

Abstract
Background & Aims
Metastatic gastrointestinal neuroendocrine tumors (NETs) are frequently refractory to chemotherapy. Chemoresistance in various malignancies has been attributed to cancer stem cells (CSCs). We sought to identify gastrointestinal neuroendocrine CSCs (N-CSCs) in surgical specimens and a NET cell line and to characterize novel N-CSC therapeutic targets.
Methods
Human gastrointestinal NETs were evaluated for CSCs using the Aldefluor assay. In vitro sphere-forming assay was performed on primary NET cells. CNDT2.5, a human midgut carcinoid cell line, was used for in vitro (sphere-formation) and in vivo (tumorigenicity assays) CSC studies. N-CSC protein expression was characterized using Western blotting. In vivo, systemic siRNA administration targeted Src.
Results
Using the Aldefluor assay, aldehyde dehydrogenase–positive (ALDH+) cells comprised 5.8% ± 1.4% (mean ± SEM) of cells from 19 patient samples. Though many primary cell lines failed to grow, CNDT96 ALDH+ cells formed spheres in anchorage-independent conditions, whereas ALDH− cells did not. CNDT2.5 ALDH+ cells formed spheres, whereas ALDH− cells did not. In vivo, ALDH+ CNDT2.5 cells generated more tumors, with shorter latency than ALDH− or sham-sorted cells. Compared with non-CSCs, ALDH+ cells demonstrated increased expression of activated Src, Erk, Akt, and mTOR. In vivo, anti-Src siRNA treatment of ALDH+ tumors reduced tumor mass by 91%.
Conclusions
CSCs are present in neuroendocrine tumors (NETs), demonstrated by in vitro sphere-formation and in vivo tumorigenicity assays. Src was activated in N-CSCs and represents a potential therapeutic target in gastrointestinal NETs.
Keywords: carcinoid tumors, cancer stem cells, Src family kinase inhibitor PP2, neuroendocrine tumors
Introduction
Cancer stem cells (CSCs) typically comprise 1–5% of the total tumor cell population, although some malignancies such as breast cancer (11–35%) and glioblastoma (5–30%) 1 possess a larger population. This cell population is considered primarily responsible for tumor initiation, growth, and metastasis2–5. According to the CSC theory, CSCs have properties similar to their non-malignant stem cell counterparts: self-renewal, anchorage-independent growth, and the potential for differentiation into heterogeneous tumors 6. CSCs are thought to be chemoresistant and thus are spared by cytotoxic therapy; CSCs are thereby enriched in the residual tumor and subsequently mediate tumor recurrence 5, 7–11.
Gastrointestinal neuroendocrine tumors (NETs), including carcinoid tumors, are thought to originate from enterochromaffin cells 12 and are highly resistant to chemotherapy13. We hypothesized that NETs contain a subpopulation of cells termed neuroendocrine cancer stem cells (N-CSCs) that possess activated signaling pathways that could be exploited as therapeutic targets.
One major difficulty in investigating the mechanisms of NET growth is the limited number of gastrointestinal NET cell lines14, 15. Our laboratory developed a midgut carcinoid cell line 15 and we used this cell line to identify and characterize a cell population enriched for N-CSCs using the Aldefluor assay. Additionally, we utilized freshly isolated tumor cells from 14 midgut and 5 pancreatic NET surgical specimens to investigate a CSC population in NET tumors. Finally, we characterized mediators that can be targeted by available therapeutics.and found the Src and mTor pathways to be activated. Since the mTor inhibitor everolimus has already been validated in clinical trials16–18, we chose to target Src, since agents are available for the transition to clinical studies.
Materials and Methods
Human Tissue Specimens and Cell Lines
Patients undergoing NET resection at the University of Texas MD Anderson Cancer Center (UTMDACC) were identified. After informed consent was obtained under an Institutional Review Board-approved protocol, a portion of resected tumor was harvested under sterile conditions in the pathology suite and placed on ice in Dulbecco’s modified Eagle’s medium supplemented with F12 solution (DMEM/F12, Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (FBS), with care to avoid contaminating adjacent normal tissue. The specimen was transported to our laboratory, mechanically dissociated with sterile scalpels and digested for 1 hour with 10 mg/ml type II collagenase (Cell Isolation Optimizing System, Worthington Biochemical Corp., Lakewood, NJ) in fresh DMEM/F12 media with 10% FBS. The resulting single cell suspension generated primary NET cultures.
The human midgut carcinoid cell line CNDT2.5 was developed in our laboratory15. Further examination of the CNDT2.5 cell line by short tandem repeat DNA analysis did not match the original patient tissue; however, because it strongly displays many NET features, it is considered to behave in a sufficiently NET-like for research purposes19. Cell lines were cultured in DMEM/F12 supplemented with 10% FBS, penicillin-streptomycin, vitamins, sodium pyruvate, L-glutamine, and non-essential amino acids, at 37°C in 5% CO2 and 95% air, as previously described20. All in vitro experiments were performed at 60% to 80% confluence.
Identifying N-CSCs Using the Aldefluor Assay
An Aldefluor kit (STEMCELL Technologies, Vancouver, CA) was used to isolate the population of cells with high aldehyde dehydrogenase (ALDH) enzymatic activity 21, 22. Cells were suspended in phosphate-buffered saline (PBS) after either trypsinization from in vitro culture or mechanical dissociation from surgical specimens and filtered through sterile 40-µm membranes to form single-cell suspensions. Red blood cells (RBCs) in the surgical samples were lysed using an RBC lysis buffer (eBioscience Inc., San Diego, CA). All Samples were subjected to the Aldefluor assay according to the manufacturer’s protocol. The cells were re-suspended in Aldefluor assay buffer containing ALDH substrate (BAAA, 1 µM) and incubated at 37°C for 30 minutes. As a negative control, an aliquot from each sample was treated with 50 mM diethyl-aminobenzaldehyde (DEAB), a specific ALDH inhibitor. The fluorescently labeled product, BODIPY-aminoacetate (BAA), was produced by cells expressing the ALDH enzyme, and quantified using fluorescence-activated cell sorting (FACS) with the fluorescein isothiocyanate (FITC)/FL1 channel and the DEAB-treated cells as a negative control. Only the brightest 0.5–1% of ALDH-positive (ALDH+) cells and most dim 0.5–1% of ALDH-negative (ALDH−) viable cells were sorted for further in vitro and in vivo studies. Control cells (“sham-sorted were generated by subjecting parental cells to flow cytometry without any sorting markers. ALDH+, ALDH−, and sham-sorted cell viability was assessed by trypan blue staining and an automated cell counter.
Sphere-Forming Assay
ALDH+, ALDH− and sham-sorted cells from fresh surgical specimens were cultured in 6-well ultra-low-attachment plates (Corning Life Sciences, Lowell, MA) at a density of 10,000 cells per well because the overall low viability of primary cells precludes the use of single-cell culture techniques. Sorted CNDT2.5 cells were plated in 96-well ultra-low-attachment plates (Corning Life Sciences) at a density of 1 viable cell per well. Cells were grown in cancer stem cell media consisting of DMEM (Invitrogen) supplemented with B27 Serum-Free Supplement (1:50; Invitrogen), 20 ng/ml epidermal growth factor, and 20 ng/ml basic fibroblast growth factor (both from R&D Systems, Minneapolis, MN) at 37°C and 5% CO2. Fresh medium was added every 3–4 days while the formation of free-floating spheres was monitored. The experiment was terminated at 21 days and the development of any spheres (>50 µm) was quantified. The experiment was done in triplicate.
