

Introduction
Implantable, three-dimensional (3D) polymer scaffolds have been drawing attention from various disciplines because of their unique properties [1–6]. There are several methods of fabricating biocompatible and biodegradable polymer scaffolds [7–12]. These implantable 3D scaffolds, made of various materials, such as lactide and glycolide polymers [1, 5, 6], poly(glycerol sebacate) [7], silk fibroin [10], and poly(propylene fumarate) [8, 13, 14], have been adapted for numerous biomedical studies. Efficient drug delivery, with a controlled drug-release manner at the targeted site, is one of the major applications for polymer scaffolds in biomedical research [15–24]. Numerous polymer scaffold candidates have been developed as drug-releasing matrices for in vitro and, potentially, in vivo use. For example, chitosan/hydroxyapatite composite scaffolds have shown a reduced initial burst, followed by a constant release of tetracycline hydrochloride in vitro [15]. Calcium alginate beads coated with silk fibroin showed improved shell stability and controlled release of BSA and FITC-Insulin model drug proteins [18]. Hydroxyapatite scaffolds have been reported to successfully release ceftriaxone drug molecules in a controlled manner due to their well-defined porous surface structure [21]. The drug delivery and release applications of hydrogel scaffolds, further developed by infusion with chitosan, have been recently reviewed [24]. Combining the advantages of each unique scaffold system may develop the optimized attributes that are critical in drug delivery applications: drug-loading capacity, controllable drug release rate; flexibility of drug loading; feasibility of drug delivery and release imaging; and quantitative assessment of loaded and released drug molecules on the scaffold.
The use of 3D poly(propylene fumarate) (PPF) scaffolds as a drug-release matrix is relatively new, but is a promising application in several aspects. First of all, the highly porous structure of PPF scaffolds provides sizable drug-loading spaces. Furthermore, a photo-crosslinked PPF scaffold is able to retain its initial porosity and mechanical properties for 18–32 weeks in vitro [25], and its degradability can be controlled by adjusting fabrication parameters, such as PPF molecular weight and photoinitiator content [26]. This ability to control PPF degradation may benefit certain applications that require the extended drug release. Although PPF scaffolds are generally recognized for their potential use in bone replacement [27, 28], PPF scaffolds have other advantages such as their biocompatibility, tunable porous surface area, and degradation time, which may promote them as an optimized platform to release drugs in a controlled manner. However, there are also a few innate limitations of the PPF scaffold for drug delivery applications. The direct incorporation of water-soluble drug molecules into the scaffold is challenging due to the hydrophobicity of PPF. Loaded drugs might be degraded by certain fabrication processes, for instance, the salt porogen leaching step [29]. Therefore, an alternative approach for PPF scaffolds to carry over a drug molecule is required.
There have been efforts to utilize nanoparticles as drug carriers and to embed a drug-nanoparticle complex in a scaffold matrix [30]. However, simultaneous detection and quantification of released drug-nanoparticles from these scaffold matrices has not been studied in detail, since most of the detection methodologies rely on only a single method of detection (e.g., fluorescence imaging or absorption spectroscopy), which may be difficult to apply to the detection of discrete nanoparticles and drug molecules.
Rapid improvements in magnetic resonance imaging (MRI) instrumentation and techniques have led to increased spatial resolution (up to 50–100 μm for rodent in vivo imaging). In addition, a variety of novel nanoparticles designed for MRI can enhance the MRI contrast even further, making it possible to image cellular and molecular events non-invasively and co-register these events with 3D anatomical structures. Recently, MRI was also suggested for studying drug release non-invasively from liposomes [31] using iron oxide-based nanoparticles and gadolinium-based agents [31, 32]. In addition to the more traditional MRI contrast mechanisms, which rely on the longitudinal relaxation (T1) and transverse relaxation (T2) of water protons [33], a new type of MRI contrast has been developed that relies on direct chemical exchange of protons with bulk water. A variety of organic and organo-metallic compounds have a sufficient number of protons (called “exchangeable protons”) with suitable chemical exchange rates and chemical shifts to be detected sensitively. Once a sample is inside the magnetic field, these exchangeable protons can be “magnetically tagged” using a radiofrequency pulse, called a saturation pulse, which is applied at the exchangeable proton’s resonance frequency. The tagged protons exchange with the protons of surrounding water molecules, and, consequently, reduce the MRI signal from the water protons and enhance the MRI contrast. The exchangeable protons are replaced with fresh protons and the same saturation process is repeated. After several seconds of this process, the effect becomes larger and larger (a so-called “saturation amplification”), and very low concentrations of agents can be detected through the water signal. Hence, these agents are called Chemical Exchange Saturation Transfer (CEST) contrast agents[34]. The main advantage of CEST-MRI is that bio-organic molecules, such as proteins, can be used for increasing the MRI contrast [35–37]. These can be used to image controlled drug release with minimal invasiveness in real time.
Here we report on the use of porous PPF scaffolds loaded with doxorubicin (DOX)-coated iron oxide and manganese oxide nanoparticles as a vehicle for sustained anti-cancer drug release. Using nanoparticles as a drug carrier contributes to more efficient loading of drug molecules onto the PPF scaffold. It also allows monitoring the release of drug-nanoparticle complexes from the PPF scaffold surface via changes in MRI contrast and absorption spectra in a media containing scaffold pieces. In addition, as a proof-of-concept experiment, it was demonstrated that the diamagnetic CEST contrast agent, protamine sulfate (PS), can also be used directly, without nanoparticle carriers, to monitor release from the PPF scaffolds by MRI. This report about drug-delivering 3D PPF scaffolds, with a sustained release rate and bimodal imaging (fluorescence and magnetic resonance) capabilities, suggests this new system may potentially be used in various biomedical applications.
