

Abstract
Background & Aims
Acute pancreatitis increases morbidity and mortality from organ necrosis by mechanisms that are incompletely understood. Dendritic cells (DCs) can promote or suppress inflammation, depending on their subtype and context. We investigated the roles of DC in development of acute pancreatitis.
Methods
Acute pancreatitis was induced in CD11c.DTR mice using caerulein or L-arginine; DCs were depleted by administration of diphtheria toxin. Survival was analyzed using Kaplan-Meier analysis.
Results
Numbers of MHC II+CD11c+DC increased 100-fold in pancreas of mice with acute pancreatitis, to account for nearly 15% of intra-pancreatic leukocytes. Intra-pancreatic DC acquired an immune phenotype in mice with acute pancreatitis; they expressed higher levels of MHC II and CD86 and increased production of interleukin-6, membrane cofactor protein (MCP)-1, and tumor necrosis factor (TNF)-α. However, rather than inducing an organ-destructive inflammatory process, DC were required for pancreatic viability; the exocrine pancreas died in mice that were depleted of DC and challenged with caerulein or L-arginine. All mice with pancreatitis that were depleted of DC died from acinar cell death within 4 days. Depletion of DC from mice with pancreatitis resulted in neutrophil infiltration and increased levels of systemic markers of inflammation. However, the organ necrosis associated with depletion of DC did not require infiltrating neutrophils, activation of NF-κB, or signaling by mitogen-activated protein kinase or TNF-α.
Conclusions
DC are required for pancreatic viability in mice with acute pancreatitis and might protect organs against cell stress.
Keywords: immune response, regulation, suppression, MCP-1, IL-6
Introduction
Acute pancreatitis has a considerable medical and economic impact. In the United States, this disease results in over 200,000 hospital admissions annually at a cost of over 2 billion dollars (1). While most patients will have an uncomplicated course, 10-20% of cases are categorized as severe, resulting from organ necrosis, and are often associated with localized infection, systemic sepsis, and multi-organ failure. Acute pancreatitis carries a 2% overall mortality rate which increases to 10-30% in cases of pancreatic necrosis (2). These data demand an enhanced understanding of the pathophysiology of acute pancreatitis in order to implement more effective treatments.
Acute pancreatitis usually results from either alcohol abuse or obstructing gallstones, which are postulated to cause intra-acinar activation of proteolytic enzymes leading to intense pancreatic inflammation. The inflammatory mediators in this disease have been partially delineated in animal models mimicking human pancreatitis (3, 4). Massive leukocyte infiltration is one of the early events of pancreatitis, and neutrophils play a distinctly important role in its pathophysiology (5). Neutrophil recruitment worsens pancreatic injury (6) and, conversely, neutrophil depletion lessens tissue injury associated with acute pancreatitis (7). Similarly, CD4+ T cells are required for pancreatic acinar injury in rodent models of acute pancreatitis (8). Importantly, Gukovsky et al. (9) demonstrated that the nuclear transcription factor NF-κB, a key regulator of inflammation, is activated early in pancreatitis. Furthermore, NF-κB has recently been shown to regulate expression of Activator Protein 1 which modulates the chemokine cascade after pancreatic insult (10). The CXC-ELR family of chemoattractants, including CXCL1 and CXCL2, further regulate the influx of inflammatory cells in acute pancreatitis (11).
Dendritic cells (DC) have emerged as important participants in inflammation. As instructors of the T cell response, DC are the most powerful antigen-presenting cell in the immune system. Upon exposure to bacterial or viral infection or inflammatory stimuli, immature DC become activated and drive both adaptive and innate immune responses (12). Pattern recognition receptors on the DC surface, the most well-understood being toll-like receptors (TLRs), recognize pathogen-associated molecular patterns (PAMPs) on foreign antigens as well as damage-associated molecular patterns (DAMPs) released by cellular injury (13). Immature DC become activated to maturity by the binding of PAMPs or DAMPs which may be released secondary to tissue damage, or by cytokines generated during the inflammatory response. Mature DC process and present antigen to T cells via MHC complexes. This classical model dictates very tightly regulated pathways to DC activation, as mature DC are central to the development of inflammation and effective immunogenic responses.
Recent work by our group and others has shown a central role for DC in a number of organ-specific inflammatory diseases. We reported that DC tightly regulate the extent of intrahepatic inflammation after liver insult from hepatotoxins via their production of TNF-α (14). Bamboat et al. (15) also reported that DC modulate the extent of liver injury after ischemic insult via their production of IL-10. DC have also been assigned a central role in modulating the severity of systemic sepsis (16, 17). Our preliminary investigations in the current study revealed a marked increase in the numbers of intra-pancreatic DC in acute pancreatitis. Based on this, we postulated an important role for DC in modulating intra-pancreatic inflammation and thus determining the extent of endocrine and exocrine pancreatic injury.