Western Blotting
Whole-cell protein samples were isolated when cells reached 70% to 80% confluence. Cells were solubilized in 20 mM Tris-HCl (pH 8.0), 137 mM NaCl, 1% Triton X-100, 1 mM Na3VO4, 2 mM ethylenediaminetetraacetic acid (EDTA), and one complete Mini Protease Inhibitor Cocktail Tablet (Roche Diagnostics, Indianapolis, IN) per 10 ml of lysis buffer. Cell lysates were separated on sodium dodecyl sulfate polyacrylamide gels at 8–15% concentration (determined by the target protein size) and transferred to polyvinylidene difluoride membranes (GE Healthcare, Piscataway, NJ). Membranes were probed with primary antibodies overnight at 4°C, washed three times for 10 minutes in Tris-buffered saline with 0.1% Tween-20 and probed with secondary antibody for 1 hour at room temperature. After incubation and three washes, immunostained proteins were detected using a chemiluminescence kit (Thermo Scientific, Waltham, MA). To confirm equal loading, membranes were re-probed with vinculin antibody.
Antibodies
The following primary antibodies were used for Western blotting: anti-pS2448-mTOR, anti-mTOR, anti-pS473-Akt, anti-Akt, anti-pY416-Src (all from Cell Signaling Technology, Danvers, MA), anti-Src (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Erk1/2 (Oncogene Science, Cambridge, MA), and anti-vinculin (Sigma-Aldrich, St. Louis, MO).
In Vivo Tumorigenicity Study
All animal studies were conducted under protocols approved by the institutional Animal Care and Use Committee. Male nude mice, 6–8 weeks old, were purchased from the National Cancer Institute at Frederick Center for Cancer Research (Frederick, MD) and maintained under specific pathogen-free conditions. All animal experiments met the requirements of UTMDACC Animal Care Facility and National Institutes of Health (NIH) guidelines on animal care and use.
We evaluated in vivo tumorigenicity of ALDH+ cells by subcutaneous injection of ALDH+, ALDH−, or sham-sorted CNDT2.5 cells, at various inoculums (100; 1,000; 10,000; or 100,000 cells), suspended in 50 µl PBS mixed with 50 µl of basement membrane extract (Trevigen, Gaithersburg, MD) into right flanks of nude mice (5 per group) . Tumors (firm masses larger than the original volume of injection, determined by blinded observer) were monitored weekly for 12 weeks, at which point, the mice were sacrificed, tumors were harvested and hematoxylin and eosin (H&E) staining was performed to confirm the characteristic CNDT2.5 morphology, utilizing standard procedures 23, 24. This experiment was repeated three times.
In vitro Src Inhibition
CNDT2.5 cells were plated in 10-cm plates (Corning Life Sciences) and allowed to adhere overnight. The next day, each subpopulation was treated with nothing, DMSO, or 10 µM PP2 for 48 hours in 10% FBS, DMEM/F12 media. After 48 hours, cells were harvested and subjected to the Aldefluor assay to evaluate the percentage of ALDH+ cells.
In vitro cellular proliferation was determined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich), as described previously25. CNDT2.5 cells were sorted into subpopulations (ALDH+, ALDH−, and sham-sorted), and plated at 2,000 cells per well in a 96-well plate. The cells adhered overnight, and were then treated with nothing, DMSO, and 10 µM PP2. At time points 0, 24, 48, and 72 hours, 40 µl of MTT solution was added to each well, and the plates were incubated for 1 hour at 37°C. 100 µl of DMSO was added, and colorimetric analysis was performed using a microplate reader to evaluate the effect of PP2 on the growth rate of the three subpopulations of cells.
Reagents
4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2, EMD Biosciences, Gibbstown, NJ) is a commercially available Src family kinase inhibitor. Dimethyl sulfoxide (DMSO, Sigma-Aldrich) was the dissolving agent for PP2, and was used as a negative control for all in vitro and in vivo PP2 studies.
Preparation of siRNA-Containing Liposomes
For in vivo Src-inhibition studies, liposomes containing anti-Src (5’-GGCTGAGGAGTGGTATTTT-3’) and non-targeting siRNA sequences (5’-TTCTCCGAACGTGTCACGT-3’) (Invitrogen, Carlsbad, CA) were prepared as previously described 26. Briefly, siRNA oligonucleotides to Src target sequences (siSrc) or scrambled non-targeting control sequences (siControl) were mixed with 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) (Avanti Polar Lipids, Alabaster, AL) at a ratio of 10:1 (w:w) DOPC:siRNA and lyophilized. Immediately before in vivo administration, lyophilized preparations were hydrated in Dulbecco’s PBS without calcium or magnesium at a concentration of 5 µg of siRNA per 100 µl of Dulbecco’s PBS.
In Vivo Src Targeting
We evaluated the effects of Src inhibition on established tumors derived from unsorted CNDT2.5 and CNDT2.5 ALDH+ cells. ALDH− cells were unable to generate tumors by 12 weeks and could not be studied. CNDT2.5 cells sorted using the Aldefluor assay and FACS. Aliquots of 1,000,000 CNDT2.5 ALDH+ cells and 4,000,000 unsorted parental CNDT2.5 cells were injected subcutaneously into 20 mice per population. Tumor growth was monitored biweekly and when mean tumor volume reached 150 mm3, tumor-bearing mice were intraperitoneally injected twice weekly with either siControl-DOPC or siSrc-DOPC nanoliposomes (5 µg of siRNA was administered in 100 µl of Dulbecco’s PBS per treatment, as noted above). Experiments were terminated when mice became moribund (lethargy, decreased grooming, or weight loss) or when an average tumor volume of 1 cm3 was reached. Mice were killed by CO2 asphyxia, and final tumor mass and volume (1/2 × width2 × length) were recorded.
Statistical Analysis
Data are presented as means, unless otherwise noted. Differences in means were defined using 95% confidence intervals. In vitro and in vivo statistical significance was determined using either Fisher’s exact tests (for comparisons of incidence) or Mann-Whitney tests (for comparisons of means), as indicated. In in vivo siRNA experiments, 10 mice in each group were used, as directed by power analysis to detect a 50% reduction in tumor size or weight, with a beta error of 3%. The non-parametric Mann-Whitney test analyzed in vivo siRNA experiments for statistical differences. Patient sample ALDH activity data was analyzed using the Grubbs test for outliers; statistical outliers were not included in the calculations. All statistical tests were two-sided. p values <0.05 were statistically significant (GraphPad Prism®).
Results
Identifying CSCs in Human Gastrointestinal NET Specimens
The Aldefluor assay enriches for CSCs22, 27 using the functional cytosolic enzyme, ALDH 28, 29. ALDH enhances self-renewal of hematopoietic stem cells30 and mediates chemoresistance in tumor cells 31. 19 NET specimens (14 midgut carcinoid tumors and 5 pancreatic NETs) were assessed for CSCs using the Aldefluor assay. FACS was used to isolate viable single cells, from freshly isolated NET specimens, with ALDH (as judged by FITC) expression greater than the top 0.1% of DEAB-treated negative control cells (Fig. 1). The ALDH+ cell subpopulations ranged from 0.5% to 20.1% in carcinoid tumors and from 0.2 to 5.9% in pancreatic NETs (Table 1). Interestingly, we found the presence of a higher percentage of ALDH+ cells (mean ± SEM: 5.8% ± 1.4%) in human surgical specimens compared to the established tumor cell line CNDT2.5 (Fig. 1). Midgut carcinoids seemed to have a larger ALDH+ subpopulation (mean: 7.1% ± 2.8%) than pancreatic NETs (mean: 2.3% ± 1.0%), but this result was not significantly significant (p = 0.12 by the Mann Whitney U-test) (Table 1). One midgut primary carcinoid tissue sample with an ALDH+ subpopulation of 37.7% was an outlier by the Grubbs test and was omitted from data analysis.
