensile strength (Black et al., 2008). During the final stages of wound healing, the relative expression levels of collagen shifts from a predominant expression of collagen type III to a predominant expression of collagen type I. The appropriate synthesis and degradation of collegen is under precise regulation of the MMPs (Davis and Saunders, 2006). When MMP activity is excessively high, inappropriate collagen deposition and deformed collagen structure arises (Ballas and Davidson, 2001). Our results are consistent with other studies in which in vivo exposure to TCDD has been shown to significantly alter the expression of genes involved in collagen synthesis, disposition and organization (i.e., MMPs and TIMPS) in the heart, kidney (Kung et al., 2009) and in the zebrafish model, during fin regeneration (Mathew et al., 2009). These events are thought to underlie TCDD’s adverse effects on ECM remodeling and elevated/mislocalization of collagen in these systems.
Macrophages play a pivotal role in integrating the inflammatory and regenerative processes that occur during proper wound healing via their expression of cytokines and chemokines (Eming et al., 2007a, Broughton et al., 2006, Goren et al., 2009). Our findings that exposure to TCDD increases the macrophage population within the wounded cutaneous tissue (Figure 5) is consistent with a number of observations obtained from in vivo studies which have examined the impact of TCDD exposure in other tissue types using different in vivo experimental paradigms. For example, TCDD given to mice as a single i.p. dose (similar to the i.p. dose of TCDD used in the current study) was found to increase the expression of the macrophage marker (F4/80) as well as the expression of monocyte chemoattractant protein 1 (MCP1) in the thymus, heart, liver, kidney and adipose tissue at seven days following exposure (Vogel et al., 2007). In addition, acute administration of TCDD at a dose higher than that used in the current study was found to induce both hepatocellular damage and an influx of F4/80 positive inflammatory cells (Pande et al., 2005). Finally, in antigen-challenged mice, exposure to TCDD was found to increase macrophage production of both IL1 and TNF (Clark et al., 1991, Moos et al., 1997). It has been previously proposed that TCDD may exert its in vivo proinflammatory effect by enhancing the ability of macrophages to increase production of IL-like cytokines (i.e., TNFα, TNFβ, IL1α and IL1β) (Moos et al., 1997, Pande et al., 2005). It is also possible that TCDD enhances the recruitment of macrophages to the site of injury via enhanced expression of chemokines such as MCP1. An interesting observation in our current study (data not shown) is that the TCDD-induced increase in macrophage cell numbers within the wounded tissue appears to coincide with the formation of the adipose tissue. In animal models of liver disease, the major source of MCP1 expression is the fat storing cells (Czaja et al., 1994). Thus, it is possible that during cutaneous wound healing, the highly lipophilic TCDD present in the adipose tissue, increases the expression of MCP1 which then acts to recruit the macrophages into the wound bed.
Using a cutaneous wounding model, we have demonstrated that exposure to an environmental contaminant, TCDD, is capable of disrupting the wound healing process. Given that these wound healing processes are relevant to a number of chronic human disease states, such as cancer, our future efforts will utilize additional murine models to determine whether exposures to environmental AHR agonists may similarly impinge on these pathophysiological processes within the context of specific diseases.
Acknowledgements
We are very grateful to the Steve Post and Eric Smart laboratories for their guidance in the wound healing protocols, to Linda Zimmerman for her assistance with the microscopy and immunohistochemistry and to Donna Gilbreath for her assistance with graphics. We also thank Dr. Robert Hadley and Dr. Michael Piascik for their insights and assistance in editing. This work was supported by grants from the National Institute of Environmental Health (R01 ES 014849 and R01 ES011295).
