

Abstract
Upon stimulation by histamine, human vascular endothelial cells (EC) shed a soluble form of tumour necrosis factor receptor 1 (sTNFR1) that binds up free TNF, dampening the inflammatory response. Shedding occurs through proteolytic cleavage of plasma membrane-expressed TNFR1 catalysed by TNF-α converting enzyme (TACE). Surface expressed TNFR1 on EC is largely sequestered into specific plasma membrane microdomains, the lipid rafts/caveolae. The purpose of this study was to determine the role of these domains in TACE-mediated TNFR1 shedding in response to histamine. Human umbilical vein endothelial cells derived EA.hy926 cells respond to histamine via H1 receptors to shed TNFR1. Both depletion of cholesterol by methyl-β-cyclodextrin and small interfering RNA knockdown of the scaffolding protein caveolin-1 (cav-1), treatments that disrupt caveolae, reduce histamine-induced shedding of membrane-bound TNFR1. Moreover, immunoblotting of discontinuous sucrose gradient fractions show that TACE, such as TNFR1, is present within low-density membrane fractions, concentrated within caveolae, in unstimulated EA.hy926 endothelial cells and co-immunoprecipitates with cav-1. Silencing of cav-1 reduces the levels of both TACE and TNFR1 protein and displaces TACE, from low-density membrane fractions where TNFR1 remains. In summary, we show that endothelial lipid rafts/caveolae co-localize TACE to surface expressed TNFR1, promoting efficient shedding of sTNFR1 in response to histamine.
Keywords: TACE, caveolin-1, lipid rafts, TNF-receptor, histamine, inflammation, endothelium
Introduction
Binding of TNF to a variety of cell types activates signaling pathways that regulate several different processes of medical significance such as apoptosis, inflammation, immunity and metabolism and has been implicated in the pathogenesis of several diseases [1, 2]. Although two different TNFRs have been identified [3], the majority of EC responses appear to be activated through TNFR1 (p55-TNFR, CD120a) [4]. TNFRs are ubiquitously expressed in most cultured cell types, but expression is highly regulated in tissues. For example, in human kidney and heart, TNFR1 is largely confined to vascular EC. Under specific circumstances, the shedding of membrane TNFR1 (mTNFR1) act by reducing the amount of mTNFR1 and produces a soluble 27–30 kD form of the receptor (sTNFR1) which compete with mTNFR1 to reduce cell sensitivity to TNF [5, 6]. Interestingly, both in human umbilical vein (HUV)EC and in the EAhy.926 endothelial cell line, the Golgi apparatus plays an important role in this scenario, functioning as a intracellular reservoir of TNFR1 from which it can be translocated to the plasma membrane and shed in response to stimuli [6-8] such as histamine. The generation of sTNFR1 is triggered by TACE (also known as ADAM-17 or CD156b) [9-11], a member of the ADAMs (A Disintegrin And Metalloprotease) family of proteases. TACE is widely expressed in a variety of tissues and shows very little sequence similarity to other ADAMs family members [12]. The mechanism of ectodomain shedding is not unique to TNFR1 and it has been reported that a wide range of inflammatory cell surface proteins operate as soluble form [13]. Regarding TACE, it has been recently proposed that the transmembrane domain (TM) is fundamental for efficient cleavage and its amino-acidic sequence can specifically regulate the shedding of a subset of substrate [14]. In other words, the mechanism of ectodomain shedding represents an additional post-translational regulatory step, which controls the expression of surface molecules. Specifically, the importance of TNFR1 shedding has been demonstrated in patients affected by unexplained episodes of fever and severe localized inflammation who show impaired cleavage of membrane TNFR1 with consequent reduced shedding of the antagonistic soluble receptor [15, 16]. Notably, the plasma membrane of mammalian cells contains small lipid microdomains enriched for cholesterol and glycosphingolipids called lipid rafts [17]. In certain cell types, such as EC, fibroblasts and muscle cells, lipid rafts can be clustered into 50- to 100-nm flask-shaped invaginations called caveolae [18, 19]. These organelles may comprise up to 50% of the plasma membrane of ECs in situ [20-22] but are rapidly lost when cells are placed in culture [23], allowing lipid rafts to be dispersed within the plasma membrane. We previously demonstrated that caveolae are retained in the EC-derived cell line EA.hy926, facilitating the study of these organelles in vitro [24]. Caveolae are organized by the cav family of small cholesterol-binding scaffolding proteins, including cav-1, cav-2 and the muscle-specific cav-3 [25-27] that contain a conserved structural motif [28]. Caveolae were initially implicated in endocytosis and transcytosis of macromolecules across the EC lining of blood vessels [29], and it has been appreciated that these organelles contribute to cell signaling by clustering together various cell surface receptors [28], facilitating receptor cross talk [28, 30]. Caveolae also play a role in the endocytosis of membrane proteins independently of clathrin-coated pits [31-33]. We previously demonstrated that TNFR1 on the plasma membrane localizes to caveolae in EA.hy926 cells and that pharmacological extraction of cholesterol, which reduces the number of caveolae, blocks ligand-induced TNFR1 internalization [24]. More recently, we found that internalization appears to be independent of cav-1 as it is unaffected by small interfering RNA (siRNA) knockdown of this protein, a treatment that reduces caveolar number but leaves lipid rafts intact [8]. Reduction in cav-1 expression also reduced total TNFR1 protein expression, although the effects on plasma membrane levels were variable. It has been reported that the sheddase activity of TACE is concentrated in lipid rafts and inhibitors of this enzyme increase TNF and TNFRs in lipid rafts [34]. Furthermore, other studies demonstrated that lipid rafts play an important role in the shedding of a variety of different molecules [35, 36]. In this study, we extended these findings and propose a key function of cav-1 in both the retaining of TACE within the caveolar network and the shedding of TNFR1 given that the disruption of caveolae inhibits histamine and H1R-mediated sTNFR1 release.