Materials and Methods
Animals
Male C57BL/6 (H-2Kb) and CD45.1 (B6.SJL-Ptprca/BoyAiTac) mice (6–8 weeks old) were purchased from Taconic Farms (Germantown, NY). CD11b.DTR (18) and CD11c.DTR (19) mice were purchased from Jackson Laboratory (Bar Harbor, Maine). Mice were housed in a pathogen-free environment and fed standard chow. Mouse serum was collected by retro-orbital bleeding. To effect macrophage or DC depletion, respectively, CD11b.DTR and CD11c.DTR mice were treated with a single intraperitoneal (i.p.) dose of diphtheria toxin (DPT; 4 ng/g; Sigma-Aldrich, Saint Louis, MO). Animal procedures were approved by the New York University School of Medicine Institutional Animal Care and Use Committee.
Models of Acute Pancreatitis and In Vivo Experiments
Pancreatitis was induced using a regimen of seven hourly i.p. injections of caerulein (50 μg/kg; Sigma-Aldrich) for two consecutive days before sacrifice 12 hours later unless otherwise specified. Alternatively, i.p. administration of two doses of L-arginine (40 mg/kg; Sigma-Aldrich) at hourly intervals was used to induce pancreatic injury (3, 4). In selected experiments, bone marrow chimeric animals were created by irradiating C57BL/6 mice (100 Gy) before i.v. bone marrow transfer (1×107) from CD45.1 or CD11c.DTR donors. In vivo TNF-α blockade was accomplished using MP6-XT22 (200 μg/day). IL-6 blockade was accomplished using MP5-20F3 (200 μg/day). MIP-1α blockade was accomplished using clone 39624 (300 μg/day; all R&D Systems, Minneapolis, MN). NF-κB blockade was accomplished using both cell permeable inhibitors of the NF-κB p50 domain which prevents its nuclear translocation (NF-κB SN50) and the NEMO binding domain (NBD) inhibitor (both 1mg/kg/day) which prevents binding of NEMO to the IKK (IκB) – kinase complex (both EMD4Biosciences, Gibbstown, NJ). MAP Kinase (MAPK) blockade was accomplished using PD98059 (2.5mg/kg/day; Invivogen). To deplete Gr1+ cells or CD4+ T cells, RB6-8C5 or GK1.5 (Monoclonal Antibody Core Facility, Sloan-Kettering Institute, New York, NY) were employed, respectively, as described (20, 21). In vivo plasmacytoid DC depletion was accomplished using either anti-mPDCA-1 (500 μg; Miltenyi, Bergisch Gladbach, Germany) or 120G8 (200 μg; Imgenex, San Diego, CA) (22, 23). Serum levels of pancreatic enzymes and glucose were measured using the Olympus AU400 Chemistry Analyzer (Center Valley, PA).
Statistics
Data is presented as mean +/- standard error of mean. Survival was measured according to the Kaplan-Meier method. Statistical significance was determined by the Student's t test and or log-rank test using GraphPad Prism 5 (GraphPad Software, La Jolla, CA). P-values < 0.05 were considered significant.
See additional Supplemental Materials and Methods
Results
DC expand in acute pancreatitis
To assess the significance of DC in acute pancreatitis, we first tested whether the intra-pancreatic MHC II+CD11c+ DC population expands after pancreatic insult from 14 caerulein injections over a 36-hour period. We found that while DC were rare in the normal pancreas, the total number of intra-pancreatic DC increased markedly in acute pancreatitis, reaching a peak at 72 hours after beginning caerulein challenge (Figure 1A). Intra-pancreatic DC numbers returned to normal by 7 days after beginning caerulein injections. The total number of other leukocyte subgroups also increased markedly in acute pancreatitis; however, there was a disproportional increase in DC (Figure 1B). In particular, the fraction of intra-pancreatic MHC II+CD11c+ DC expanded from a baseline of 1-3% to nearly 15% of all CD45+ intra-pancreatic leukocytes (Figure 1C-E). Conversely, the number of splenic DC remained constant in acute pancreatitis, suggesting that DC expansion is a pancreas-specific phenomenon (Figure 1C).
Figure 1. Intra-pancreatic DC expand in acute pancreatitis.