Figure 1. ALDH+ N-CSCs are present in human surgical specimens.
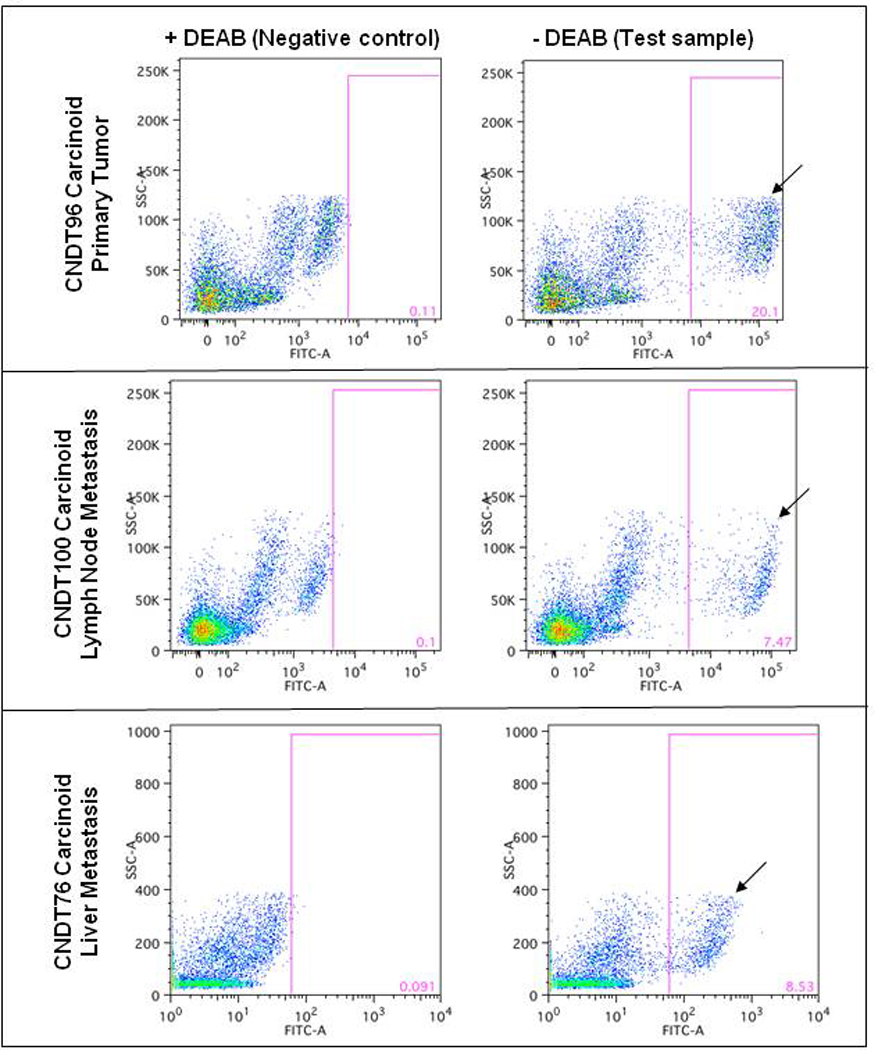
Single cell suspensions from surgical specimens were analyzed for the presence of ALDH+ cells. The ALDH+ cells are indicated by a black arrow. The gates are set for analytical purposes only and were not used for sorting.
Table 1.
ALDH+ population in human NET specimens.
| Samples | n | Mean % ALDH+ |
Range | |
|---|---|---|---|---|
| All | 19 | 5.8b | 0.2 – 20.1b | |
| Midgut | Total | 14 | 7.1a,b | 0.5 – 20.1b |
| carcinoid | Primary | 7 | 7.8b | 0.5 – 20.1b |
| Liver metastasis | 3 | 10.4 | 3.8 – 19.8 | |
| Lymph node | ||||
| metastasis | 4 | 3.8 | 0.8 – 8.5 | |
| Pancreatic | ||||
| NET | Total | 5 | 2.3a | 0.2 – 5.9 |
| Primary | 1 | 3.2 | 3.2 | |
| Liver metastasis | 4 | 2 | 0.2 – 5.9 |
p = 0.12 by Mann Whitney U test
Dataset omits one Grubbs test outlier (37.7% ALDH+ cells).
Identifying CSCs in a Gastrointestinal NET Cell Line
Flow cytometric analysis revealed a subpopulation of ALDH+ cells ranging from 1–3% in CNDT2.5 cells (Fig. 2). These cells were isolated using FACS for all subsequent studies.
Figure 2. ALDH+ N-CSCs are present in the NET cell line CNDT2.5.
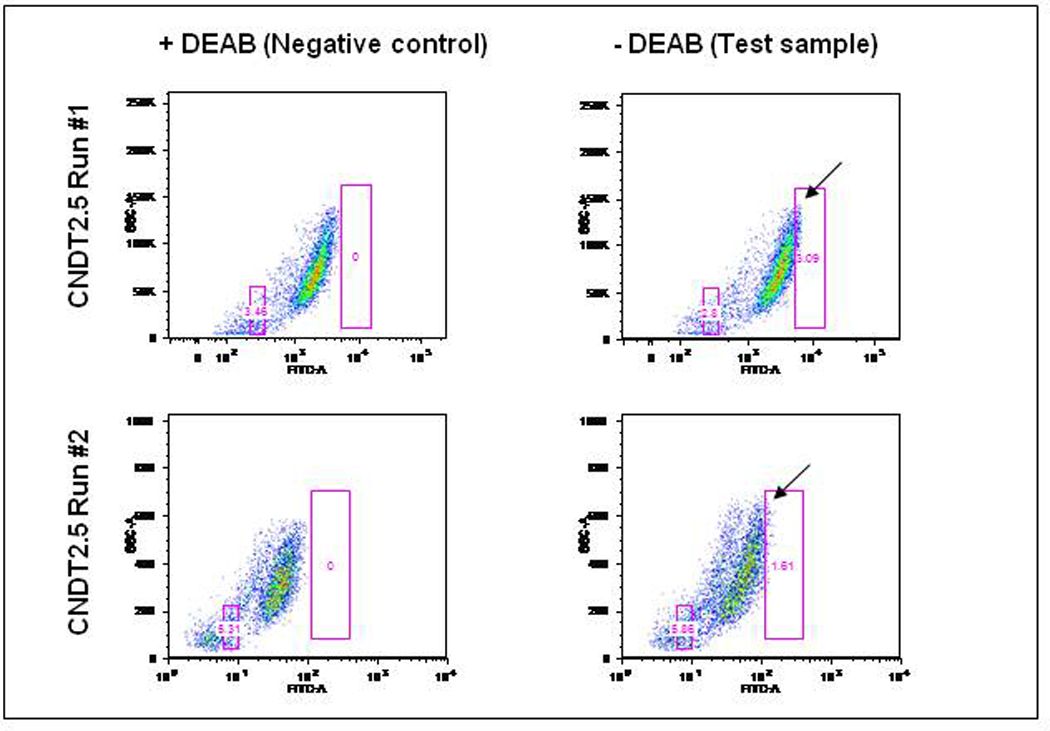
The CNDT2.5 ALDH+ population ranged from 1.6 to 3% of viable cells, indicated by a black arrow. The boxes in these figures are the sorting gates used to isolate ALDH+ and ALDH− populations in future experiments.