Materials and methods
Evaluation of TNFR1 surface expression by cytofluorimetric analysis
Replicate cultures of EA.hy926 cells were subjected to specific treatment in six-well plates. For evaluating the involvement of protein-G to both the shedding and surface expression of TNFR1, cells were pre-incubated in the presence of the indicated doses of Pertussis toxin (Calbiochem-Novabiochem, Nottinghamshire, UK), Ptx for 1 hr at 37°C. Cells were harvested with 2.5 g/l trypsin 0.2 g/l ethylenediaminetetraacetic acid (EDTA) in Hanks’ balanced salt solution (HBSS) without phenol red, at 37°C and stained with anti-human CD120a (1 μg/106 cells) or with isotype control antibodies, respectively, for 1 hr at 4°C. Cells were washed once by centrifugation at 1000 r.p.m. in HBSS containing 1% bovine serum albumin (BSA) and incubated with streptavidin phycoerythrin for 30 min. at 4°C. After three additional washes, cells were resuspended in HBSS without BSA and acquired using CyAn™ ADP Analyser 9 colour (Beckman Coulter Inc., Miami, FL). Fluorescence of 20 × 103 cells/sample was acquired and analyzed using Summit v4.3 software.
Ca2+ imaging
EA.hy926 were cultivated onto 35-mm dishes in medium containing 3.5 μM fura-2-AM (Invitrogen Corporation, CA, USA) for 1 hr at 37°C, and then rinsed with Krebs–Henseleit–Hepes buffer (140 mM Na+, 5.3 mM K+, 132.4 mM Cl−, 0.98 mM PO42−, 1.25 mM Ca2+, 0.81 mM Mg2+, 5.5 mM glucose and 20 mM Hepes) supplemented with 0.2% fatty acid free BSA or with HBSS. Each dish was placed into a culture chamber at 37°C on the stage of an inverted fluorescence microscope (TE2000E; Nikon), connected to a cooled charge-coupled device camera (512B Cascade; Roper Scientific, Tucson, AZ, USA). Samples were illuminated alternately at 340 and 380 nm using a random access monochromator (Photon Technology International, NJ, USA) and emission was detected using a 510 nm emission filter. Images were acquired (1 ratio image/s) using Metafluor software (Universal Imaging Corporation, Downington, PA, USA). Calibration was obtained at the end of each experiment by maximally increasing intracellular Ca2+-dependent fura-2-AM fluorescence with 5 μM ionomycin, followed by recording minimal fluorescence in Ca2+-free medium. [Ca2+]i was calculated as previously described [37].
Quantitative RT-PCR analysis
Total RNA was extracted with the TRIZOL Reagent (Invitrogen), according to the manufacturer’s instructions. Briefly, 3 μg of RNA was converted into single-stranded DNA by a standard 20 μl RT reaction with the TaqMan RT Reagent (Applied Biosystems, CA, USA). Real-time quantitative RT-PCR was performed on a 7500 Real-Time PCR System (Applied Biosystems). Briefly, the cDNA generated from the reverse transcription reactions was amplified by PCR with the SYBR Green JumpStart Taq ReadyMix (Sigma-Aldrich, St. Louis, MO, USA) in a total volume of 25 μl according to the manufacturer’s instructions. The primers used were as follows: TNFR1, 5′-AATGCCGAAAGGAAATGGGTCAGG-3′ and 5′-TGCACACGGTGTTCTGTTTCTCCT-3′; TACE, 5′-AGTGCAGTGACAGGAACAGTCCTT-3′ and 5′-GGACACGCCTTTGCAAGTAGCATT-3′; β-Actin, 5′-TGCACCACACCTTCTACAATGA-3′ and 5′-CAGCCTGGATAGCAACGTACAT-3′. The level of messenger analysed was expressed as relative fold change versus the β-actin messenger RNA and analysed by means of the 7500 System Software (Applied Biosystems).
Design and transfection of cav-1 siRNA duplexes
siRNA duplex oligonucleotides against the coding sequence of human cav-1 cDNA (NM_001753) were synthesized and purchased by Integrated DNA Technologies (Coralville, IA, USA). We selected two target sequences as follows: 5′-ACCAGAAGGGACACACAGUdTdT-3′- (sense-#538) and 5′-ACUGUGACGAAAUACUGGUdTdT-3′ (sense #542). Transfection of 80 pmol siRNA in EA.hy926 was carried out by using oligofectamine (0.3%, v/v; Invitrogen), according to the manufacturer’s instructions. Fresh medium was added 5 hrs after transfection and experiments were conducted for 72 hrs. Non-targeting control siRNA-A (Santa Cruz, CA, USA) was used as control.
Isolation of caveolae-enriched membranes
Purification of caveolae-enriched membrane fractions was performed as previously described [24, 38] with minor modifications. In brief, cells were washed twice with ice-cold Dulbecco’s PBS and scraped with 25 mM 2-(N-Morpholino) ethanesulfonic acid (MES) hydrate buffer pH 6.5 containing 0.15M NaCl, 5 mM EDTA and 0.5% Triton X-100 [MES-Buffer Saline, (MBS)] with protease inhibitors (10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM sodium orthovanadate, 10 mM NaF, 1 mM Pefabloc) and left 20 min. on ice. Three wells of a six-well plate were pooled together and the resultant lysate was subjected to 15 strokes in a Dounce homogenizer and centrifuged for 10 min. at 2000 r.p.m. at 4°C to remove nuclei. Clarified post-nuclear supernatants were combined with 90% (w/v) sucrose prepared in MBS, transferred to the bottom of a Beckman 2 ml ultracentrifuge tubes and overlaid gently with 1 ml of 35% and 0.6 ml of 5% sucrose, respectively. The resulting 5–40% discontinuous sucrose gradients were centrifuged 18 hrs at 35,000 r.p.m. at 4°C. After centrifugation, 10 fractions of 200 μl each were harvested from the top to the bottom of the gradients and analysed either by SDS-PAGE and immunoblotting.