Abbreviations
- AHR
aryl hydrocarbon receptor
- CYP1A1
cytochrome P4501A1
- CYP1B1
cytochrome P4501B1
- DAPI
4’,6-diamidino-2-phenylindole
- ECM
extracellular matrix
- H&E
hematoxylin and eosin
- IF
immunofluorescence
- IL-1α
interleukin 1 alpha
- IL-1β
interleukin 1 beta
- IL-4
interleukin 4
- IL-10
interleukin 10
- IL-12
interleukin 12
- MCP1
monocyte chemoattractant protein 1
- MMPs
matrix metalloproteinases
- MPO
myeloperoxidase
- TIMPS
tissue inhibitors of MMPs
- TGFβ
transforming growth factor beta
- TNFα
tumor necrosis factor alpha
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aragon AC, Goens MB, Carbett E, Walker MK. Perinatal 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure sensitizes offspring to angiotensin II-induced hypertension. Cardiovasc Toxicol. 2008;8:145–154. doi: 10.1007/s12012-008-9023-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas CB, Davidson JM. Delayed wound healing in aged rats is associated with increased collagen gel remodeling and contraction by skin fibroblasts, not with differences in apoptotic or myofibroblast cell populations. Wound Repair Regen. 2001;9:223–237. doi: 10.1046/j.1524-475x.2001.00223.x. [DOI] [PubMed] [Google Scholar]
- Black LD, Allen PG, Morris SM, Stone PJ, Suki B. Mechanical and failure properties of extracellular matrix sheets as a function of structural protein composition. Biophys J. 2008;94:1916–1929. doi: 10.1529/biophysj.107.107144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braiman-Wiksman L, Solomonik I, Spira R, Tennenbaum T. Novel insights into wound healing sequence of events. Toxicol Pathol. 2007;35:767–779. doi: 10.1080/01926230701584189. [DOI] [PubMed] [Google Scholar]
- Broughton G, 2nd, Janis JE, Attinger CE. The basic science of wound healing. Plast Reconstr Surg. 2006;117:12S–34S. doi: 10.1097/01.prs.0000225430.42531.c2. [DOI] [PubMed] [Google Scholar]
- Cavalcante FS, Ito S, Brewer K, Sakai H, Alencar AM, Almeida MP, Andrade JS, Jr., Majumdar A, Ingenito EP, Suki B. Mechanical interactions between collagen and proteoglycans: implications for the stability of lung tissue. J Appl Physiol. 2005;98:672–679. doi: 10.1152/japplphysiol.00619.2004. [DOI] [PubMed] [Google Scholar]
- Clark GC, Taylor MJ, Tritscher AM, Lucier GW. Tumor necrosis factor involvement in 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated endotoxin hypersensitivity in C57BL/6J mice congenic at the Ah locus. Toxicol Appl Pharmacol. 1991;111:422–431. doi: 10.1016/0041-008x(91)90247-c. [DOI] [PubMed] [Google Scholar]
- Czaja MJ, Geerts A, Xu J, Schmiedeberg P, Ju Y. Monocyte chemoattractant protein 1 (MCP-1) expression occurs in toxic rat liver injury and human liver disease. J Leukoc Biol. 1994;55:120–126. doi: 10.1002/jlb.55.1.120. [DOI] [PubMed] [Google Scholar]
- Davis GE, Saunders WB. Molecular balance of capillary tube formation versus regression in wound repair: role of matrix metalloproteinases and their inhibitors. J Investig Dermatol Symp Proc. 2006;11:44–56. doi: 10.1038/sj.jidsymp.5650008. [DOI] [PubMed] [Google Scholar]
- Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol. 2007a;127:514–525. doi: 10.1038/sj.jid.5700701. [DOI] [PubMed] [Google Scholar]
- Eming SA, Werner S, Bugnon P, Wickenhauser C, Siewe L, Utermohlen O, Davidson JM, Krieg T, Roers A. Accelerated wound closure in mice deficient for interleukin-10. Am J Pathol. 2007b;170:188–202. doi: 10.2353/ajpath.2007.060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Kampfer H. Excisional wound healing: an experimental approach. In: DiPietro LA, Burns AL, editors. Methods in molecular medicine. vol. 78. Humana Press; 2003. pp. 3–15. [DOI] [PubMed] [Google Scholar]