Self-renewal and Growth of ALDH+ Cells Cultured in Anchorage-Independent Conditions
Freshly isolated gastrointestinal NET cells and CNDT2.5 cells were sorted into three subpopulations (ALDH+, ALDH−, and sham-sorted) by FACS and subjected to a sphere-forming assay to establish their capacity for anchorage-independent proliferation and confirm the enrichment of CSCs. Cells from fresh surgical samples were plated in sphere-forming conditions and monitored for 21 days. Compared with established cell lines, these cells required increased plating density to account for their inherent overall lower viability. Spheres formed in the ALDH+ subpopulation of CNDT96, a primary midgut tumor cell line, by 8 days and continued to grow through 21 days of observation. Viable ALDH− cells from this sample were unable to survive in sphere-forming conditions and no viable cells were seen by day 8 (Fig. 3A). We attempted to study cells from six other patient samples for sphere-forming capability, but due to slow growth rates and poor NET cell survival in vitro, no spheres were observed in any of these samples. Furthermore, all attempts to propagate these cells under adherent conditions were unsuccessful, underscoring the difficulty of culturing NET cells in vitro.
Figure 3. NET ALDH+ cells form spheres.
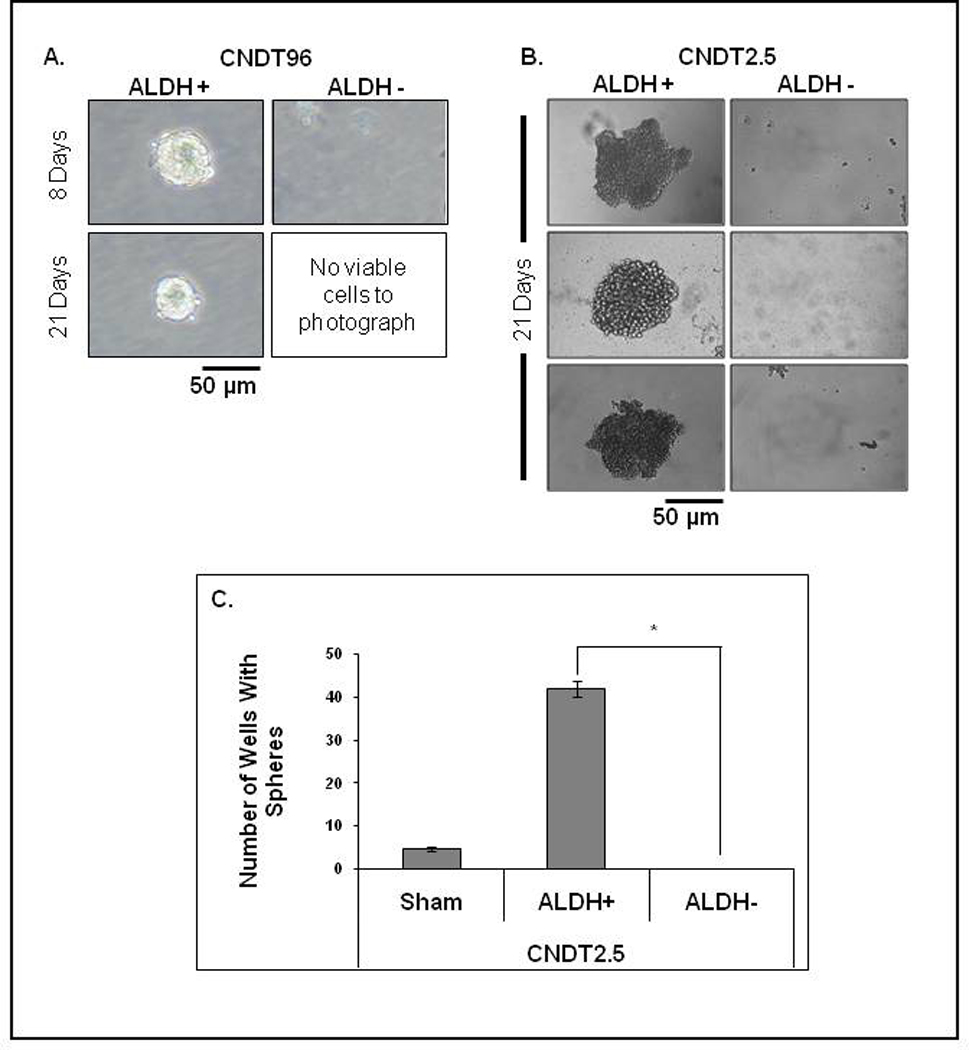
(A): CNDT96 ALDH+ cells formed spheres by 8 days that were viable at 21 days. The ALDH− cells died by day 8. (B): ALDH+ CNDT2.5 cells formed spheres whereas ALDH− cells did not. (C): CNDT2.5 spheres >50 µm on day 21. (p < 0.001, mean ± SEM).
To further validate the ALDH+ subpopulation’s CSC characteristics, the three subpopulations (ALDH+, ALDH−, and sham-sorted) of CNDT2.5 cells were grown in 96-well ultra-low-attachment plates at a density of 1 viable cell per well in stem cell media. The cells’ ability to form spheres was determined on day 21. CNDT2.5 ALDH+ cells formed numerous spheres (Fig. 3B), but no spheres were detected in the ALDH− plates (Fig. 3C, p < 0.001). An average of 42 ± 2 wells (out of 96) formed spheres in the ALDH+ group. The sham-sorted parental cells formed spheres with similar morphologies as those of the ALDH+ cells (data not shown), but the efficiency of sphere formation (4.7 ± 0.7) was significantly lower than that of the ALDH+ cells.
In Vivo Tumorigenicity Assay of Aldefluor-Sorted Cells
To validate that the ALDH+ carcinoid cell subpopulation is enriched for CSCs, we compared in vivo tumorigenicity of ALDH+, ALDH−, and sham-sorted cells. 100, 1,000, 10,000, and 100,000 ALDH+, ALDH−, and sham-sorted CNDT2.5 cells were injected subcutaneously into the flanks of nude mice (with 5 mice per group). Tumor growth was monitored for 12 weeks. The ALDH+ cells generated more tumors than ALDH− or sham-sorted cells for any given cell concentration (Table 2). Subcutaneous injection of 100,000 ALDH+ cells formed tumors in 4/5 mice versus 0/5 from ALDH− cells (p=0.048, by Fisher’s exact test). The ALDH+ group demonstrated the shortest latency period with 100,000 ALDH+ cells forming visible tumors by week 3, while 100,000 sham-sorted cells required 6 or 7 weeks to form tumors. H&E staining revealed that the tumors derived from ALDH+ cells resembled tumors formed by injection of the parental CNDT2.5 cells (Supplement 1).
Table 2.