Immunoblotting
For immunoblotting experiments, scrape-harvested cells were extracted in 150 μl of lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, 2% SDS and 1 mM Pefabloc) for 15 min. on ice. Unbroken cells and debris were spun down by centrifugation at 2000 r.p.m. for 10 min. at 4°C. Twenty micrograms of cell lysate was fractionated by SDS-PAGE, transferred to a nitrocellulose membrane (Trans-Blot Transfer Medium, Bio-Rad, Hercules, CA, USA) and subjected to immunoblotting with primary antibody followed by incubation with HRP-conjugated secondary antibody. Detection of the bound antibody was performed by enhanced chemiluminescence (Pierce Biotechnology, Rockford, IL, USA). For immunoblotting analysis of sTNFR1, cells were treated with indicated chemicals diluted in 1.6 ml of OPTI-MEM. Culture media were collected and concentrated about 50 times using Vivaspin ultrafiltration spin columns (Sartorius Stedim Biotech GmbH, Goettingen, Germany), and samples were analysed by SDS-PAGE electrophoresis and immunoblotting. Intensities of the bands for specific proteins of interest were quantified and normalized to the intensity of the band for β-actin with Scion Image application software (Scion Corporation, Frederick, MD, USA).
Reagents and antibodies
Mouse anti-TNFR1 and H1R were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Mouse anti-human TNFR1-B1 employed for cytofluorimetric analysis was from BD Transduction Laboratories (Lexington, KY, USA). Cav-1 and TACE antibodies were from Cell Signaling (Beverly, MA, USA). Actin antibody was from Sigma-Aldrich. Anti-mouse R-phycoerythrin were from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Enhanced chemiluminescence used in immunoblotting was from Pierce (Pierce, Rockford, IL, USA). Oligofectamine and Opti-MEM were purchased from Invitrogen. All other chemicals were from Sigma-Aldrich.
Results
Modulation of surface expression of TNFR1 by histamine H1 receptor
In human umbilical vein endothelial cells (HUVEC), histamine causes TACE-mediated shedding of mTNFR1, which in turn desensitizes TNFR1-mediated responses [6]. Although HUVEC recapitulate many features of EC in situ, they rapidly lose their caveolae in culture [23], limiting the investigation of these organelles in vitro. Interestingly, we have recently reported that the EA.hy926 endothelial cell line [39] not only retains caveolae but also expresses higher surface TNFR1 level than HUVEC, which renders this cell line an appropriate model for studying the role of caveolae in vitro. Moreover, of the four known histamine receptors identified [40], EA.hy926 appear to express only H1R and H2R [41]. To evaluate which histamine receptor was involved in TNFR1 cleavage we performed immunoblotting analysis on culture medium collected by both unstimulated- and histamine-stimulated cells in the presence or absence of H1R antagonist mepyramine. Our data suggest that the shedding of TNFR1 was prevented by pre-incubation of cells with mepyramine, suggesting that H1R was primarily involved in the cleavage of TNFR1. As expected, stimulation of cells with 2-3-trifluoromethylphenyl-histamine (TMPH), the specific H1R agonist, did also induce receptor shedding (Fig. 1A). Fluorescence activated cell sorting (FACS) analysis confirmed that both histamine and TMPH stimulation reduced the expression of TNFR1 on the plasma membrane (Fig. 1B), and that this effect was prevented by mepyramine (Fig. 1C), further supporting the involvement of only H1R in this mechanism. TACE has been reported as the major ADAM responsible for the cleavage of membrane TNFR1 [9-11]. Therefore, we next employed the TACE inhibitor TAPI-0 to confirm the contribution of TACE- to H1R-mediated TNFR1 shedding as reported in (Fig. 1D). We conclude that EA.hy926 cells, such as HUVEC, respond to histamine through H1R to induce TACE-mediated shedding of TNFR1 from the plasma membrane generating a soluble form of the receptor (sTNFR1).
Fig 1.
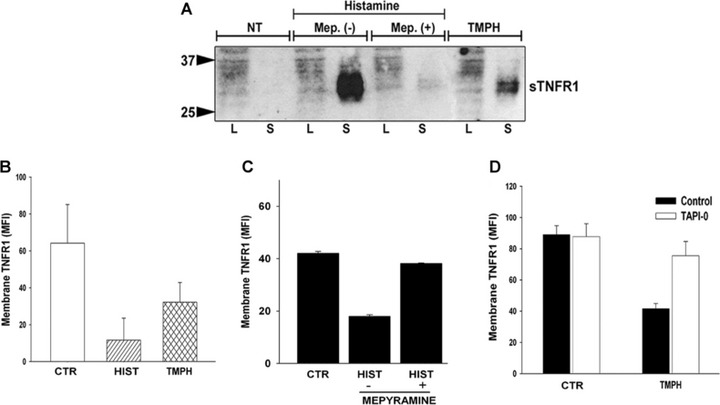
Effect of histamine receptors on TNFR1. (A) Cells were cultured in the presence or absence of mepyramine as described in Materials and methods prior to histamine treatment for 30 min. Concentrated medium from treated cells were analysed by SDS-PAGE and immunoblotted with TNFR1 antibody. TMPH was used as specific agonist of H1R. NT represent cells stimulated with vehicle alone. Data are representative of three different experiments. (B) Effect of histamine and TMPH on the expression of surface TNFR1. (C) Effect of mepyramine on histamine-induced down-regulation of membrane TNFR1. (D) Inhibition of TMPH-induced down-regulation of membrane bound TNFR1 in the presence of TACE inhibitor, TAPI-0.