(A) Mice were challenged with caerulein at hourly intervals seven times daily for two days. The number of intra-pancreatic DC was determined at various time points starting from immediately before the first injection. (B) Pancreas-infiltrating leukocytes from caerulein treated mice were characterized at 48 hours after initiation of caerulein treatment and their numbers compared with saline-treated controls. (C) The fraction of DC among intra-pancreatic and splenic CD45+ leukocytes was compared in saline (Ctl) and caerulein (C) treated animals. (D) Frozen pancreatic sections were stained using antibodies directed against MHC II and visualized by immunofluorescence. (E) Intra-pancreatic CD45+CD11c+ cells in control and caerulein-treated mice were measured at 48 hours by flow cytometry. (F) The number of intra-pancreatic CFSE+ cells was determined at 48 hours after injection of saline or CFSE-labeled BMDC. Experiments were repeated more than three times using 2-5 mice per data point (***p<0.001).
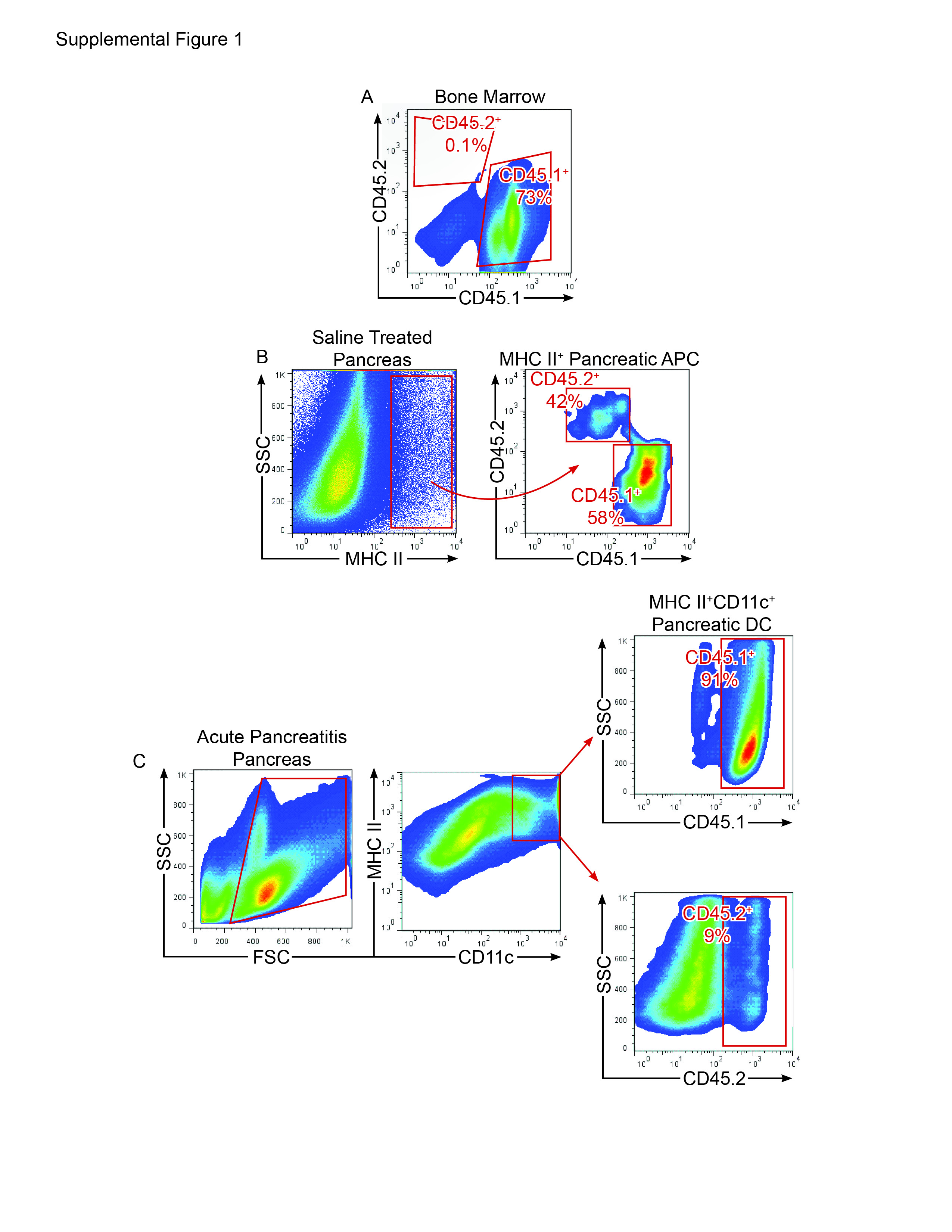
The origins of intra-pancreatic DC in acute pancreatitis are not entirely certain given the experimental limitations of tracking DC in situ. However, we found that 48 hours after adoptive transfer of CFSE-stained BM-DC to mice undergoing caerulein challenge, a large number of transferred BM-DC were found in the pancreas, suggesting that DC derived from the bone marrow may have a tendency to migrate to the pancreas in inflammatory conditions (Figure 1F). To further explore the issue of DC origins in pancreatitis, we made C57BL/6 mice chimeric using bone marrow from CD45.1 mice. Two weeks later, when bone marrow leukocytes were almost entirely CD45.1+ (Supplemental Figure 1A) but only approximately 50% of intra-pancreatic antigen-presenting cells were CD45.1+ (Supplemental Figure 1B), acute pancreatitis was induced. We found that in caerulein-challenged animals, the vast majority of expanded intra-pancreatic DC were CD45.1+, suggesting a bone-marrow origin of pancreatic DC in acute pancreatitis (Supplemental Figure 1C).