Tumor incidence of ALDH+, ALDH−, and sham-sorted CNDT2.5 cells.
| Number of CNDT2.5 cells injected |
Tumor incidence at 12 weeks |
|||
|---|---|---|---|---|
| ALDH+ | ALDH− | Sham-Sorted | ||
| 100 | 1/5 | 0/5 | 0/5 | |
| 1,000 | 2/5 | 0/5 | 1/5 | |
| 10,000 | 2/5 | 0/5 | 1/5 | |
| 100,000 | 4/5* | 0/5* | 1/5 | |
| Total | 9/20 | 0/20 | 3/20 | |
(* p-value: 0.048 by Chi-Square Test, ALDH+ vs ALDH−)
Signaling Pathway Analysis in ALDH+ and ALDH− Subpopulations
N-CSCs and non-tumorigenic daughter cells differ in phenotype and tumorigenic potential. According to the CSC hypothesis, genes regulating self-renewal are likely to be differentially expressed between these two subpopulations. To potentially exploit these differences in future clinical trials, we sought to evaluate signaling proteins in CSC related pathways to which there are existing therapeutic agents; specifically levels of activated and total Src, Erk, Akt, and mTOR.
We analyzed these pathways in the ALDH+ and ALDH− cell subpopulations by Western blot (Fig. 4A) and found the Src pathway to be upregulated in the ALDH+ group. Since Src is activated in various hematologic stem cells and its inhibition results in significant leukemic cell growth inhibition32–34 we evaluated the in vitro effects of Src inhibition on our putative N-CSCs using PP2, a known Src inhibitor 35, 36. PP2 was used as in vitro Src inhibitor since it has immediate effects of Src activity, while other in vitro manipulations (gene knockdown) requires longer cell culturing and additional treatments that, over time, would inherently alter and dilute the ALDH+ cell population (CSC division leads to non-CSC daughter cells). CNDT2.5 cells treated with DMSO or PP2 for 48 hours showed a significant decrease in the ALDH+ subpopulation in the PP2 treatment group compared to control cells (Fig. 4B, p < 0.001). Additionally, when ALDH+, ALDH−, and sham-sorted cells were subjected to DMSO or PP2 treatment followed by MTT assay, the ALDH+ subpopulation showed a significant reduction in cell number over a period of 72 hours compared to ALDH− and sham-sorted cells (Fig. 4C, p < 0.001). The sham-sorted group was also affected by treatment with PP2, as expected. However, there was no significant decrease in cell number in the ALDH− group.
Figure 4. Src inhibition targets ALDH+ cells.
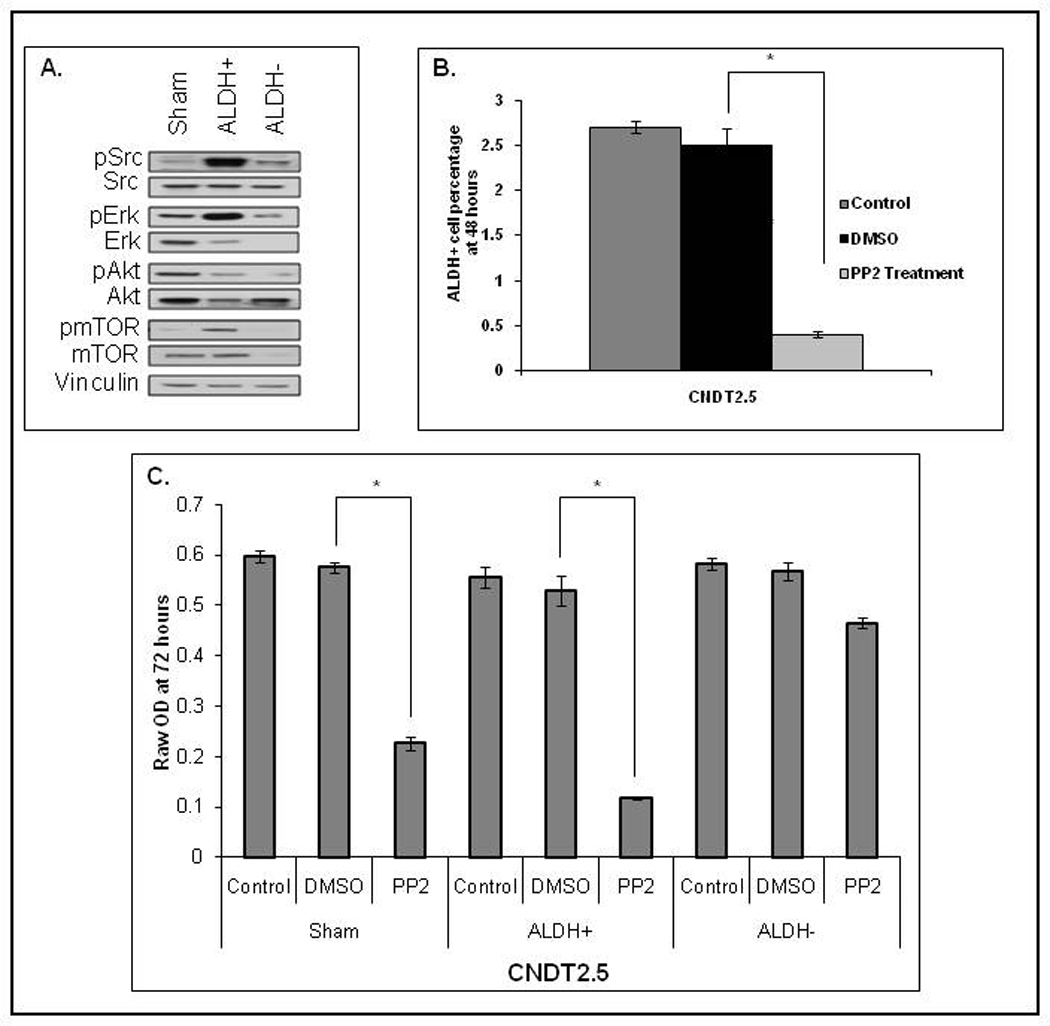
(A): Src, Erk, Akt, and mTOR were activated in CNDT2.5 ALDH+ cells. (B): CNDT2.5 cells treated with PP2 (Src inhibitor) had fewer ALDH+ cells than the DMSO control (p < 0.0001). (C): Src inhibition markedly decreases proliferation of ALDH+ CNDT2.5 cells, compared to ALDH− cells (p < 0.0001).
In Vivo Targeting of Src Using siRNA
After validating the similar histology between ALDH+ and parental derived xenografts (Supplement 1) and Src siRNA’s effectiveness in vitro (Fig. 5A), 1,000,000 ALDH+ and 4,000,000 parental CNDT2.5 cells were subcutaneously injected into 20 mice. The increased number of parental cells was needed to account for the inherent decreased tumorigenicity and growth rates of parental cells compared to ALDH+ cells. Tumor-bearing mice received biweekly treatments with either siControl or siSrc DOPC-conjugated liposomes. The siSRC group showed marked inhibition of tumor growth compared to the siControl-DOPC group, with 20% of the tumors completely regressing upon treatment with siSrc-DOPC (Fig. 5B–C). At the time of euthanasia, parental cell derived tumors were drastically reduced or disappeared and ALDH+ tumors showed a 91% reduction in total tumor mass in the siSrc-DOPC group compared to the siControl-DOPC group (Fig. 5D–E, p < 0.001).
Figure 5. In vivo Src inhibition reduces NET tumor size.