H2R counteracts constitutive shedding of TNFR1
Because EA.hy926 cells also express H2R, which in addition to H1R functions as natural receptor of histamine, we wondered whether this histamine receptor was also involved in the ectodomain shedding of TNFR1. To this purpose, we employed amthamine (AMTHA), the specific H2R agonist. We found that incubation of cells with AMTHA alone did not affect the shedding of TNFR1 but rather appeared to reduce its constitutive release as sTNFR1 (Fig. 2A). This result suggested that signaling activated by H2R, possibly through the accumulation of cAMP, may perhaps counteract the release of sTNFR1. In fact, unlike H1R, which has been showed to induce a rise of cytosolic Ca2+ concentration [42-44], H2R typically appears to function by increasing adenylate cyclase (AC) activity and accumulation of cAMP [40]. To test this hypothesis, we stimulated cells with forskolin, a receptor-independent activator of AC and evaluated the shedding of TNFR1. Forskolin appeared to diminish both constitutive and TMPH-stimulated release of sTNFR1 (Fig. 2B), without significant changes of surface exposed TNFR1 (Fig. 2C). In addition, we investigated the possible involvement of the inhibitory Gi protein by employing pertussis toxin. Our data showed that pre-incubation of cells with Ptx slightly reduced the constitutive release of sTNFR1 (Fig. 2D) while showing a non-significant change of TNFR1 surface expression (Fig. 2E), confirming our findings obtained with forskolin. Moreover, histamine-induced shedding of TNFR1 was not affected by Ptx, indicating that histamine H1 and H2 receptors are not Gi-protein linked. These findings suggest that the fate of TNFR1 shedding induced by TACE in EA.hy926 cells may be further regulated by a functional interplay between signaling activated by both histamine H1 and H2 receptors.
Fig 2.
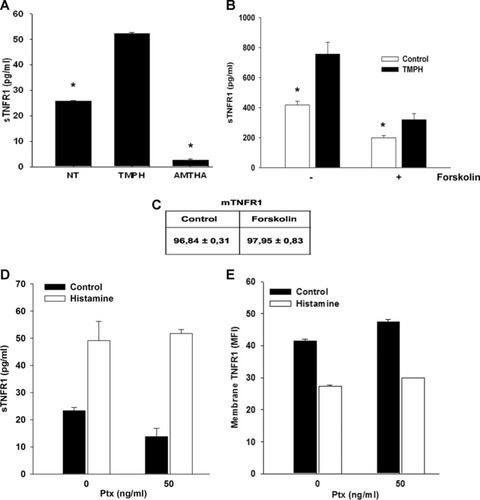
Forskolin reduced both constitutive and TMPH-stimulated release of TNFR1. (A) Effect of 500 μM AMTHA and 100 μM TMPH on sTNFR1 release. (B) Cells were pre-incubated with 10 μM forskolin or with vehicle alone prior to TMPH treatment and soluble TNFR1 was measured by ELISA assay. (C) Values indicate the mean fluorescence intensity measured by FACS analysis of membrane TNFR1 in both forskolin- and vehicle-treated cells. Effect of Ptx treatment on both sTNFR1 release (D) and membrane TNFR1 expression. Data are representative of two independent experiments (*P < 0.05).
Effects of disrupting caveolae on histamine-induced shedding of TNFR1
We have previously shown that TNFR1 and cav-1 associate with caveolae in EA.hy926 cells and that this association as well as ligand-induced TNFR1 internalization is disrupted by extraction of cholesterol with methyl-β-cyclodextrin (MbCD) [24]. MbCD functions by extracting cholesterol from the plasma membrane, which results fundamental for the correct formation of lipid rafts microdomains. Therefore, we wondered whether MbCD could affect TMPH-induced release of sTNFR1. ELISA analysis showed that pre-treatment of cells with MbCD decreased TMPH-induced release of sTNFR1 (Fig. 3A), and FACS analysis confirmed that cholesterol depletion did also reduce the capability of TMPH to decrease the expression of exposed TNFR1 (Fig. 3B). Nevertheless, to determine whether the effect of cholesterol extraction was on a proximal signaling event, we examined H1R-dependent calcium responses [40] in the presence of MbCD and found that cholesterol depletion did not interfere with TMPH-induced calcium mobilization (Fig. 3C), suggesting that action of MbCD may involve downstream events, possibly affecting the process of shedding itself.
Fig 3.
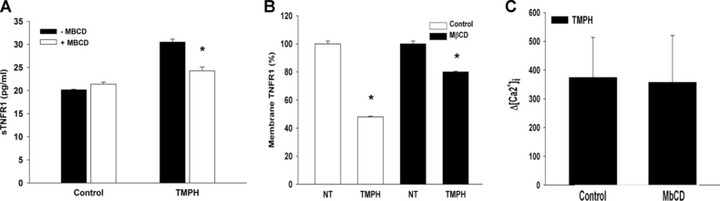
Evaluation of MbCD effect on TNFR1 shedding and Ca2+ mobilization. (A) ELISA analysis of sTNFR1 from culture media of cells pre-incubated with 5 mM MbCD for 30 min. and then stimulated with TMPH. (B) FACS analysis showing the effect of MbCD on expression of surface TNFR1 in response to TMPH. (C) Effect of cholesterol depletion by MbCD on TMPH-induced calcium mobilization (*P < 0.05).
Cav-1 down-regulation inhibits TMPH-induced TNFR1 shedding
In certain cell types, the expression of the scaffolding protein cav-1 induces the clustering of lipid rafts into vesicle-like organelles called caveolae. To this regard, we investigated the contribution of cav-1 to TNFR1 shedding by evaluating the behaviour of cav-1 silenced cells in response to histamine and TMPH. We designed two different siRNA duplex oligonucleotides targeting the coding sequence of human cav-1 cDNA and tested their silencing efficiency by immunoblotting (Fig. 4A). Interestingly, we found that reducing cav-1 expression by RNAi did affect both histamine- and TMPH-induced release of sTNFR1 (Fig. 4B), suggesting that cav-1 may play an important role in the regulation of TACE activity. In addition, similar to what we had seen with MbCD, cav-1 knockdown did not impair TMPH-induced calcium mobilization (Fig. 4C). These data support the hypothesis that the mechanism of calcium release induced by TMPH and TNFR1 shedding are functionally uncoupled.
Fig 4.