Pancreatic DC gain a distinct immune phenotype in acute pancreatitis
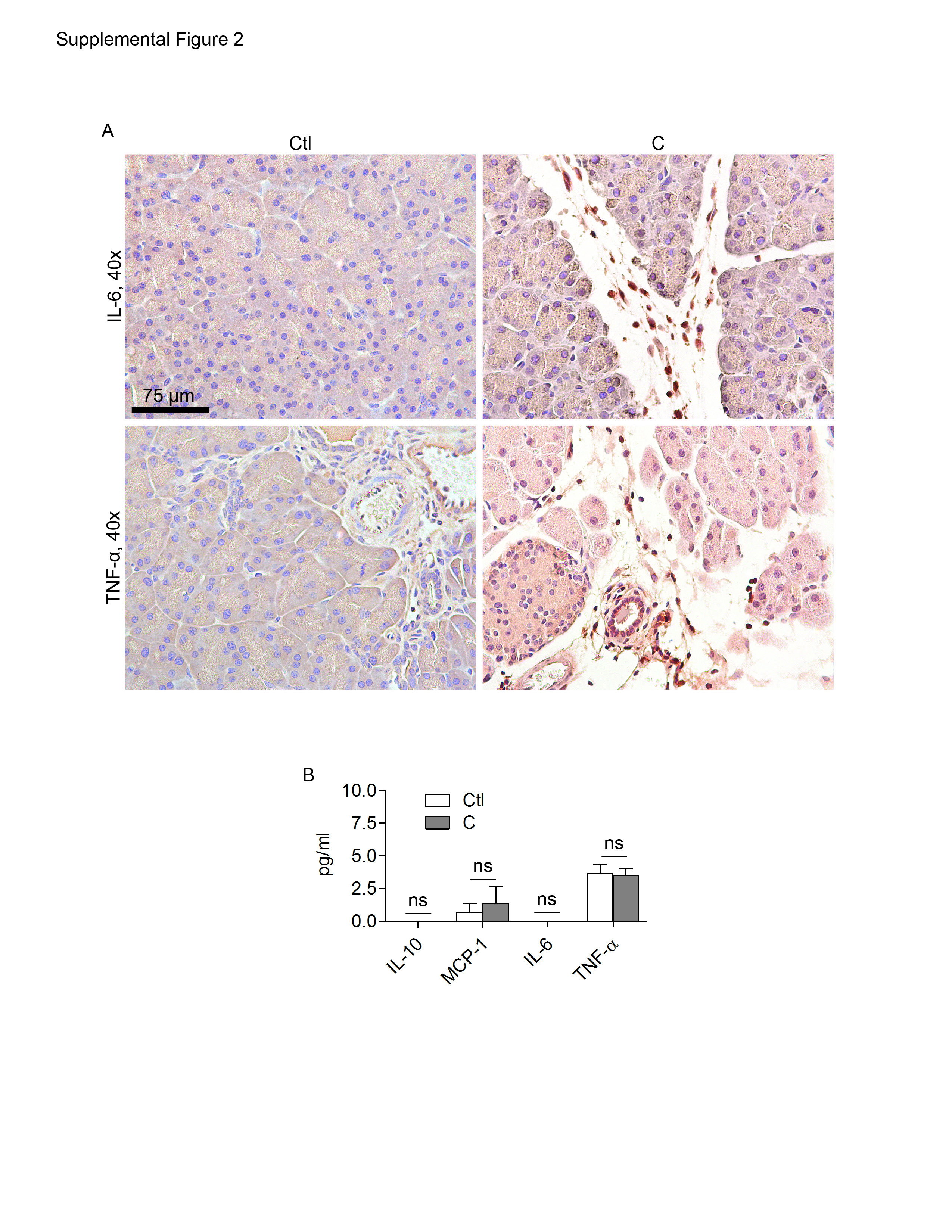
To determine whether intra-pancreatic DC are in an activated state in acute pancreatitis, we compared the surface phenotype of pancreatic DC from caerulein-induced pancreatitis to those of saline treated mice. Pancreatic DC in acute pancreatitis were markedly more mature than controls, expressing higher levels of MHC II, CD40, CD80, and CD86 (Figure 2A). In addition, the DC population underwent a mild myeloid shift expressing slightly higher levels of CD11b and lower CD8α (Figure 2A). Conversely, spleen DC surface phenotype was unaltered in acute pancreatitis (Figure 2B). In acute pancreatitis, pancreatic inflammatory cells expressed TNF-α and IL-6 (Supplemental Figure 2A). To determine whether pancreatic DC become pro-inflammatory after pancreatic insult, we measured their production of cytokines. Pancreatic DC produced markedly elevated levels of activating cytokines in acute pancreatitis, including TNF-α (Figure 2C, D), MCP-1 (Figure 2C), IL-6 (Figure 2C, D), IL-17a (Figure 2D), and IFN-γ (Figure 2D). Conversely, pancreatic DC production of IL-10 and TGF-β, a regulatory cytokine, was unchanged in pancreatitis (Figure 2C). Furthermore, in consort with our previous findings, spleen DC production of inflammatory mediators was unchanged in acute pancreatitis (Supplemental Figure 2B).
Figure 2. Intra-pancreatic DC are mature and pro-inflammatory in acute pancreatitis.

(A) Pancreatic and (B) splenic DC were assayed for surface marker expression by flow cytometry. Median fluorescence index is indicated below each histogram. (C, D) Intra-pancreatic DC production of various inflammatory mediators was measured (C) in cell culture supernatant and (D) using intracellular cytokine analysis. Data are representative of experiments repeated three times and performed in triplicate using 2-5 mice per group (*p<0.05, **p<0.01, ***p<0.001).