Materials and Methods
Synthesis and characterization of iron oxide nanoparticles (IONP) and manganese oxide nanoparticles (MONP)
IONP and MONP with amine functional groups on their surfaces were prepared using silane conjugation chemistry and a surface exchange method previously reported elsewhere [38, 39]. Briefly, both nanoparticles, each dispersed in non-polar organic solvent, were synthesized by the thermal decomposition of a Fe-oleate and Mn-oleate complex [40]. Then, a coating of IONP and MONP with porous silica was achieved using a sol-gel reaction of tetraethyl orthosilicate (TEOS) in an aqueous solution containing CTAB and Fe3O4 (or MnO) nanoparticles. First, Fe3O4 (or MnO) nanoparticles in chloroform were poured into 1 mL of 0.55 M aqueous cetyltrimethylammonium bromide (CTAB) solution, and the resulting solution was stirred vigorously for 30 min. Forming an oil-in-water microemulsion, the mixture was heated up to 60ºC and kept at that temperature for 10 min under stirring in order to evaporate the chloroform. The resulting transparent solution of Fe3O4(or MnO)/CTAB was added to a mixture of 9 mL of water and 60 μL of 2 M NaOH solution, and the mixture was heated up to 70ºC under stirring, followed by the addition of 0.1 mL of tetraethylorthosilicate (TEOS) and 0.6 mL of ethylacetate [39]. In the middle of the reaction, for amine functionalization of the surface, 10 μL of APTES was added and the solution was stirred for 3 h. The porous silica-coated Fe3O4 (or MnO) nanoparticles were washed with ethanol to remove unreacted species, and then were redispersed in 5 mL of ethanol. A JEM 2100 transmission electron microscope (JEOL, Tokyo, Japan) was used to characterize the size and shape of synthesized nanoparticles.
Doxorubicin coating of nanoparticles
The anti-cancer drug doxorubicin (Dox) was attached to IONP and MONP by its electrostatic interaction with the negatively charged porous silica surface of the nanoparticles. 1 mg/mL of each nanoparticle batch was dispersed in PBS, and 1 mM Dox was mixed with the nanoparticles and incubated for 1 hr. Next, 9 mL of PBS was added to the Dox and nanoparticle mixture and centrifuged at 3000g for 5 min to precipitate the Dox-coated nanoparticles. After the supernatant was aspirated, the Dox-coated nanoparticles were washed two more times with phosphate buffered saline (PBS). Finally, the Dox-nanoparticle solution was resuspended in 1 mL of PBS. The amount of Dox coated on the nanoparticles was calculated by measuring the difference in absorption at 480 nm between the Dox stock solution and the supernatant after 1 h incubation with the nanoparticles.
PPF scaffold fabrication and characterization
PPF was synthesized as previously described [8, 41]. Purified PPF with a number average molecular weight of 1,260 Da was used for porous scaffold fabrication. PPF was first mixed with 0.5 wt% of a photoinitiator, bis(2,4,6-trimethylbenzoyl) phenylphosphine oxide (BAPO, Ciba chemical), and NaCl crystals (>500 μm) were added to the PPF/BAPO mixture as a porogen. The PPF/BAPO/NaCl mixture was packed into glass cylinder molds of 6.3 mm in diameter and radiated with UV light for 2 hr for photo-crosslinking. Crosslinked PPF was retrieved and cut into disks of 5 mm in thickness. Salt crystals were leached out in water for 72 hr and porous PPF scaffolds were air-dried overnight.
Dox-nanoparticle incorporation into the PPF scaffold
Two methods were adapted in order to incorporate the Dox-nanoparticles into the PPF scaffold matrix. In the surface-coating method, Dox-nanoparticles were adsorbed onto the surface of the PPF scaffold. Drops of 0.1 mM of Dox-IONP or Dox-MONP were placed on the surface of PPF scaffolds with a 6.3 mm of diameter and a 5.0 mm thickness. The coated scaffolds were dried in a vacuum chamber for 30 min. The presence of the Dox coating on the PPF scaffolds was confirmed by UV light (350 nm) excitation, generating orange (560 nm) emission from Dox on the surface of the scaffolds.
The pre-mixing method was performed by adding Dox-nanoparticles (0.1 mM) to the PPF/BAPO/NaCl mixture before photo-cross-linking. A mixture of Dox-nanoparticles and PPF was thoroughly stirred to obtain a well-distributed Dox-nanoparticles. Next, the PPF/particle mixture was processed to fabricate porous scaffolds in the manner described above.
Time-lapse study of Dox-nanoparticles release from PPF scaffolds
PPF scaffolds bearing Dox-nanoparticles on the surface, by either the surface-coating or pre-mixing method, were placed in 2 mL tubes containing PBS or cell culture media. Tubes were kept at 37ºC in an incubator, and 10 μL of supernatant in each tube was then sampled at the designated time point for the analysis of MRI contrast. Tubes were stored in an incubator until each time point was reached. Three sets of independent samples were tested at each time point.
MRI analysis of PPF scaffolds and released Dox-nanoparticles
MR images of the PPF scaffolds in solution or the supernatants of a sample containing a PPF scaffold coated with Dox-nanoparticles were obtained at different time points using an 11.7 T Bruker Avance system equipped with a 15 mm birdcage RF coil. T1 and T2 relaxation times were measured using a modified MSME protocol [42]. The typical settings for T2 measurements were as follows: 128 × 128 matrix size; 3000 ms repetition time (TR); echo time (TE); series of 6.4, 12.8, 19.2, 25.6, 32.0, 38.4, 44.8, 51.2, 57.6, 64.0, 70.4, and 76.8 (ms), number of averages=2. The typical settings for T1 measurements were as follows: matrix size 64 × 64 pixels, 6.4 ms TE; and TR series of 10000, 6000, 4000, 3000, 1500, 1000, 800, 400, and 200 (ms). All data processing was performed using custom-made codes in Matlab (ver. R2009a, Mathworks, Natick, MA).