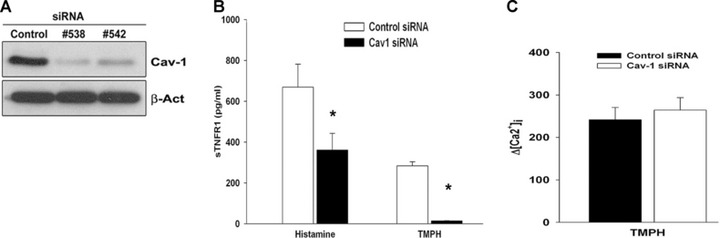
Involvement of cav-1 in the release of TMPH- and histamine-induced release of sTNFR1. (A) Immunoblotting of cav-1 expression showing the efficiency of the two cav-1 dsRNA sequences used for RNAi experiments in EA.hy926 cell line. (B) EA.hy926 cells were first subjected to cav-1 knockdown as described in Materials and methods and then treated with either histamine or TMPH for 30 min. The amount of sTNFR1 was measured by ELISA assay from cell culture supernatants. (C) Cav-1 knockdown did not affect TMPH-induced calcium mobilization. Bars show the increase of calcium mobilization after stimulation with TMPH. Data are representative of results of three independent experiments (*P < 0.05).
Sub cellular localization of H1R and TACE in EA.hy926 cells
Despite the limitations in the use of cav-1 silencing to quantify shedding, it is still useful as an approach to study receptor localization because it disrupts caveolae while dispersing rather than causing loss of low density lipid rafts. Some G protein coupled receptors, the category to which H1R belongs, localize to lipid rafts [45, 46] and TACE has also been localized to lipid rafts in some cell types [34]. Disruption of lipid rafts and caveolae by pharmacological depletion of cholesterol has been also reported to affect the release of ADAMs substrates [47-49]. Therefore, we next evaluated the distribution of both H1R and TACE by means of sucrose gradient fractionation in EC. Under our experimental procedures, H1R was mainly excluded from low-density membranes in either case (Fig. 5A). By contrast, TACE was extensively detected along the sucrose gradient obtained from cells transfected with the control dsRNA duplex (Fig. 5B, upper). In contrast, cav-1 silencing reduced TACE expression in high-density membranes and its localization within the low-density membranes (Fig. 5B, lower). Moreover, we found that immunoprecipitation of cav-1 followed by immonoblotting with anti-TACE antibody revealed the interaction between these two proteins that was also unchanged after histamine treatment (Fig. 5C). Although we did not investigate the origin of this association, this result suggest that cav-1 expression may be significant for sequestrating TACE within the caveolar network, placing the enzyme in proximity to its substrate. Interestingly, we showed that cav-1 silencing reduced TNFR1 protein but not mRNA expression in EA.hy926 cells, although it did not cause TNFR1 to dissociate from (now dispersed) lipid rafts [8]. We therefore evaluated if cav-1 silencing affected TACE protein expression. Besides confirming the overall reduction of TNFR1 induced by cav-1 down-regulation [8] (Fig. 6A), we found that TACE protein expression, but not its mRNA level, was also decreased in cav-1 knockdown cells (Fig. 6B), indicating that the level of TNFR1 shedding can be regulated by the reduction of both TACE and TNFR1.
Fig 5.
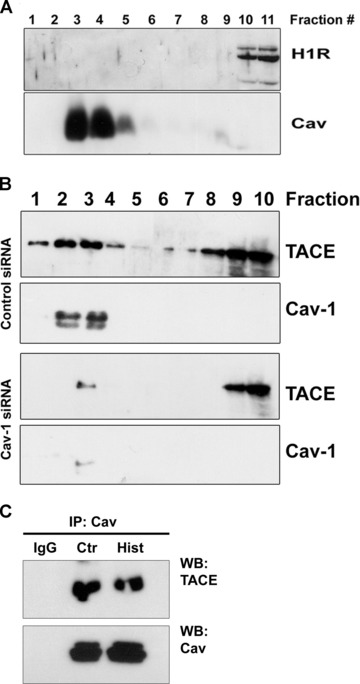
Effect of cav-1 knockdown on proteins distribution and transcripts level. (A) Sucrose gradient fractionation was performed to isolate low buoyant fractions enriched in caveolin proteins. Immunoblotting of fractions harvested from the top (low sucrose density) to the bottom (high sucrose density) show that H1R was excluded from caveolin-enriched fractions. (B) In resting cells, TACE appeared to localize both with low and high sucrose density membranes. By contrast, cav-1 down-regulation by RNAi induced displacement of TACE mainly from the caveolin-enriched membranes (lower panels). (C) Cav-1 immunoprecipitates were immunoblotted for TACE to evaluate protein–protein interaction. IgG served as negative control.
Fig 6.
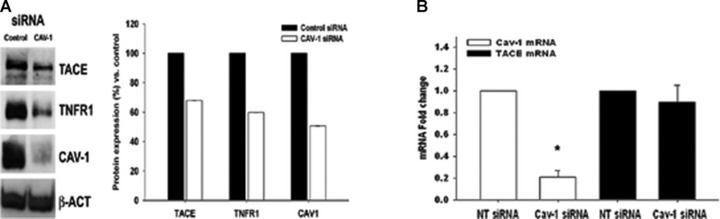
Effect of cav-1 RNAi on both TACE and TNFR1 protein expression. (A) Immunoblotting analysis showing the effect of cav-1 knockdown on TACE, TNFR1 and cav-1 protein expression. β-Actin (indicated as β-Act) was used as equal loading control (left). Densitometric analysis of replicate immunoblotting experiment showing the effect of cav-1 RNAi on TACE, TNFR1 and cav-1 protein expression (right). (B) Effect of cav-1 silencing on both cav-1 and TACE mRNA transcripts (*P < 0.05).