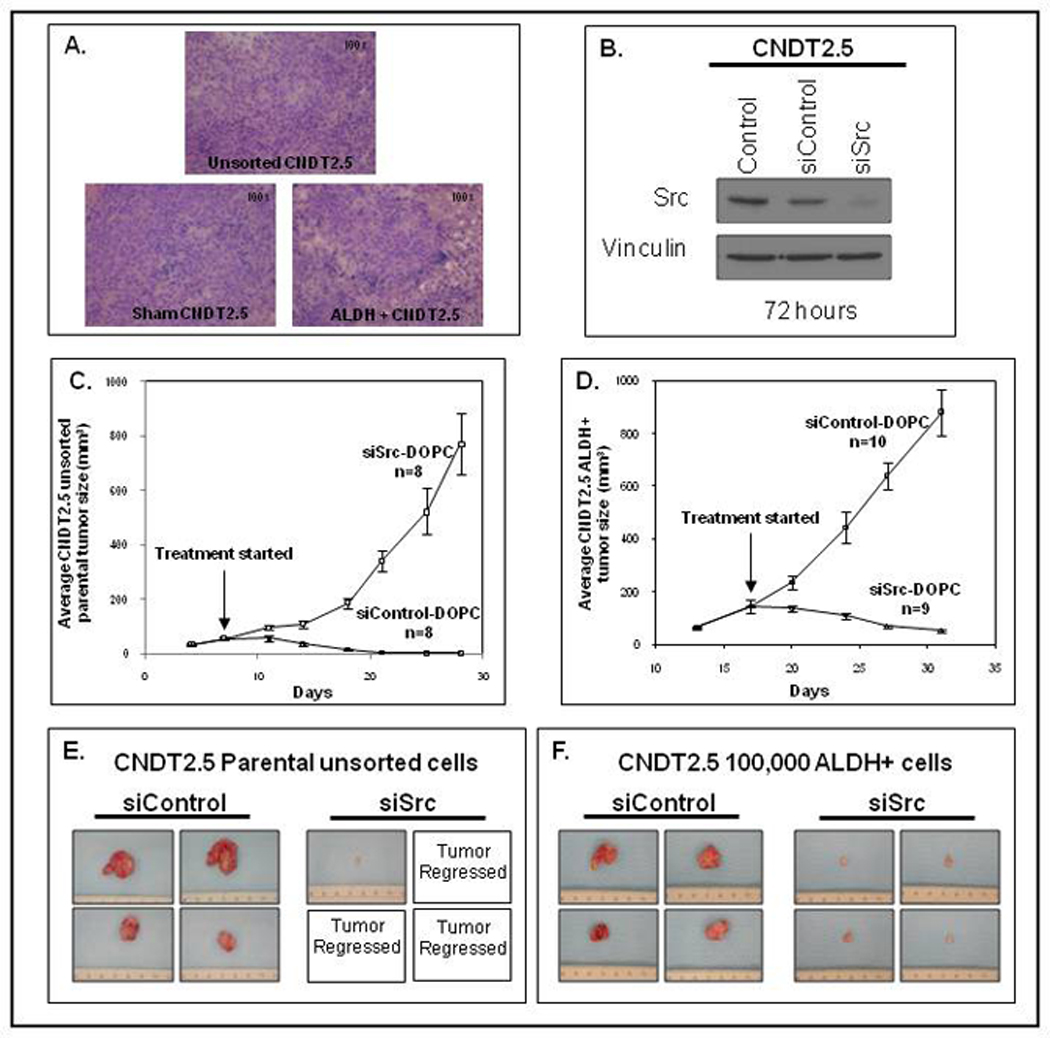
(A): Src-siRNA knock-down of CNDT2.5 Src expression. (B, C): Src-SiRNA (siSrc-DOPC) treatment decreased average CNDT2.5 tumor size in vivo (p < 0.001) in tumors generated by unsorted CNDT2.5 cells and tumors generated by ALDH+ CNDT2.5 cells. (D, E): Total tumor mass was greatly decreased by siSrc-DOPC-treatment in tumors generated from unsorted parental cells and resulted in a 91% total volume reduction compared to the siControl-DOPC-treated group in ALDH+ derived tumors.
Aldefluor Assay on ALDH+ Tumors after siRNA Treatment
Finally, we sought to evaluate the effects of siRNA treatment on the percentage of ALDH+ cells in xenografts. Following in vivo siRNA treatment, three tumors from each group (siControl-DOPC and siSrc-DOPC) were harvested and subjected to the Aldefluor assay. In the tumors derived from ALDH+ CNDT2.5 cells, the siControl-DOPC group had 0.77 ± 0.03% ALDH+ cells, compared to 0.40 ± 0.06% ALDH+ cells in the siSrc-DOPC group (p < 0.001). These results suggested that siSrc-DOPC treatment targeted CSCs and therefore may be responsible for the impaired tumor growth in the siSrc group.
Discussion
We demonstrate that ALDH activity may be a marker of CSCs in gastrointestinal NETs and CNDT2.5. The Aldefluor assay was particularly suited for identifying potential N-CSCs because it is a functional readout of CSC characteristics. Common CSC surface markers, such as CD44 and CD13337,38,39 used in epithelial malignancies were non-specific in our cells, with nearly all CNDT2.5 cells binding to CD44, and CD133 not labeling any cells. Since these markers were unable to isolate population-CSCs, we chose the Aldefluor assay as a functional CSC assay. The other common functional assay, the side-population assay led to cell death and thus it was not feasible to use this method of enriching for CSCs.
In vitro tumorigenicity assays were limited due to small patient samples and the inherent difficulty of long-term NET cell culture; however the samples for which proliferation did occur showed increased sphere formation in the ALDH+ subpopulation. CNDT2.5 sphere-formation and in vivo tumorigenicity assays confirmed that the Aldefluor assay selected for an N-CSC-enriched subpopulation, consistent with studies in other tumor types40. Similar to other reports21, 22 only ALDH+ CNDT2.5 cells were capable of anchorage independent growth in ultra-low-attachment conditions. Additionally, only ALDH+ cells were capable of initiating tumor growth that recapitulated unsorted CNDT2.5 xenograft tumor histology. The ALDH− cells were able to proliferate in vitro at a rate similar to sham and ALDH+ sorted CNDT2.5 cells, demonstrated by our MTT results, indicating that the sphere and tumor forming ability of ALDH+ cells is due to a CSC phenotype rather than variations in viability or proliferation of the cell populations.
Along with increased tumorigenic potential, CSCs possess an innate ability to escape the cytotoxic effects of conventional therapy 8, 41–43 by employing drug transporters 41–43 and enhanced DNA repair mechanisms 8, which could contribute to the relative chemoresistance of carcinoid tumors. Recent phase I and II trials have shown that the Src inhibitor, dasatinib (Bristol-Myers Squibb, New York, NY), has potential therapeutic benefits in numerous solid malignancies49. Given the availability of this drug and its potential effects in other tumor types, we decided to further examine Src expression in the NET cell subpopulations. We noted a markedly higher activated Src expression in ALDH+ CNDT2.5 subpopulations compared to ALDH− populations. Our results demonstrated that Src inhibition decreased the percentage of viable ALDH+ cells and selectively inhibited the growth of ALDH+ cells. Furthermore, anti-Src siRNA inhibited the in vivo growth of tumors derived from ALDH+ cells. These in vitro and in vivo results from CNDT2.5 cells confirm that Src is a potential therapeutic target for N-CSCs.