MRI quantification of released Dox-nanoparticles
The amount of Dox-nanoparticles released from the scaffold was calculated by measuring the MR contrast in the solution at each time point. A decreased T2 relaxation time in the sampled solution from a tube containing a Dox-IONP-coated PPF scaffold was due to released IONP. Using a standard curve relating the amount of nanoparticles and the 1/T2 relaxation rate, the amount of nanoparticles at a certain incubation time point was calculated. In a similar manner, the amount of Dox-MONP in the solution was calculated using a standard curve between the mass of MONP and the 1/T1 relaxation rate. The release of Dox-nanoparticles was confirmed by measuring the Dox absorption spectra of the sampled solution at each time point. The intensity of the signature Dox absorption peak at 480 nm was used to estimate the amount of Dox in the solution.
Drug efficacy test against PC12 cells
Scaffolds carrying drug nanoparticles were incubated with a monolayer of PC12 cells in six-well plates. After 48 hr incubation, the culture medium (100 μL) was removed from each well. Cell Titer-Blue solution (Promega, Madison, WI) (100 μL) was added to each well and the plates were incubated for 3 hr. The uorescence was quantified with an excitation wavelength of 560 nm and an emission wavelength of 590 nm. The percent of cell survival was calculated by normalizing the uorescence intensity of the samples to the fluorescence intensity of the control.
Attachment of protamine sulfate to PPF scaffolds
Protamine sulfate (PS, Sigma-Aldrich, St. Louis, MO) in PBS (1 mg/mL) was surface-coated onto a PPF scaffold by adsorption following the procedure as described above. PPF scaffolds with protamine sulfate (PS) were incubated in a tube with PBS at 37ºC, and 10 μl of supernatant from each tube was obtained at pre-determined time points for CEST imaging and quantification of released CEST molecules at each time point. A 5 mg/mL protamine sulfate solution sample was also used as positive control.
CEST MRI quantification of released protamine sulfate in solution
All samples described above were transferred into capillaries (inner diameter: 1.1 mm) and immobilized in a home-made high-throughput MR sample holder [43]. CEST MRI images were acquired at 310 K using an 11.7 T Bruker Avance system equipped with a 15 mm sawtooth RF coil. Each series of MRI studies, including z-spectra and B0 mapping acquisition, took 1 hour and 8 min. A modified RARE sequence (TR=6.0 sec, effective TE = 16.5 ms, RARE factor =16, slice thickness=1 mm, FOV=14×14 mm, matrix size=128×32, resolution= 0.11×0.44 mm2, and NA=2), including a magnetization transfer (MT) module (one CW pulse, saturation power = 4.7 μT, pulse duration 4sec) was used to acquire CEST-weighted images from −5 ppm to 5 ppm (step= 0.2 ppm) around the water resonance (0 ppm). The absolute water resonant frequency shift was measured using a modified Water Saturation Shift Reference (WASSR) method[44], using the same parameters as in CEST imaging, except TR=1.5 sec, saturation pulse duration= 500 ms, saturation power =0.5 μT, and a sweep from −3 ppm to 3 ppm (step= 0.1ppm). Data processing was performed using custom-written scripts in Matlab. Z-spectra were calculated from the mean of ROI for each sample after the pixelwise B0 correction using WASSR spectra [43]. CEST contrast was quantified using MTRasym = (S−Δω – S+Δω)/ S(−5ppm) and was computed at each offset, Δω. The amount of PS was measured using a bicinchoninic acid (BCA) assay according to the manufacturer’s instructions (Thermo Scientific (Pierce) 23227).
Drug loading capacity estimation
A PPF scaffold piece was placed in a 2 mL centrifuge tube, and 100 μl of either 0.1 mM Dox-IONP or Dox-MONP was dropped onto the scaffold. After the Dox-nanoparticle suspension was completely dried and coated on the scaffold, the scaffold piece was removed and 1 mL of PBS was added to the tube to dissolve any residual Dox-nanoparticles. The amount of residual Dox in PBS was calculated using the absorption spectra of Dox molecules as described above. The amount of loaded drug was calculated by subtracting the residual amount from the initial amount of Dox molecules. The loading capacity was determined as the weight of the loaded drug/weight of the scaffold × 100% [45] (Supplementary Material, Table S2). The loading capacity of PS on a scaffold was estimated by measuring the difference between the initial loading amount of PS (5 mg) to the scaffold and residual amount of PS left in a tube. The residual PS in a tube was re-dissolved in PBS, and then measured using a BCA assay.
Results and Discussion
Synthesis and characterization of IONP and MONP
The overall procedure for developing a drug-releasing PPF scaffold system is illustrated in Figure 1.
Figure 1. Schematic illustration of the procedure for fabrication of PPF scaffolds and approaches for loading drug-coated nanoparticles.

Drug-coated nanoparticles were released in the medium from the scaffold. The release kinetics was studied by measuring the amount of nanoparticles in the solution using MRI contrast. The efficacy of the drug-coated nanoparticles released in the medium was measured by co-culturing the scaffolds with cells.
Both IONP and MONP were surface-modified with porous silica to enhance their water-solubility and minimize leakage of the core ions. Representative transmission electron microscopy (TEM) images of both types of nanoparticles showed well-dispersed, spherically shaped particles with an average particle diameter of 40 and 70 nm for IONP and MONP, respectively (Figure 2B).
Figure 2. Characterization of IONP and MONP.