- Furness PN, Rogers-Wheatley L, Harris KP. Semiautomatic quantitation of macrophages in human renal biopsy specimens in proteinuric states. J Clin Pathol. 1997;50:118–122. doi: 10.1136/jcp.50.2.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goren I, Allmann N, Yogev N, Schurmann C, Linke A, Holdener M, Waisman A, Pfeilschifter J, Frank S. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am J Pathol. 2009;175:132–147. doi: 10.2353/ajpath.2009.081002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Hsu EL, Yoon D, Choi HH, Wang F, Taylor RT, Chen N, Zhang R, Hankinson O. A proposed mechanism for the protective effect of dioxin against breast cancer. Toxicol Sci. 2007;98:436–444. doi: 10.1093/toxsci/kfm125. [DOI] [PubMed] [Google Scholar]
- Ivnitski-Steele I, Walker MK. Inhibition of neovascularization by environmental agents. Cardiovasc Toxicol. 2005;5:215–226. doi: 10.1385/ct:5:2:215. [DOI] [PubMed] [Google Scholar]
- Kerkvliet NI, Oughton JA. Acute inflammatory response to sheep red blood cell challenge in mice treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): phenotypic and functional analysis of peritoneal exudate cells. Toxicol Appl Pharmacol. 1993;119:248–257. doi: 10.1006/taap.1993.1066. [DOI] [PubMed] [Google Scholar]
- Kung T, Murphy KA, White LA. The aryl hydrocarbon receptor (AhR) pathway as a regulatory pathway for cell adhesion and matrix metabolism. Biochem Pharmacol. 2009;77:536–546. doi: 10.1016/j.bcp.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK. Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol B. 2005;175:221–230. doi: 10.1007/s00360-005-0483-3. [DOI] [PubMed] [Google Scholar]
- Marlowe JL, Puga A. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J Cell Biochem. 2005;96:1174–1184. doi: 10.1002/jcb.20656. [DOI] [PubMed] [Google Scholar]
- Mathew LK, Simonich MT, Tanguay RL. AHR-dependent misregulation of Wnt signaling disrupts tissue regeneration. Biochem Pharmacol. 2009;77:498–507. doi: 10.1016/j.bcp.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland-Mancini MM, Funk HM, Paluch AM, Zhou M, Giridhar PV, Mercer CA, Kozma SC, Drew AF. Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J Immunol. 184:7219–7228. doi: 10.4049/jimmunol.0901929. [DOI] [PubMed] [Google Scholar]
- Moos AB, Oughton JA, Kerkvliet NI. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on tumor necrosis factor (TNF) production by peritoneal cells. Toxicol Lett. 1997;90:145–153. doi: 10.1016/s0378-4274(96)03838-6. [DOI] [PubMed] [Google Scholar]
- Pande K, Moran SM, Bradfield CA. Aspects of dioxin toxicity are mediated by interleukin 1-like cytokines. Mol Pharmacol. 2005;67:1393–1398. doi: 10.1124/mol.105.010983. [DOI] [PubMed] [Google Scholar]
- Peng TL, Chen J, Mao W, Song X, Chen MH. Aryl hydrocarbon receptor pathway activation enhances gastric cancer cell invasiveness likely through a c-Jun-dependent induction of matrix metalloproteinase-9. BMC Cell Biol. 2009;10:27. doi: 10.1186/1471-2121-10-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Swanson HI. Activation of the aryl hydrocarbon receptor by TCDD inhibits senescence: a tumor promoting event? Biochem Pharmacol. 2009;77:681–688. doi: 10.1016/j.bcp.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CF, Nishimura N, Sciullo E, Wong P, Li W, Matsumura F. Modulation of the chemokines KC and MCP-1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice. Arch Biochem Biophys. 2007;461:169–175. doi: 10.1016/j.abb.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Wenstrup RJ, Florer JB, Davidson JM, Phillips CL, Pfeiffer BJ, Menezes DW, Chervoneva I, Birk DE. Murine model of the Ehlers-Danlos syndrome. col5a1 haploinsufficiency disrupts collagen fibril assembly at multiple stages. J Biol Chem. 2006;281:12888–12895. doi: 10.1074/jbc.M511528200. [DOI] [PubMed] [Google Scholar]
- Yoshizawa K, Heatherly A, Malarkey DE, Walker NJ, Nyska A. A critical comparison of murine pathology and epidemiological data of TCDD, PCB126, and PeCDF. Toxicol Pathol. 2007;35:865–879. doi: 10.1080/01926230701618516. [DOI] [PMC free article] [PubMed] [Google Scholar]