Discussion
ADAM family of metalloproteases has been linked to diverse biological processes leading different groups to explore novel potential regulators which can activate or inhibit these proteins both under physiological and pathological conditions [12, 50, 51]. Because TNF and TNF receptors are targets of TACE/ADAM17, understanding the mechanism which regulate TACE activity may represents an important target for modulating the inflammatory response. In this study, we extended our previous works on the relationship between TNFR1 and caveolae [8, 24] and reported the contribution of cav-1 to TACE-mediated shedding of the membrane-bound TNFR1. Histamine evokes a variety of effects in different systems such as nervous, endocrine and cardiovascular by binding to four different receptors. However, the availability of specific agonists and antagonists [52] represents an important tool for studying the contribution of histamine receptors in different cell types. Notably, it has been reported that primary human ECs, such as HUVEC, rapidly loose caveolae after few cell doubling in culture [23], limiting the investigation of caveolae in primary EC. Therefore, we employed the permanent cell line EA.hy926 established by fusing primary HUVEC with a thioguanine-resistant clone of A549, which shows functions characteristic of human vascular endothelium [39]. Unlike cultured HUVEC, EA.hy926 retain an extensive caveolar network and express higher level of surface TNFR1 compared to primary cells, which make them an appropriate model to explore the contribution of caveolae to TNFR1 signaling in vitro. To this regard, we found that similarly to what had been reported in HUVEC [6], histamine also stimulated TACE-mediated TNFR1 shedding in EA.hy926. Moreover, by employing the specific H1R antagonist mepyramine, we show that this mechanism was specifically mediated through the recruitment of H1 receptor and the contribution of TACE was confirmed by pre-treatment of cells with TACE inhibitor TAPI-0. Importantly, H1 and H2 receptors usually coexist in blood vessels [53, 54] and are also co-expressed in EA.hy926 [41], allowing us to investigate the contribution of H2R to the shedding of TNFR1. We found that stimulation of histamine H2 receptor with AMTHA not only failed to induce TNFR1 shedding, but also appeared to diminish the constitutive release of the cleaved TNFR1. Given that H2R recruitment was reported to induce cAMP accumulation via Gs-protein activation [55, 56], we hypothesized that the second messenger may function by counteracting H1R-induced shedding of membrane TNFR1. Our data, in fact, demonstrate that forskolin-mediated activation of AC reduced TMPH-induced sTNFR1 accumulation, which suggests a contribution of cAMP on TACE-mediated receptor shedding. Forskolin is generally used to induce intracellular cAMP accumulation by directly activating AC bypassing H2R recruitment. Moreover, additional experiments performed in the presence of Ptx, which catalyses ADP-ribosylation at cysteine residue of Giα blocking the AC inhibitory pathway, indicate that histamine H1 and H2 receptors are not Gi-protein linked in EA.hy926 cells. Therefore, our data indicate the involvement of cAMP in the inhibition of TNFR1 shedding, although other signaling might affect TACE activity in response to AMTHA. In fact, it has been reported that H2R stimulation also affect other second messengers, such as arachidonic acid, and phospholipase A2 that may in turn disturb TACE-mediated shedding [56]. Although the mechanism of H2R-mediated TACE inhibition remains unknown, it is possible that the mechanism of TNFR1 shedding might be affected by opposing responses activated by H1R and H2R stimulation.
A key finding of this report is that both cholesterol depletion with MbCD and knockdown of cav-1 by RNAi markedly reduced H1R-dependent TNFR1 shedding, and neither treatments reduced calcium mobilization caused by H1R signaling. This suggests that the mechanism of shedding itself may be independent of calcium mobilization induced by H1R. Notably, cav-1 silencing reduces the expression level of TNFR1 [8], highlighting the importance of cav-1 for receptor stability. On the other hand, cholesterol depletion which pharmacologically disrupt caveolae/lipid rafts would only induce surface redistribution of TNFR1 which is also sufficient to affect the shedding mechanism itself, possibly by destabilizing specific protein interactions. To this regard, we demonstrated that short MbCD treatment prevented co-localization of cav-1 and TNFR1 [24]. We have also showed that intrinsic signaling by TNFR1 is not inhibited by these treatments [8], suggesting that localization of the receptor to caveolae may be important for interactions with other caveolar proteins. Because TNFR1 shedding is mediated by TACE, which has been reported to localize to lipid rafts [34], structures that are concentrated within caveolae, we hypothesized that caveolae/lipid rafts may be important for regulating the compartmentalization of TACE and its substrates. By employing a sucrose gradient fractionation technique, we found that TACE localized to cholesterol (e.g. lipid rafts micro domains) and cav-enriched domains, which in EA.hy926 EC are largely confined to caveolae. In contrast, H1R appeared to be restricted to high-density membranes that does not contains lipid raft domains. These findings allow to exclude a direct interaction between H1R and TACE. Interestingly, sucrose gradient fractionation performed on of cav-1 silenced cells showed that knock down of cav-1 displaced TACE from the light-density fractions and apparently reduced its expression within high-density membrane fractions. However, although TNFR1 similar to TACE can be co-imunoprecipitated with cav-1 from EA.hy926 cell lysates, cav-1 knockdown does not displace TNFR1 from (now dispersed) lipid rafts [8]. Data generated in HUVEC at early passages demonstrated similar effects of cav-1 knockdown on histamine-induced shedding (data not shown) even though at lower extent. This phenomenon is not unexpected because primary HUVEC rapidly lose their caveolae in culture and for unknown reason show a smaller amount of exposed TNFR1 compared to EAhy.926. In conclusion, data reported here suggest that cav-1 may play several crucial roles in the shedding of TNFR1 from EC, including maintenance of caveolae network, co-localizing TNFR1 and TACE within caveolae and sustaining higher levels of both proteins. The reduction in the expression of both TNFR1 and TACE followed by cav-1 silencing may be thus the cause of the reduced shedding of TNFR1 receptor, which suggests the importance of cav-1 for maintaining this mechanism controlled. In other word, caveolae/lipid rafts would play a crucial structural role preserving receptor cross-talk rather than regulating receptor signalling. In conclusion, our data suggest that specific targeting of caveolae signalling platform may represent an innovative approach to control endothelial cell functions and might have prospective for management of inflammatory disorders.
Acknowledgments
The authors thank Prof. Fioretta Palombi for critical reading and Dr. Fabrizio Padula for technical assistance with flow cytometry.
Funding source: This work was supported by the “Rientro dei Cervelli” Grant Program from Ministero dell’Universit‡ e della Ricerca (MIUR) to AD, from Italian Ministry Health Grant 2007 to EZ, from PRIN 2008 to AF, and a grant from the U.S. National Institutes of Health (HL036003) to JSP.