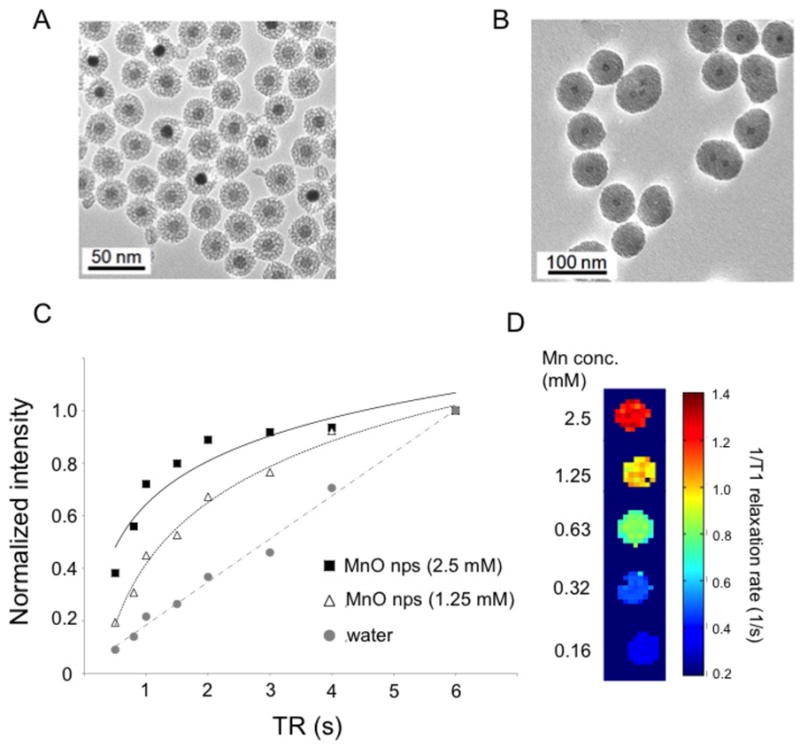
TEM images of IONP (A) and MONP (B). The mean diameter of the nanoparticles was 40 and 70 nm for IONP and MONP, respectively. (C) Plot of normalized MR intensity versus repetition time for MONP. (D) 1/T1-weighted MR color map of MONP.
TEM imaging revealed that both IONP and MONP were stable in water and did not form aggregates. While the IONP have superparamagnetic characteristics, thereby shortening the transverse relaxation (T2) of water protons, the MONP are paramagnetic, shortening the water proton longitudinal relaxation (T1) [42, 46]. As described in Figure 2C and 2D, MONP shortens the T1 in a concentration-dependent manner with a T1 relaxivity (R1) of 0.61 mM−1s−1 at 11.7 T.
Fabrication and characteristics of the porous PPF scaffold
The porous PPF scaffold has a large surface area that can retain a significant number of molecules. The porosity and pore size of the scaffold was relatively high, while maintaining the physical matrix so that the scaffold could be degraded gradually in solution at physiological conditions.
The porous structure of the PPF scaffold is clearly shown in SEM images (Figure 3A). Premixed nanoparticles (Figure 3B) and surface-coated nanoparticles (Figure 3C) and their aggregates on the PPF porous scaffold are all clearly visible in the SEM images. It is notable that the PPF scaffold is clearly visible on T2-weighted MR images (Figure 3D), which indicates the potential for their use in in vivo imaging.
Figure 3. Characterization of the scaffold.

(A) SEM image showing the surface of the two control scaffolds. The left panel shows the porous structure; the right panel the clean surface. (B) SEM image showing the attachment of nanoparticles on the scaffold mixed with the nanoparticles. Large aggregates of nanoparticles have been adsorbed to the surface of the PPF scaffolds. (C) SEM image of Dox-carrying nanoparticles, which have been attached to the surface of the scaffolds after the scaffolds are coated with the particles. (D) MRI of a sample tube containing several small pieces of PPF scaffold (5 mm thickness) in PBS. Black arrow: porous structured PPF scaffold. White arrow: capillary containing water as a control.
Surface modification of the PPF scaffold with Dox-coated magnetic nanoparticles
To examine the drug releasing capability of the PPF scaffold system, a chemotherapeutic agent, Doxorubicin (Dox), was incorporated into the PPF scaffolds via nanoparticles. Dox was coated onto the surface of the nanoparticles, and the Dox-nanoparticle complexes were subsequently incorporated into the PPF scaffolds, either by electrostatic attachment or by pre-mixing into the PPF resin before cross-linking in the fabrication process (Supplementary Material, Figure S2A and S2B). PPF scaffolds that were either surface coated or pre-mixed with Dox-nanoparticles were incubated in PBS, and the PBS supernatant was sampled to analyze the trace of released nanoparticles in the solution (Supplementary Material, Figure S2C). The presence of Dox was confirmed by measuring fluorescence from the surface of the PPF scaffolds (Supplementary Material, Figure S2D). The loading capacity of Dox on the surface of single PPF scaffold was estimated as 9.18 % for the Dox-IONP and 8.81 % for the Dox-MONP (Supplementary Material, Table S2).
Quantification of released drug-nanoparticles
Figure 4A–D shows the release of the Dox-coated nanoparticles for the PPF scaffolds after incubation for 48 h, with MRI. It can be seen that the Dox-coated IONP shorten the T2 of the surrounding solution, indicating release of the drug (Figure 4A, B). Similar results were observed for the Dox-MONP coated scaffold (Figure 4C, D) where the released drug led to a reduction in the T1 of the surrounding solution
Figure 4. Quantification of drug release using MRI signal changes.