DC are necessary for pancreas viability after injury
Since DC expand, mature, and become pro-inflammatory in acute pancreatitis, we postulated that they contribute significantly to intra-pancreatic inflammation and end-organ injury. To test this hypothesis, we employed CD11c.DTR mice (19), in which transient DC depletion can be effected for 48 hours (Supplemental Figure 3). Mice were either depleted of DC (C-DC) or mock-depleted before caerulein challenge. Remarkably, rather than lessening the severity of pancreatitis, DC depletion resulted in almost complete death of the exocrine pancreas (Figure 3A, B). Furthermore, all C-DC mice died of acinar cell death within four days of commencing caerulein injections (Figure 3C). Importantly, the severe effects on the pancreas associated with DC depletion were not related to either DPT treatment or the CD11c.DTR transgenic model, as DPT treated C57BL/6 or CD11b.DTR (macrophage depleted) mice did not develop exacerbated injury (Figure 3D). Similarly, CD11c.DTR mice challenged with caerulein but not depleted of DC did not progress to acinar cell death (Figure 3D).
Figure 3. DC depletion in acute pancreatitis results in pancreatic necrosis.

(A-C) Mice were challenged for two days with caerulein after DC depletion or mock depletion. (A) Pancreata were analyzed using H&E. (B) The percentage of non-viable acinar cells was determined by examining 10 HPF per mouse (mean 5 mice per group). (C) Cohorts of mice (4-5 per group) were not sacrificed but instead analyzed for survival according to the Kaplan-Meier method. (D) Diffuse acinar cell death was specific to DC depletion and not mouse strain or DPT treatment. Notably, treatment of CD11b.DTR mice with caerulein and DPT to deplete macrophages did not result in exacerbated injury. Mouse strain is indicated in parentheses preceded by treatment (***p<0.001).