Authors’ contributions
A.D. designed, performed the research and wrote the paper, A.F. designed the research and wrote the paper, J.S.P. designed the research, B.E. and C.G. performed the research and E.Z. critically read the paper.
Conflict of interest
The authors have no financial conflicts of interest.
References
- 1.Chen G, Goeddel DV. TNF-R1 signaling: abeautiful pathway. Science. 2002;296:1634–5. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 2.Liu Z-G. Molecular mechanism of TNF signaling and beyond. Cell Res. 2005;15:24–7. doi: 10.1038/sj.cr.7290259. [DOI] [PubMed] [Google Scholar]
- 3.Loetscher H, Schlaeger EJ, Lahm HW, et al. Purification and partial amino acid sequence analysis of two distinct tumour necrosis factor receptors from HL60 cells. J Biol Chem. 1990;265:20131–8. [PubMed] [Google Scholar]
- 4.Slowik MR, De Luca LG, Fiers W, et al. Tumour necrosis factor activates human endothelial cells through the p55 tumour necrosis factor receptor but the p75 receptor contributes to activation at low tumour necrosis factor concentration. Am J Pathol. 1993;143:1724–30. [PMC free article] [PubMed] [Google Scholar]
- 5.Aderka D, Engelmann H, Shemer-Avni Y, et al. Variation in serum levels of the soluble TNF receptors among healthy individuals. Lymphokine Cytokine Res. 1992;11:157–9. [PubMed] [Google Scholar]
- 6.Wang J, Al-Lamki RS, Zhang H, et al. Histamine antagonizes tumour necrosis factor (TNF) signaling by stimulating TNF receptor shedding from the cell surface and Golgi storage pool. J Biol Chem. 2003;278:21751–60. doi: 10.1074/jbc.M212662200. [DOI] [PubMed] [Google Scholar]
- 7.Bradley JR, Thiru S, Pober JS. Disparate localization of 55-kd and 75-kd tumour necrosis factor receptors in human endothelial cells. Am J Pathol. 1995;146:27–32. [PMC free article] [PubMed] [Google Scholar]
- 8.D’Alessio A, Kluger MS, Li JH, et al. Targeting of tumour necrosis factor receptor 1 to low density plasma membrane domains in human endothelial cells. J Biol Chem. 2010;285:23868–79. doi: 10.1074/jbc.M110.122853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–33. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 10.Moss ML, Jin SL, Milla ME, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–6. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 11.Reddy P, Slack JL, Davis R, et al. Functional analysis of the domain structure of tumour necrosis factor-alpha converting enzyme. J Biol Chem. 2000;275:14608–14. doi: 10.1074/jbc.275.19.14608. [DOI] [PubMed] [Google Scholar]
- 12.Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008;29:258–89. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hooper NM, Karran EH, Turner AJ. Membrane protein secretases. Biochem J. 1997;321:265–79. doi: 10.1042/bj3210265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Perez L, Pan Z, et al. The transmembrane domain of TACE regulates protein ectodomain shedding. Cell Res. 2007;17:985–98. doi: 10.1038/cr.2007.98. [DOI] [PubMed] [Google Scholar]
- 15.McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–44. doi: 10.1016/s0092-8674(00)80721-7. [DOI] [PubMed] [Google Scholar]
- 16.Galon J, Aksentijevich I, McDermott MF, et al. TNFRSF1A mutations and autoinflammatory syndromes. Curr Opin Immunol. 2000;12:479–86. doi: 10.1016/s0952-7915(00)00124-2. [DOI] [PubMed] [Google Scholar]
- 17.Helms JB, Zurzolo C. Lipids as targeting signals: lipid rafts and intracellular trafficking. Traffic. 2004;5:247–54. doi: 10.1111/j.1600-0854.2004.0181.x. [DOI] [PubMed] [Google Scholar]
- 18.Yamada E. The fine structure of the gall bladder epithelium of the mouse. J Biophys Biochem Cytol. 1955;1:445–58. doi: 10.1083/jcb.1.5.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stan RV, Roberts WG, Predescu D, et al. Immunoisolation and partial characterization of endothelial plasmalemmal vesicles (caveolae) Mol Biol Cell. 1997;8:595–605. doi: 10.1091/mbc.8.4.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bruns RR, Palade GE. Studies on blood capillaries. I. General organization of blood capillaries in muscle. J Cell Biol. 1968;37:244–76. doi: 10.1083/jcb.37.2.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simionescu M, Simionescu N, Palade GE. Morphometric data on the endothelium of blood capillaries. J Cell Biol. 1974;60:128–52. doi: 10.1083/jcb.60.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johansson BR. Size and distribution of endothelial plasmalemmal vesicles in consecutive segments of the microvasculature in cat skeletal muscle. Microvasc Res. 1979;17:107–17. doi: 10.1016/0026-2862(79)90400-x. [DOI] [PubMed] [Google Scholar]
- 23.Vasile E, Qu H, Dvorak HF, et al. Caveolae and vesiculo-vacuolar organelles in bovine capillary endothelial cells cultured with VPF/VEGF on floating Matrigel-collagen gels. J Histochem Cytochem. 1999;47:159–67. doi: 10.1177/002215549904700205. [DOI] [PubMed] [Google Scholar]
- 24.D’Alessio A, Al-Lamki RS, Bradley JR, et al. Caveolae participate in tumour necrosis factor receptor 1 signaling and internalization in a human endothelial cell line. Am J Pathol. 2005;166:1273–82. doi: 10.1016/S0002-9440(10)62346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang Z, Scherer PE, Okamoto T, et al. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem. 1996;271:2255–61. doi: 10.1074/jbc.271.4.2255. [DOI] [PubMed] [Google Scholar]