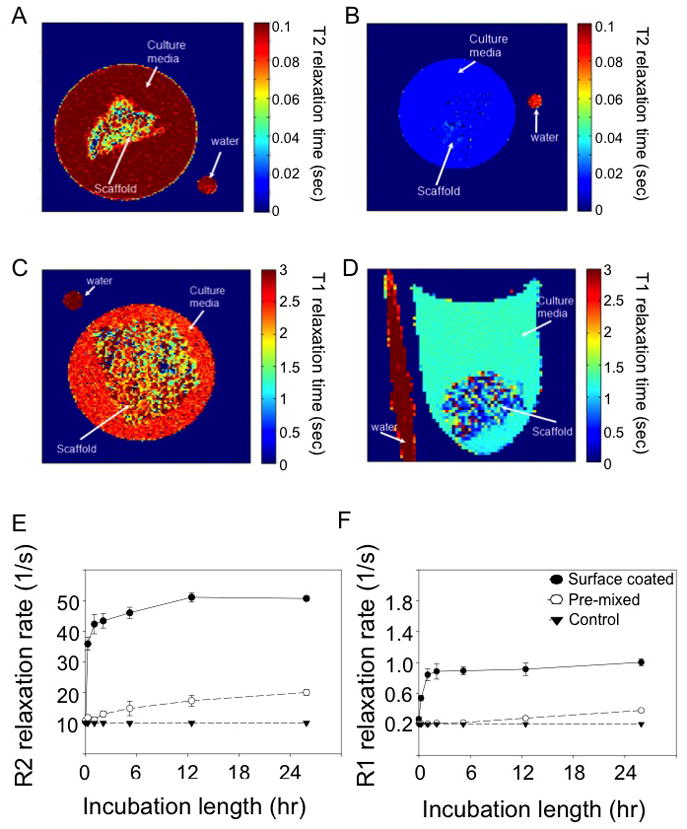
T2 relaxation map of control scaffold (A), and PPF scaffolds coated with IONP (B). T1 relaxation map of control scaffold (C) and PPF scaffolds coated with MONP (D). All the maps were acquired after 48 h incubation at 37ºC in PBS. Plots of 1/T2 (E) and 1/T1 (F) relaxation in the solution versus the length of scaffold incubation. Both 1/T2 and 1/T1 shortening were proportional to the amount of Dox-coated nanoparticles that had been released from each scaffold.
Close monitoring of the release by sampling the surrounding solution at different time points reveals the different magnitudes of signal change for pre-mixed and surface-coated particles (Figure 4E, F). The surface coating method of incorporating nanoparticles to the scaffold released more particles faster than the pre-mixing approach at short time periods (within 24h incubation). The ratio between mixed and coated can be further used to fine-tune the release rate of the drug in order to fit different treatment regimes.
Calculating the release rate
The release profiles of Dox-IONP or Dox-MONP were calculated based on the relaxivity data obtained from the MR images (Supplementary Material, Figure S3). The drug release kinetics for the coated Dox-IONP or Dox-MONP were compared with the modeling results (supporting information) and showed a steep increase in the amount of released particles at an early incubation time period (up to 24 h). At later times, the profile maintains a constant level of particle mass until 30 days after incubation.
UV-VIS absorption spectroscopy was performed to quantify the amount of Dox in the solution, either free or attached to the nanoparticles, after either IONP or MONP were released from each PPF scaffold carrying the nanoparticles (Supplementary Material, Figure S4). The release profiles of both Dox-IONP and Dox-MONP in Figure S4 were comparable to the profiles of the nanoparticles released from the scaffolds as measured with MRI in Figure S3. UV-VIS absorption spectra confirmed the appearance of Dox absorption (490 nm) in the solution due to its release from the scaffold. The amount of solution released Dox reached 592.2 ng in 1 ml of PBS after 48 h incubation (for Dox-MONP) as measured by using a fluorescence intensity of released Dox molecules in the solution. (Supplementary, Material Figure S4). The results showed an increasing amount of Dox, corresponding to the trend of released nanoparticles in the solution (after the same period of incubation (Supplementary Material, Figure S4). Cell viability assay demonstrated that incorporation of Dox into either IONP or MONP had no effect on the anti-cancer activity of the drug (Supplementary Material, Figure S5).
Quantifying protein release from PPF scaffolds
In order to demonstrate the versatility of the PPF scaffolds in regulating the release of drugs and the feasibility of using PPF scaffolds in other systems, we studied the kinetics of the release of protamine sulfate (PS), a small cationic protein, as a model system for protein and polypeptide release. Protamine has a high content of arginine residues, each with two exchangeable protons that can be detected via chemical exchange saturation transfer (CEST) MRI [47]. The guanidine proton exchanges at 1.8 ppm from the water peak and the amide proton exchanges 3.6 ppm.
PS was coated on the surface of the PPF scaffold by adsorption. The loading capacity of PS molecules on the scaffold was estimated as 8.15% (Supplementary Material, Table S2). The MTRasym (measure of the CEST contrast) increased with incubation time at the irradiation frequency offset of 1.8 ppm (Figure 5). Based on the plot in Figure 5a–b, sustained release was observed for the first 24 hours.
Figure 5. Release of PS from PPF scaffolds, as measured with CEST MRI.

(A) Plot of MTRasym versus irradiation frequency offset. The solution surrounding the PPF scaffold coated with PS was sampled at different time points: red solid circle (
 ): 6 hours; green solid square (
): 6 hours; green solid square (
 ): 12 hours; black open triangle (△): 24 hours; blue open circle (
): 12 hours; black open triangle (△): 24 hours; blue open circle (
 ): 72 hours. Solid line=5 mg/mL protamine sulfate; dashed line=PBS. (B) The MTRasym at 1.8 ppm shows an increase with incubation time due to the release of PS from the PPF scaffolds. Inset: MTRasym map showing CEST signal intensity corresponding to the plot in B0, which provides direct visualization of the released PS into the solution. (C) Samples at different time points were measured with CEST MRI (red circle,
): 72 hours. Solid line=5 mg/mL protamine sulfate; dashed line=PBS. (B) The MTRasym at 1.8 ppm shows an increase with incubation time due to the release of PS from the PPF scaffolds. Inset: MTRasym map showing CEST signal intensity corresponding to the plot in B0, which provides direct visualization of the released PS into the solution. (C) Samples at different time points were measured with CEST MRI (red circle,
 ) and BCA assay (black hyphen symbol -; average ± standard deviation, n=3). (D) The mass of PS released from the 3 scaffold disks (mean weight of disk= 41.8±0.54 mg, as in C above) placed in 1 mL of PBS as calculated using a standard curve. Plots in C and D represent data from two independent measurements.