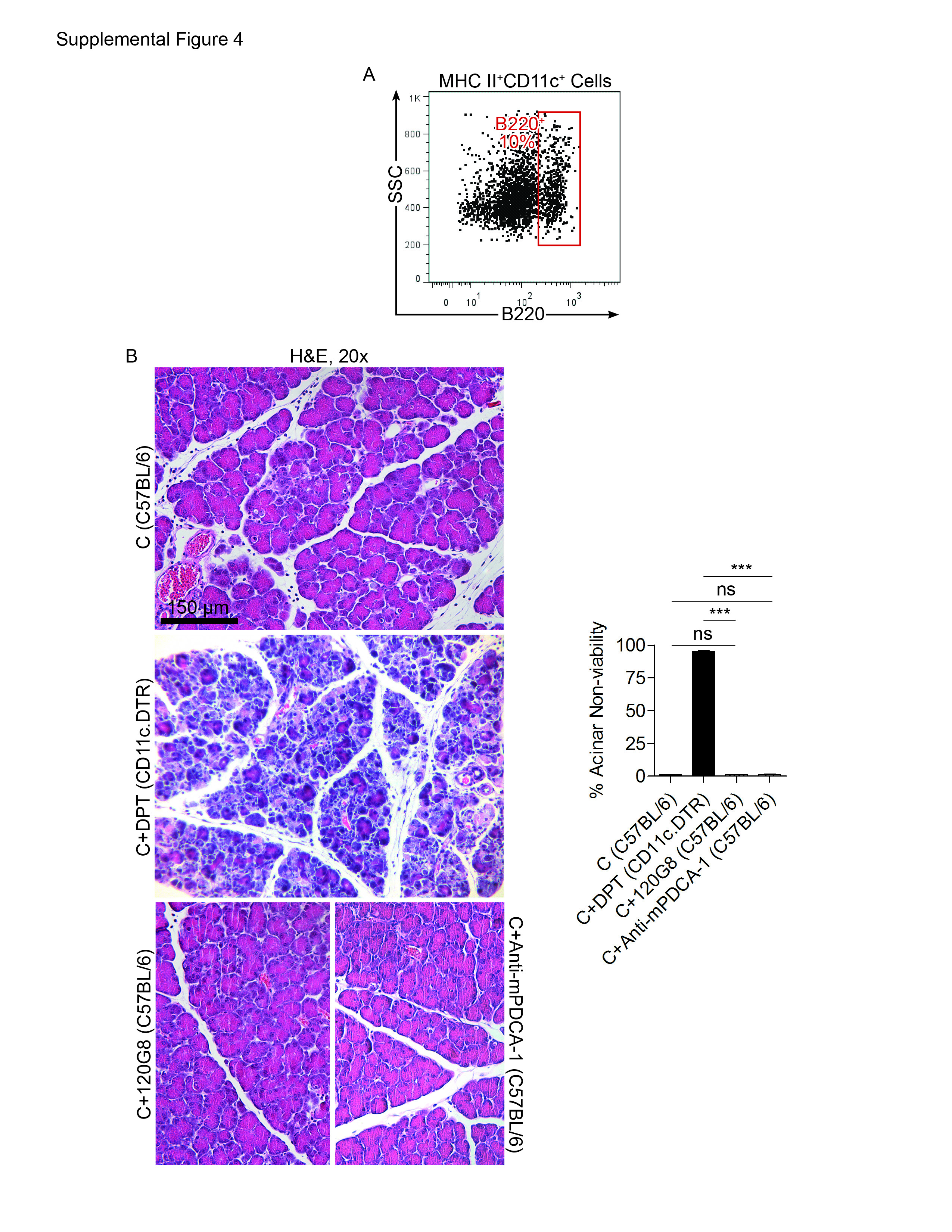
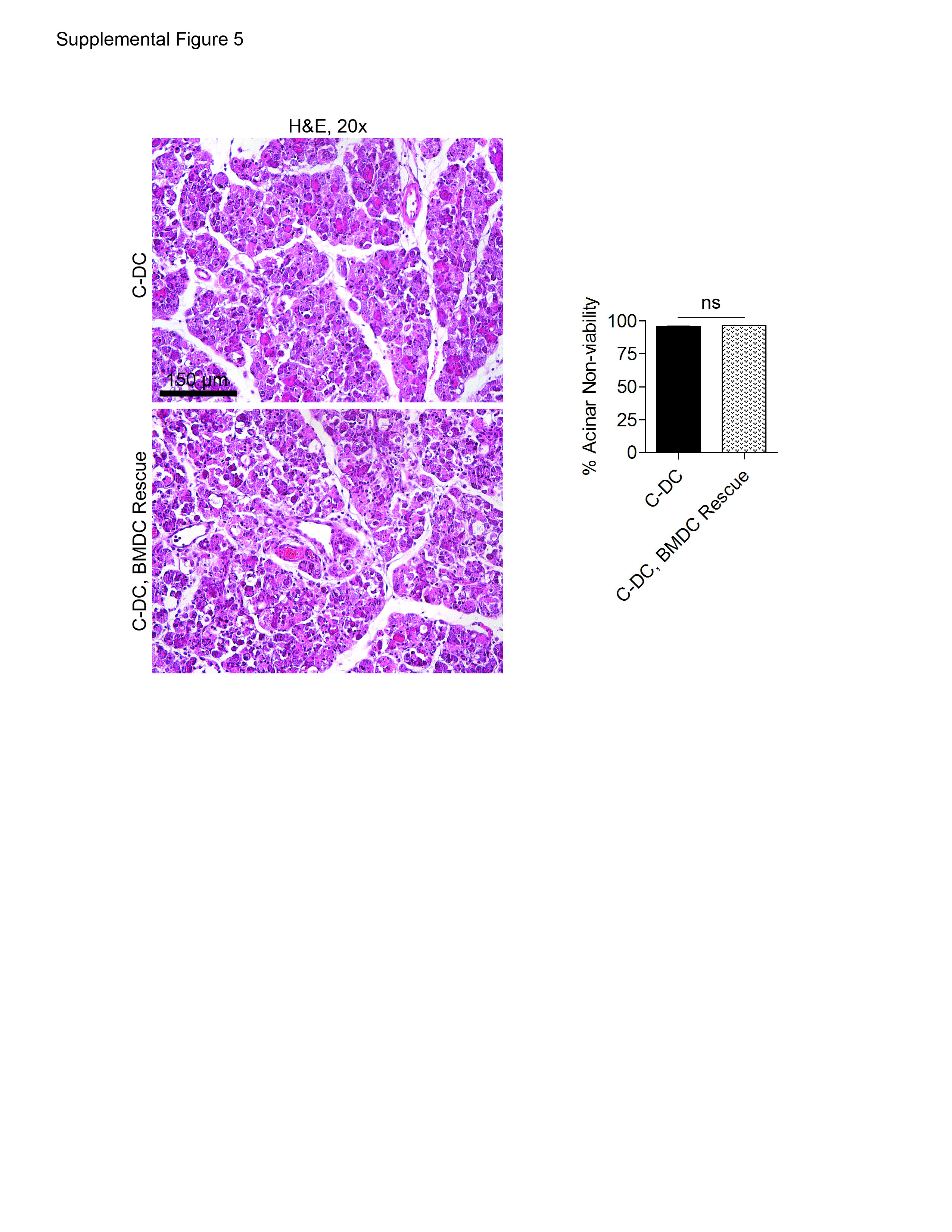
Inflammation can result in expansion of immature plasmacytoid DC (pDC) (24). We found that approximately 10% of intra-pancreatic DC in pancreatitis had the B220+ plasmacytoid phenotype (Supplemental Figure 4A). To investigate whether the pDC subpopulation was required for protection, we selectively depleted pDC using either Anti-mPDCA-1 or 120G8. However, regardless of model of depletion, pDC depletion did not result in exacerbated pancreatic injury in acute pancreatitis (Supplemental Figure 4B). Notably, adoptive transfer of BM-DC or spleen DC were unable to rescue the pancreata of caerulein-challenged DC depleted mice (Supplemental Figure 5).
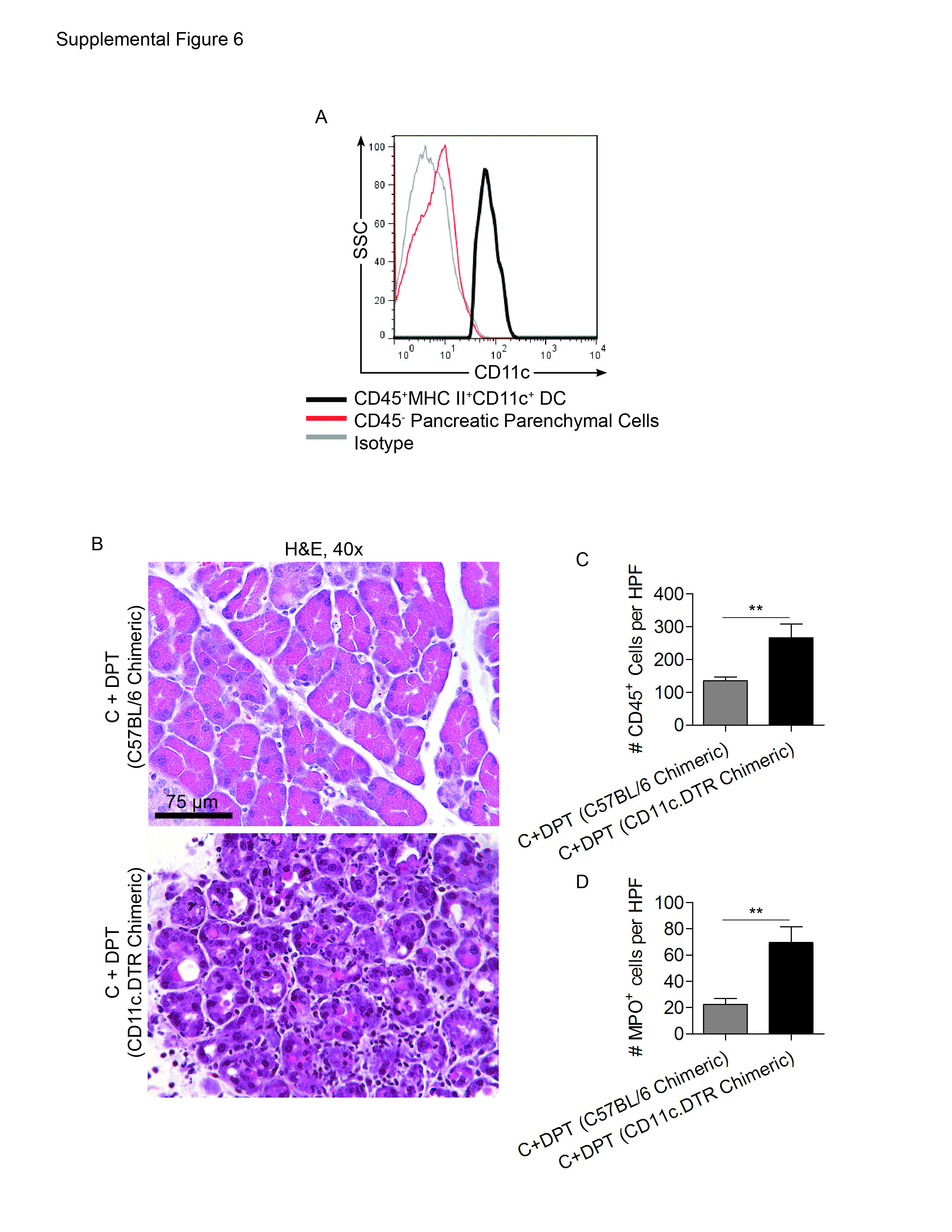
To confirm that pancreatic parenchymal cells were not inadvertently targeted in depleting DC in CD11c.DTR mice, we tested their expression of CD11c. However, CD45- pancreatic cells did not express CD11c (Supplemental Figure 6A). To further exclude direct targeting of pancreatic acini or ducts in CD11c.DTR mice, we created CD11c.DTR bone marrow chimerics by irradiating C57BL/6 mice and transferring CD11c.DTR bone marrow cells. For controls, we created