- 26.Glenney JR, Jr, Soppet D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. Proc Natl Acad Sci USA. 1992;89:10517–21. doi: 10.1073/pnas.89.21.10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scherer PE, Okamoto T, Chun M, et al. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci USA. 1996;93:131–5. doi: 10.1073/pnas.93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–67. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 29.Anderson RG. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- 30.Lisanti MP, Scherer PE, Tang Z, et al. Caveolae, caveolin and caveolin-rich membrane domains: a signalling hypothesis. Trends Cell Biol. 1994;4:231–5. doi: 10.1016/0962-8924(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 31.Sigismund S, Woelk T, Puri C, et al. Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci USA. 2005;102:2760–5. doi: 10.1073/pnas.0409817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aguilar RC, Wendland B. Endocytosis of membrane receptors: two pathways are better than one. Proc Natl Acad Sci USA. 2005;102:2679–80. doi: 10.1073/pnas.0500213102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumari S, Mg S, Mayor S. Endocytosis unplugged: multiple ways to enter the cell. Cell Res. 2010;20:256–75. doi: 10.1038/cr.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tellier E, Canault M, Rebsomen L, et al. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp Cell Res. 2006;312:3969–80. doi: 10.1016/j.yexcr.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 35.Gil C, Cubi R, Aguilera J. Shedding of the p75NTR neurotrophin receptor is modulated by lipid rafts. FEBS Lett. 2007;581:1851–8. doi: 10.1016/j.febslet.2007.03.080. [DOI] [PubMed] [Google Scholar]
- 36.Schuster B, Meinert W, Rose-John S, et al. The human interleukin-6 (IL-6) receptor exists as a preformed dimer in the plasma membrane. FEBS Lett. 2003;538:113–6. doi: 10.1016/s0014-5793(03)00154-6. [DOI] [PubMed] [Google Scholar]
- 37.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–50. [PubMed] [Google Scholar]
- 38.Sargiacomo M, Sudol M, Tang Z, et al. Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J Cell Biol. 1993;122:789–807. doi: 10.1083/jcb.122.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA. 1983;80:3734–7. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill SJ, Ganellin CR, Timmerman H, et al. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol Rev. 1997;49:253–78. [PubMed] [Google Scholar]
- 41.Li H, Burkhardt C, Heinrich UR, et al. Histamine upregulates gene expression of endothelial nitric oxide synthase in human vascular endothelial cells. Circulation. 2003;107:2348–54. doi: 10.1161/01.CIR.0000066697.19571.AF. [DOI] [PubMed] [Google Scholar]
- 42.Carson MR, Shasby SS, Shasby DM. Histamine and inositol phosphate accumulation in endothelium: cAMP and a G protein. Am J Physiol. 1989;257:L259–64. doi: 10.1152/ajplung.1989.257.4.L259. [DOI] [PubMed] [Google Scholar]
- 43.Jacob R, Merritt JE, Hallam TJ, et al. Repetitive spikes in cytoplasmic calcium evoked by histamine in human endothelial cells. Nature. 1988;335:40–5. doi: 10.1038/335040a0. [DOI] [PubMed] [Google Scholar]
- 44.Rotrosen D, Gallin JI. Histamine type I receptor occupancy increases endothelial cytosolic calcium, reduces F-actin, and promotes albumin diffusion across cultured endothelial monolayers. J Cell Biol. 1986;103:2379–87. doi: 10.1083/jcb.103.6.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ostrom RS, Insel PA. The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br J Pharmacol. 2004;143:235–45. doi: 10.1038/sj.bjp.0705930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chini B, Parenti M. G-protein coupled receptors in lipid rafts and caveolae: how, when and why do they go there. J Mol Endocrinol. 2004;32:325–38. doi: 10.1677/jme.0.0320325. [DOI] [PubMed] [Google Scholar]
- 47.Zimina EP, Bruckner-Tuderman L, Franzke CW. Shedding of collagen XVII ectodomain depends on plasma membrane microenvironment. J Biol Chem. 2005;280:34019–24. doi: 10.1074/jbc.M503751200. [DOI] [PubMed] [Google Scholar]
- 48.Matthews V, Schuster B, Schutze S, et al. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE) J Biol Chem. 2003;278:38829–39. doi: 10.1074/jbc.M210584200. [DOI] [PubMed] [Google Scholar]
- 49.von Tresckow B, Kallen KJ, von Strandmann EP, et al. Depletion of cellular cholesterol and lipid rafts increases shedding of CD30. J Immunol. 2004;172:4324–31. doi: 10.4049/jimmunol.172.7.4324. [DOI] [PubMed] [Google Scholar]
- 50.Duffy MJ, McKiernan E, O’Donovan N, et al. The role of ADAMs in disease pathophysiology. Clin Chim Acta. 2009;403:31–6. doi: 10.1016/j.cca.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 51.Duffy MJ, McKiernan E, O’Donovan N, et al. Role of ADAMs in cancer formation and progression. Clin Cancer Res. 2009;15:1140–4. doi: 10.1158/1078-0432.CCR-08-1585. [DOI] [PubMed] [Google Scholar]
- 52.Huang JF, Thurmond RL. The new biology of histamine receptors. Curr Allergy Asthma Rep. 2008;8:21–7. doi: 10.1007/s11882-008-0005-y. [DOI] [PubMed] [Google Scholar]
- 53.Matsuki T, Ohhashi T. Endothelium and mechanical responses of isolated monkey pulmonary veins to histamine. Am J Physiol. 1990;259:H1032–7. doi: 10.1152/ajpheart.1990.259.4.H1032. [DOI] [PubMed] [Google Scholar]
- 54.Chipman P, Glover WE. Histamine H2-receptors in the human peripheral circulation. Br J Pharmacol. 1976;56:494–6. doi: 10.1111/j.1476-5381.1976.tb07463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gantz I, Schaffer M, DelValle J, et al. Molecular cloning of a gene encoding the histamine H2 receptor. Proc Natl Acad Sci USA. 1991;88:429–33. doi: 10.1073/pnas.88.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Traiffort E, Ruat M, Arrang JM, et al. Expression of a cloned rat histamine H2 receptor mediating inhibition of arachidonate release and activation of cAMP accumulation. Proc Natl Acad Sci USA. 1992;89:2649–53. doi: 10.1073/pnas.89.7.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]