) and BCA assay (black hyphen symbol -; average ± standard deviation, n=3). (D) The mass of PS released from the 3 scaffold disks (mean weight of disk= 41.8±0.54 mg, as in C above) placed in 1 mL of PBS as calculated using a standard curve. Plots in C and D represent data from two independent measurements.
In order to validate the CEST MRI measurements, imaging data from the first 12 hours were compared with protein measurements, using a BCA protein assay. Samples at eight consecutive time points were obtained from the solution containing a PPF scaffold coated with PS. PS in a range of 0–1.5 mg (in 1 mL of PBS) was used as a standard curve (Supplementary Material, Figure S6). Figure 5C illustrates the release profile of PS in the first 12 hours, as was measured both with CEST MRI and with the BCA assay. An excellent correlation (R2=0.997) was found for the two methods (Supplementary Material, Figure S7). Figure 5D shows the calculated mass of PS released into the solution, as calculated using the standard curves (Supplementary Material, Figure S7). These findings indicate that CEST MRI represents a novel technique that yields results similar to a conventional biochemical protein assay. Consequently, CEST MRI can be used to monitor release from the scaffold without intermediate metallic compounds.
MRI has been used extensively in the last two decades for anatomical, functional, and dynamic imaging. Rapid improvements in MRI instrumentation and techniques have led to increased spatial resolution (up to 50–100 μm for rodent imaging in vivo). The detection of drug carrying nanoparticles using MRI relies on the contrast in the MRI signal between the tissue of interest and its surrounding tissue, which can be further enhanced using contrast agents. These agents can be used for monitoring drug release from transplanted scaffolds with an anticipated resolution of 100 μm. However, in vivo, unlike in a tube, the diffusion rate of these agents will be most likely be more restricted by the extracellular matrix. On the other hand, immune cells can facilitate the removal of certain agents from the tissue. Therefore, further detailed characterization of the behavior of the different agents in vivo is required for in vivo translation.
Conclusions
This study shows that biodegradable 3D porous PPF scaffolds can be used for sustained drug release. Furthermore, a released drug (Dox) can be monitored both with MRI and with optical imaging, which allows quantification of the release rate without compromising the drug’s cytotoxic effect. Here we also demonstrate, for the first time, that CEST MRI can be used to monitor the release of bio-polymers (such as proteins) directly and in real time. This new approach has the potential to help monitor the controlled release of drugs, growth factors, and cytokines, which is particularly important for tailoring individual treatments.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R21 EB008769 (AAG), R21 NS065284 (AAG), R21 EB005252 (JWMB), K01 EB006394 (MTM), and NSF CBET 0448684 (JPF). JC acknowledges the National Institute of Standards and Technology (NIST) for the SRM development grant. JC thanks Ms. Aehee Shin at the Maryland Institute College of Art for assistance with the graphics. The authors also thank Dr. Vytas Reipa (NIST) and Dr. Peter C.M. van Zijl for their advice and Mr. Segun M. Bernard and Ms. Anna E. Munsey for technical assistance.
Footnotes
Detailed description of the material and methods, modeling, porosity, and pore size analysis, MR imaging parameters, 1/T1 and 1/T2 calculations, CEST imaging parameters, quantification of released Dox and PS, and drug loading efficiency calculation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thorrez L, Shansky J, Wang L, Fast L, VandenDriessche T, Chuah M, Mooney D, Vandenburgh H. Growth, differentiation, transplantation and survival of human skeletal myofibers on biodegradable scaffolds. Biomaterials. 2008;29:75–84. doi: 10.1016/j.biomaterials.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan G, Mooney DJ. New materials for tissue engineering: towards greater control over the biological response. Trends Biotechnol. 2008;26:382–392. doi: 10.1016/j.tibtech.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Kretlow JD, Klouda L, Mikos AG. Injectable matrices and scaffolds for drug delivery in tissue engineering. Adv Drug Deliv Rev. 2007;59:263–273. doi: 10.1016/j.addr.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 4.Holland TA, Mikos AG. Biodegradable polymeric scaffolds. Improvements in bone tissue engineering through controlled drug delivery. Adv Biochem Eng Biotechnol. 2006;102:161–185. doi: 10.1007/b137205. [DOI] [PubMed] [Google Scholar]
- 5.Shea LD, Wang D, Franceschi RT, Mooney DJ. Engineered bone development from a pre-osteoblast cell line on three-dimensional scaffolds. Tissue Eng. 2000;6:605–617. doi: 10.1089/10763270050199550. [DOI] [PubMed] [Google Scholar]
- 6.Freed LE, Vunjak-Novakovic G, Biron RJ, Eagles DB, Lesnoy DC, Barlow SK, Langer R. Biodegradable polymer scaffolds for tissue engineering. Biotechnology (N Y) 1994;12:689–693. doi: 10.1038/nbt0794-689. [DOI] [PubMed] [Google Scholar]