Although we validated the presence of N-CSCs in vitro with CNDT2.5 and identified this population in surgical specimens, it was difficult to validate our in vitro and in vivo data using cells from fresh surgical specimens because of the limited amount of tissue obtained and the inherent difficulty of culturing NET tumor cell lines in vitro. The rare tumors that form in mice injected with fresh NET cells developed in about 6 months making it unfeasible to perform in vivo studies with freshly derived samples. In the samples that did remain viable, there appeared to be increased sphere-forming capability and viability in low-attachment conditions; however, absolute quantification was complicated by the large number of cells per well needed to observe sphere formation. Although staining NET tissue samples for Src or ALDH would be a potential method to examine the localization and relative levels of Src in human tumors, we found the current ALDH and Src antibodies to lack the necessary specificity. Nonetheless, we demonstrated that carcinoid tumors have an inherent CSC population that may mediate resistance to systemic therapies. The trend toward a higher percentage of ALDH+ cells in midgut carcinoid tumors compared to pancreatic NETs may explain the increased chemosensitivity of pancreatic NETs relative to midgut carcinoids; this highlights the importance of CSCs as a potential therapeutic target and warrants the examination of more pancreatic samples. To our knowledge, this is the first study targeting N-CSCs in carcinoid tumors by inhibiting the Src pathway. The findings of our study offer a potential therapeutic approach that can be used to target the cells responsible for carcinoid growth.
Supplementary Material
Acknowledgements
We thank Pablo Vivas-Mejia (Department of Experimental Therapeutics) for assistance with preparation of liposomal siRNA; Karen Ramirez (Flow Cytometry and Cellular Imaging Facility, NIH Cancer Center Support Grant 5 P30 CA016672) for assistance with flow cytometry sorting; and Zach Bohannan, Sunita Patterson (both of the Department of Scientific Publications) and Rita Hernandez (Department of Surgical Oncology) for editorial assistance.
Grant Support: Supported, in part, by National Institutes of Health (NIH) T32 grant CA009599 (PG, ES, FT,) NIH Cancer Center Support Grant CA016672 (MD Anderson Cancer Center), NIH CA151668 (AKS), The Caring for Carcinoid Foundation (LME), The Raymond and Beverly Sackler Fund for the Arts and Sciences (LME), and the William C. Liedtke, Jr., Chair in Cancer Research (LME).
Abbreviations
- ALDH
aldehyde-dehydrogenase
- BAA
BODIPY-aminoacetate
- CSCs
Cancer Stem Cells
- DEAB
diethyl-aminobenzaldehyde
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphatidylcholine
- EDTA
ethylenediaminetetraacetic acid
- FACS
Fluorescence-activated Cell Sorting
- FBS
Fetal Bovine Serum
- FITC
Fluorescein Isothiacyanate
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- N-CSCs
Neuroendocrine Cancer Stem Cells
- NETs
Neuroendocrine Tumors
- NIH
National Institutes of Health
- PBS
Phosphate-Buffered Saline
- PP2
4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- RBCs
Red Blood Cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
- Puja Gaura*: Conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript
- Eric L. Sceusia*: Conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript
- Shaija Samuelb: Collection and/or assembly of data, data analysis and interpretation
- Ling Xiab: Collection and/or assembly of data, data analysis and interpretation
- Fan Fanb: Collection and/or assembly of data, data analysis and interpretation
- Yunfei Zhoub: Collection and/or assembly of data, data analysis and interpretation
- Jia Lub: Collection and/or assembly of data, data analysis and interpretation
- Federico Tozzia: Collection and/or assembly of data
- Gabriel Lopez-Beresteinc: Collection and/or assembly of data
- Pablo Vivas-Mejiac: Collection and/or assembly of data
- Asif Rashidd: Provision of study material or patients
- Jason B. Fleminga: Provision of study material or patients
- Eddie K. Abdallaa: Provision of study material or patients
- Steven A. Curleya: Provision of study material or patients
- Jean-Nicolas Vautheya: Provision of study material or patients
- Anil K. Soode: Provision of study material or patients
- James C. Yaof: Provision of study material or patients, financial support, administrative support
- Lee M. Ellisa, b: Conception and design, financial support, administrative support, data analysis and interpretation, manuscript writing, final approval of manuscript
Disclosures: Nothing to disclose
Writing Assistance: Zach Bohannan, Sunita Patterson (both of the Department of Scientific Publications) and Rita Hernandez (Departments of Surgical Oncology and Cancer Biology) provided editorial assistance.
References
- 1.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–284. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 2.Leiros GJ, Balana ME. Metastatic Cancer Stem Cells: New Molecular Targets for Cancer Therapy. Curr Pharm Biotechnol. 2011 doi: 10.2174/138920111798377094. [DOI] [PubMed] [Google Scholar]
- 3.Adhikari AS, Agarwal N, Iwakuma T. Metastatic potential of tumor-initiating cells in solid tumors. Front Biosci. 2011;16:1927–1938. doi: 10.2741/3831. [DOI] [PubMed] [Google Scholar]
- 4.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 5.Soltanian S, Matin MM. Cancer stem cells and cancer therapy. Tumour Biol. 2011 doi: 10.1007/s13277-011-0155-8. [DOI] [PubMed] [Google Scholar]
- 6.Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- 7.Al-Hajj M. Cancer stem cells and oncology therapeutics. Curr Opin Oncol. 2007;19:61–64. doi: 10.1097/CCO.0b013e328011a8d6. [DOI] [PubMed] [Google Scholar]
- 8.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 9.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26:2839–2845. doi: 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guzman ML, Swiderski CF, Howard DS, Grimes BA, Rossi RM, Szilvassy SJ, Jordan CT. Preferential induction of apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci U S A. 2002;99:16220–16225. doi: 10.1073/pnas.252462599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 12.Lechago J. Neuroendocrine cells of the gut and their disorders. Monogr Pathol. 1990:181–219. [PubMed] [Google Scholar]
- 13.Yao JC, Phan A, Hoff PM, Chen HX, Charnsangavej C, Yeung SC, Hess K, Ng C, Abbruzzese JL, Ajani JA. Targeting vascular endothelial growth factor in advanced carcinoid tumor: a random assignment phase II study of depot octreotide with bevacizumab and pegylated interferon alpha-2b. J Clin Oncol. 2008;26:1316–1323. doi: 10.1200/JCO.2007.13.6374. [DOI] [PubMed] [Google Scholar]
- 14.Yao JC. Neuroendocrine tumors. Molecular targeted therapy for carcinoid and islet-cell carcinoma. Best Pract Res Clin Endocrinol Metab. 2007;21:163–172. doi: 10.1016/j.beem.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 15.Van Buren G, 2nd, Rashid A, Yang AD, Abdalla EK, Gray MJ, Liu W, Somcio R, Fan F, Camp ER, Yao JC, Ellis LM. The development and characterization of a human midgut carcinoid cell line. Clin Cancer Res. 2007;13:4704–4712. doi: 10.1158/1078-0432.CCR-06-2723. [DOI] [PubMed] [Google Scholar]