- 7.Redenti S, Neeley WL, Rompani S, Saigal S, Yang J, Klassen H, Langer R, Young MJ. Engineering retinal progenitor cell and scrollable poly(glycerol-sebacate) composites for expansion and subretinal transplantation. Biomaterials. 2009;30:3405–3414. doi: 10.1016/j.biomaterials.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kasper FK, Tanahashi K, Fisher JP, Mikos AG. Synthesis of poly(propylene fumarate) Nat Protoc. 2009;4:518–525. doi: 10.1038/nprot.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Ayoubi R, Eliopoulos N, Diraddo R, Galipeau J, Yousefi AM. Design and fabrication of 3D porous scaffolds to facilitate cell-based gene therapy. Tissue Eng Part A. 2008;14:1037–1048. doi: 10.1089/ten.tea.2006.0418. [DOI] [PubMed] [Google Scholar]
- 10.Hofmann S, Knecht S, Langer R, Kaplan DL, Vunjak-Novakovic G, Merkle HP, Meinel L. Cartilage-like tissue engineering using silk scaffolds and mesenchymal stem cells. Tissue Eng. 2006;12:2729–2738. doi: 10.1089/ten.2006.12.2729. [DOI] [PubMed] [Google Scholar]
- 11.Karageorgiou V, Kaplan D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials. 2005;26:5474–5491. doi: 10.1016/j.biomaterials.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Ameer GA, Mahmood TA, Langer R. A biodegradable composite scaffold for cell transplantation. J Orthop Res. 2002;20:16–19. doi: 10.1016/S0736-0266(01)00074-2. [DOI] [PubMed] [Google Scholar]
- 13.Kim K, Dean D, Lu A, Mikos AG, Fisher JP. Early osteogenic signal expression of rat bone marrow stromal cells is influenced by both hydroxyapatite nanoparticle content and initial cell seeding density in biodegradable nanocomposite scaffolds. Acta Biomaterialia. 2011;7:1249–1264. doi: 10.1016/j.actbio.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim K, Yeatts A, Dean D, Fisher JP. Stereolithographic bone scaffold design parameters: osteogenic differentiation and signal expression. Tissue Eng Part B Rev. 2010;16:523–539. doi: 10.1089/ten.teb.2010.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teng SH, Lee EJ, Wang P, Jun SH, Han CM, Kim HE. Functionally gradient chitosan/hydroxyapatite composite scaffolds for controlled drug release. J Biomed Mater Res B Appl Biomater. 2009;90:275–282. doi: 10.1002/jbm.b.31283. [DOI] [PubMed] [Google Scholar]
- 16.Shi X, Wang Y, Ren L, Zhao N, Gong Y, Wang DA. Novel mesoporous silica-based antibiotic releasing scaffold for bone repair. Acta Biomater. 2009;5:1697–1707. doi: 10.1016/j.actbio.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Shi X, Wang Y, Ren L, Huang W, Wang DA. A protein/antibiotic releasing poly(lactic-co-glycolic acid)/lecithin scaffold for bone repair applications. Int J Pharm. 2009;373:85–92. doi: 10.1016/j.ijpharm.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Mandal BB, Kundu SC. Calcium alginate beads embedded in silk fibroin as 3D dual drug releasing scaffolds. Biomaterials. 2009;30:5170–5177. doi: 10.1016/j.biomaterials.2009.05.072. [DOI] [PubMed] [Google Scholar]
- 19.Koutsopoulos S, Unsworth LD, Nagai Y, Zhang S. Controlled release of functional proteins through designer self-assembling peptide nanofiber hydrogel scaffold. Proc Natl Acad Sci U S A. 2009;106:4623–4628. doi: 10.1073/pnas.0807506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson PJ, Parker SR, Sakiyama-Elbert SE. Controlled release of neurotrophin-3 from fibrin-based tissue engineering scaffolds enhances neural fiber sprouting following subacute spinal cord injury. Biotechnol Bioeng. 2009;104:1207–1214. doi: 10.1002/bit.22476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Sokanee ZN, Toabi AA, Al-Assadi MJ, Alassadi EA. The drug release study of ceftriaxone from porous hydroxyapatite scaffolds. AAPS PharmSciTech. 2009;10:772–779. doi: 10.1208/s12249-009-9265-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srinivasan B, Kumar R, Shanmugam K, Sivagnam UT, Reddy NP, Sehgal PK. Porous keratin scaffold-promising biomaterial for tissue engineering and drug delivery. J Biomed Mater Res B Appl Biomater. 2010;92:5–12. doi: 10.1002/jbm.b.31483. [DOI] [PubMed] [Google Scholar]
- 23.Pang Y, Wang X, Ucuzian AA, Brey EM, Burgess WH, Jones KJ, Alexander TD, Greisler HP. Local delivery of a collagen-binding FGF-1 chimera to smooth muscle cells in collagen scaffolds for vascular tissue engineering. Biomaterials. 2010;31:878–885. doi: 10.1016/j.biomaterials.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattarai N, Gunn J, Zhang M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv Drug Deliv Rev. 2010;62:83–99. doi: 10.1016/j.addr.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 25.Fisher JP, Holland TA, Dean D, Mikos AG. Photoinitiated cross-linking of the biodegradable polyester poly(propylene fumarate). Part II. In vitro degradation. Biomacromolecules. 2003;4:1335–1342. doi: 10.1021/bm0300296. [DOI] [PubMed] [Google Scholar]
- 26.Fisher JP, Dean D, Mikos AG. Photocrosslinking characteristics and mechanical properties of diethyl fumarate/poly(propylene fumarate) biomaterials. Biomaterials. 2002;23:4333–4343. doi: 10.1016/s0142-9612(02)00178-3. [DOI] [PubMed] [Google Scholar]
- 27.Fisher JP, Vehof JWM, Dean D, van der Waerden JPCM, Holland TA, Mikos AG, Jansen JA. Soft and hard tissue response to photocrosslinked poly(propylene fumarate) scaffolds in a rabbit model. Journal of Biomedical Materials Research. 2002;59:547–556. doi: 10.1002/jbm.1268. [DOI] [PubMed] [Google Scholar]
- 28.Vehof JW, Fisher JP, Dean D, van der Waerden JP, Spauwen PH, Mikos AG, Jansen JA. Bone formation in transforming growth factor beta-1-coated porous poly(propylene fumarate) scaffolds. J Biomed Mater Res. 2002;60:241–251. doi: 10.1002/jbm.10073. [DOI] [PubMed] [Google Scholar]
- 29.Murphy WL, Dennis RG, Kileny JL, Mooney DJ. Salt fusion: an approach to improve pore interconnectivity within tissue engineering scaffolds. Tissue Eng. 2002;8:43–52. doi: 10.1089/107632702753503045. [DOI] [PubMed] [Google Scholar]
- 30.Kim K, Fisher JP. Nanoparticle technology in bone tissue engineering. J Drug Target. 2007;15:241–252. doi: 10.1080/10611860701289818. [DOI] [PubMed] [Google Scholar]
- 31.Kato Y, Artemov D. Monitoring of release of cargo from nanocarriers by MRI/MR spectroscopy (MRS): significance of T2/T2* effect of iron particles. Magn Reson Med. 2009;61:1059–1065. doi: 10.1002/mrm.21939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Smet M, Langereis S, van den Bosch S, Grull H. Temperature-sensitive liposomes for doxorubicin delivery under MRI guidance. J Control Release. 2010;143:120–127. doi: 10.1016/j.jconrel.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Caravan P. Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem Soc Rev. 2006;35:512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- 34.Sherry AD, Woods M. Chemical Exchange Saturation Transfer Contrast Agents for Magnetic Resonance Imaging. Annu Rev Biomed Eng. 2008;10:391–411. doi: 10.1146/annurev.bioeng.9.060906.151929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou J, Payen JF, Wilson DA, Traystman RJ, van Zijl PC. Using the amide proton signals of intracellular proteins and peptides to detect pH effects in MRI. Nat Med. 2003;9:1085–1090. doi: 10.1038/nm907. [DOI] [PubMed] [Google Scholar]
- 36.McMahon MT, Gilad AA, Zhou J, Sun PZ, Bulte JW, van Zijl PC. Quantifying exchange rates in chemical exchange saturation transfer agents using the saturation time and saturation power dependencies of the magnetization transfer effect on the magnetic resonance imaging signal (QUEST and QUESP): Ph calibration for poly-L-lysine and a starburst dendrimer. Magn Reson Med. 2006;55:836–847. doi: 10.1002/mrm.20818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gilad AA, McMahon MT, Walczak P, Winnard PT, Jr, Raman V, van Laarhoven HW, Skoglund CM, Bulte JW, van Zijl PC. Artificial reporter gene providing MRI contrast based on proton exchange. Nat Biotechnol. 2007;25:217–219. doi: 10.1038/nbt1277. [DOI] [PubMed] [Google Scholar]
- 38.Lee JE, Lee N, Kim H, Kim J, Choi SH, Kim JH, Kim T, Song IC, Park SP, Moon WK, Hyeon T. Uniform Mesoporous Dye-Doped Silica Nanoparticles Decorated with Multiple Magnetite Nanocrystals for Simultaneous Enhanced Magnetic Resonance Imaging, Fluorescence Imaging, and Drug Delivery. J Am Chem Soc. 2009;132:552–557. doi: 10.1021/ja905793q. [DOI] [PubMed] [Google Scholar]
- 39.Kim J, Kim HS, Lee N, Kim T, Kim H, Yu T, Song IC, Moon WK, Hyeon T. Angewandte Chemie International Edition. 13. Vol. 47. 2008. Multifunctional Uniform Nanoparticles Composed of a Magnetite Nanocrystal Core and a Mesoporous Silica Shell for Magnetic Resonance and Fluorescence Imaging and for Drug Delivery; pp. 8438–8441. [DOI] [PubMed] [Google Scholar]
- 40.Park J, An K, Hwang Y, Park JG, Noh HJ, Kim JY, Park JH, Hwang NM, Hyeon T. Ultra-large-scale syntheses of monodisperse nanocrystals. Nat Mater. 2004;3:891–895. doi: 10.1038/nmat1251. [DOI] [PubMed] [Google Scholar]
- 41.Kim K, Dean D, Mikos AG, Fisher JP. Effect of Initial Cell Seeding Density on Early Osteogenic Signal Expression of Rat Bone Marrow Stromal Cells Cultured on Cross-Linked Poly(propylene fumarate) Disks. Biomacromolecules. 2009;10:1810–1817. doi: 10.1021/bm900240k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gilad AA, Walczak P, McMahon MT, Na HB, Lee JH, An K, Hyeon T, van Zijl PC, Bulte JW. MR tracking of transplanted cells with “positive contrast” using manganese oxide nanoparticles. Magn Reson Med. 2008;60:1–7. doi: 10.1002/mrm.21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu G, Gilad AA, Bulte JW, van Zijl PC, McMahon MT. High-throughput screening of chemical exchange saturation transfer MR contrast agents. Contrast Media Mol Imaging. 2010;5:162–170. doi: 10.1002/cmmi.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim M, Gillen J, Landman BA, Zhou J, van Zijl PC. Water saturation shift referencing (WASSR) for chemical exchange saturation transfer (CEST) experiments. Magn Reson Med. 2009;61:1441–1450. doi: 10.1002/mrm.21873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ye M, Kim S, Park K. Issues in long-term protein delivery using biodegradable microparticles. J Control Release. 2010;146:241–260. doi: 10.1016/j.jconrel.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 46.Na HB, Lee JH, An K, Park YI, Park M, Lee IS, Nam DH, Kim ST, Kim SH, Kim SW, Lim KH, Kim KS, Kim SO, Hyeon T. Development of a T1 contrast agent for magnetic resonance imaging using MnO nanoparticles. Angew Chem Int Ed Engl. 2007;46:5397–5401. doi: 10.1002/anie.200604775. [DOI] [PubMed] [Google Scholar]
- 47.McMahon MT, Gilad AA, DeLiso MA, Berman SM, Bulte JW, van Zijl PC. New “multicolor” polypeptide diamagnetic chemical exchange saturation transfer (DIACEST) contrast agents for MRI. Magn Reson Med. 2008;60:803–812. doi: 10.1002/mrm.21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.