- 16.Yao JC, Lombard-Bohas C, Baudin E, Kvols LK, Rougier P, Ruszniewski P, Hoosen S, St Peter J, Haas T, Lebwohl D, Van Cutsem E, Kulke MH, Hobday TJ, O'Dorisio TM, Shah MH, Cadiot G, Luppi G, Posey JA, Wiedenmann B. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol. 28:69–76. doi: 10.1200/JCO.2009.24.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, Abdalla EK, Fleming JB, Vauthey JN, Rashid A, Evans DB. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–3072. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 18.Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, Tomassetti P, Pavel ME, Hoosen S, Haas T, Lincy J, Lebwohl D, Oberg K. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 364:514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellis LM, Samuel S, Sceusi E. Varying opinions on the authenticity of a human midgut carcinoid cell line--letter. Clin Cancer Res. 2010;16:5365–5366. doi: 10.1158/1078-0432.CCR-10-2550. [DOI] [PubMed] [Google Scholar]
- 20.Van Buren G, 2nd, Gray MJ, Dallas NA, Xia L, Lim SJ, Fan F, Mazar AP, Ellis LM. Targeting the urokinase plasminogen activator receptor with a monoclonal antibody impairs the growth of human colorectal cancer in the liver. Cancer. 2009;115:3360–3368. doi: 10.1002/cncr.24371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camp ER, Yang A, Liu W, Fan F, Somcio R, Hicklin DJ, Ellis LM. Roles of nitric oxide synthase inhibition and vascular endothelial growth factor receptor-2 inhibition on vascular morphology and function in an in vivo model of pancreatic cancer. Clin Cancer Res. 2006;12:2628–2633. doi: 10.1158/1078-0432.CCR-05-2257. [DOI] [PubMed] [Google Scholar]
- 24.Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, Gray MJ, Cheng H, Hoff PM, Ellis LM. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006;12:4147–4153. doi: 10.1158/1078-0432.CCR-06-0038. [DOI] [PubMed] [Google Scholar]
- 25.Gray MJ, Van Buren G, Dallas NA, Xia L, Wang X, Yang AD, Somcio RJ, Lin YG, Lim S, Fan F, Mangala LS, Arumugam T, Logsdon CD, Lopez-Berestein G, Sood AK, Ellis LM. Therapeutic targeting of neuropilin-2 on colorectal carcinoma cells implanted in the murine liver. J Natl Cancer Inst. 2008;100:109–120. doi: 10.1093/jnci/djm279. [DOI] [PubMed] [Google Scholar]
- 26.Landen CN, Jr, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, Sood AK. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65:6910–6918. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- 27.Douville J, Beaulieu R, Balicki D. ALDH1 as a Functional Marker of Cancer Stem and Progenitor Cells. Stem Cells Dev. 2008 doi: 10.1089/scd.2008.0055. [DOI] [PubMed] [Google Scholar]
- 28.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duester G. Families of retinoid dehydrogenases regulating vitamin A function: production of visual pigment and retinoic acid. Eur J Biochem. 2000;267:4315–4324. doi: 10.1046/j.1432-1327.2000.01497.x. [DOI] [PubMed] [Google Scholar]
- 30.Purton LE, Bernstein ID, Collins SJ. All-trans retinoic acid enhances the long-term repopulating activity of cultured hematopoietic stem cells. Blood. 2000;95:470–477. [PubMed] [Google Scholar]
- 31.Sladek NE, Kollander R, Sreerama L, Kiang DT. Cellular levels of aldehyde dehydrogenases (ALDH1A1 and ALDH3A1) as predictors of therapeutic responses to cyclophosphamide-based chemotherapy of breast cancer: a retrospective study. Rational individualization of oxazaphosphorine-based cancer chemotherapeutic regimens. Cancer Chemother Pharmacol. 2002;49:309–321. doi: 10.1007/s00280-001-0412-4. [DOI] [PubMed] [Google Scholar]
- 32.Boettiger D, Anderson S, Dexter TM. Effect of src infection on long-term marrow cultures: increased self-renewal of hemopoietic progenitor cells without leukemia. Cell. 1984;36:763–773. doi: 10.1016/0092-8674(84)90356-8. [DOI] [PubMed] [Google Scholar]
- 33.Konig H, Copland M, Chu S, Jove R, Holyoake TL, Bhatia R. Effects of dasatinib on SRC kinase activity and downstream intracellular signaling in primitive chronic myelogenous leukemia hematopoietic cells. Cancer Res. 2008;68:9624–9633. doi: 10.1158/0008-5472.CAN-08-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Konig H, Holyoake TL, Bhatia R. Effective and selective inhibition of chronic myeloid leukemia primitive hematopoietic progenitors by the dual Src/Abl kinase inhibitor SKI-606. Blood. 2008;111:2329–2338. doi: 10.1182/blood-2007-05-092056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nam JS, Ino Y, Sakamoto M, Hirohashi S. Src family kinase inhibitor PP2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clin Cancer Res. 2002;8:2430–2436. [PubMed] [Google Scholar]
- 36.Trevino JG, Summy JM, Gray MJ, Nilsson MB, Lesslie DP, Baker CH, Gallick GE. Expression and activity of SRC regulate interleukin-8 expression in pancreatic adenocarcinoma cells: implications for angiogenesis. Cancer Res. 2005;65:7214–7222. doi: 10.1158/0008-5472.CAN-04-3858. [DOI] [PubMed] [Google Scholar]
- 37.Marhaba R, Klingbeil P, Nuebel T, Nazarenko I, Buechler MW, Zoeller M. CD44 and EpCAM: cancer-initiating cell markers. Curr Mol Med. 2008;8:784–804. doi: 10.2174/156652408786733667. [DOI] [PubMed] [Google Scholar]
- 38.Keysar SB, Jimeno A. More than markers: biological significance of cancer stem cell-defining molecules. Mol Cancer Ther. 2010;9:2450–2457. doi: 10.1158/1535-7163.MCT-10-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanders MA, Majumdar AP. Colon cancer stem cells: implications in carcinogenesis. Front Biosci. 2011;16:1651–1662. doi: 10.2741/3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alison MR, Guppy NJ, Lim SM, Nicholson LJ. Finding cancer stem cells: are aldehyde dehydrogenases fit for purpose? J Pathol. 2010;222:335–344. doi: 10.1002/path.2772. [DOI] [PubMed] [Google Scholar]
- 41.Huss WJ, Gray DR, Greenberg NM, Mohler JL, Smith GJ. Breast cancer resistance protein-mediated efflux of androgen in putative benign and malignant prostate stem cells. Cancer Res. 2005;65:6640–6650. doi: 10.1158/0008-5472.CAN-04-2548. [DOI] [PubMed] [Google Scholar]
- 42.Krishnamurthy P, Ross DD, Nakanishi T, Bailey-Dell K, Zhou S, Mercer KE, Sarkadi B, Sorrentino BP, Schuetz JD. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem. 2004;279:24218–24225. doi: 10.1074/jbc.M313599200. [DOI] [PubMed] [Google Scholar]
- 43.Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, Sorrentino BP. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 44.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 45.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 46.Chen MS, Woodward WA, Behbod F, Peddibhotla S, Alfaro MP, Buchholz TA, Rosen JM. Wnt/beta-catenin mediates radiation resistance of Sca1+ progenitors in an immortalized mammary gland cell line. J Cell Sci. 2007;120:468–477. doi: 10.1242/jcs.03348. [DOI] [PubMed] [Google Scholar]
- 47.Hadjihannas MV, Bruckner M, Jerchow B, Birchmeier W, Dietmaier W, Behrens J. Aberrant Wnt/beta-catenin signaling can induce chromosomal instability in colon cancer. Proc Natl Acad Sci U S A. 2006;103:10747–10752. doi: 10.1073/pnas.0604206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci U S A. 2007;104:618–623. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Araujo J, Logothetis C. Dasatinib: a potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat Rev. 36:492–500. doi: 10.1016/j.ctrv.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.