C57BL/6 chimerics by transferring wild-type bone marrow to irradiated mice. Six weeks later, chimeric mice were depleted of DC and challenged with caerulein. CD11c.DTR bone marrow chimerics developed severely exacerbated pancreatitis excluding direct targeting of parenchymal cells in the CD11c.DTR model (Supplemental Figure 6B-D).
Effects of DC depletion are not model specific
To confirm the relevance of our findings, we next investigated whether the non-viable pancreatic parenchyma resulting from DC depletion in the context of acute pancreatitis was restricted to the caerulein model. To test this, we employed L-arginine to induce acute pancreatitis in DC depleted CD11c.DTR mice or controls. We again observed diffusely nonviable pancreata after DC depletion in the L-arginine model of acute pancreatitis confirming that this finding is not model-specific (Figure 4).
Figure 4. DC depletion in L-arginine induced acute pancreatitis results in severe injury.

Mice were challenged with two doses of L-arginine after DC depletion or mock depletion (3 mice per group). Percent organ necrosis was calculated as above by examining 10 high powered fields (HPF) per pancreas (***p<0.001).
Time course of acinar cell death
To determine the time course of acinar death upon induction of acute pancreatitis in the context of DC depletion, we harvested pancreata at serial intervals after beginning caerulein treatment. Approximately 20% of acini were non-viable after seven caerulein doses in mice depleted of DC. The fraction of non-viable acini increased to nearly 80% after 14 treatments by histologic examination. Acinar death was nearly complete after an additional 12 hour delay and was associated with a robust inflammatory infiltrate (Figure 5A). TUNEL staining also revealed that acinar cell apoptosis increased significantly during the 12 hour lag period after cessation of caerulein treatment (Figure 5B).
Figure 5. Time course of pancreatic injury.

(A) Mice were depleted of DC and treated with various doses of caerulein over 1-2 days. Mice were sacrificed either one hour after the final dose or after a 12 hour delay and pancreata examined by H&E staining (average of 3 mice per data point). (B) Mice were treated with 1, 3, 7, or 14 doses of caerulein and sacrificed either one hour or 12 hours after the final treatment. Formalin-fixed, paraffin-embedded pancreata were then examined by TUNEL staining (*p<0.05; **p<0.01; ***p<0.001).
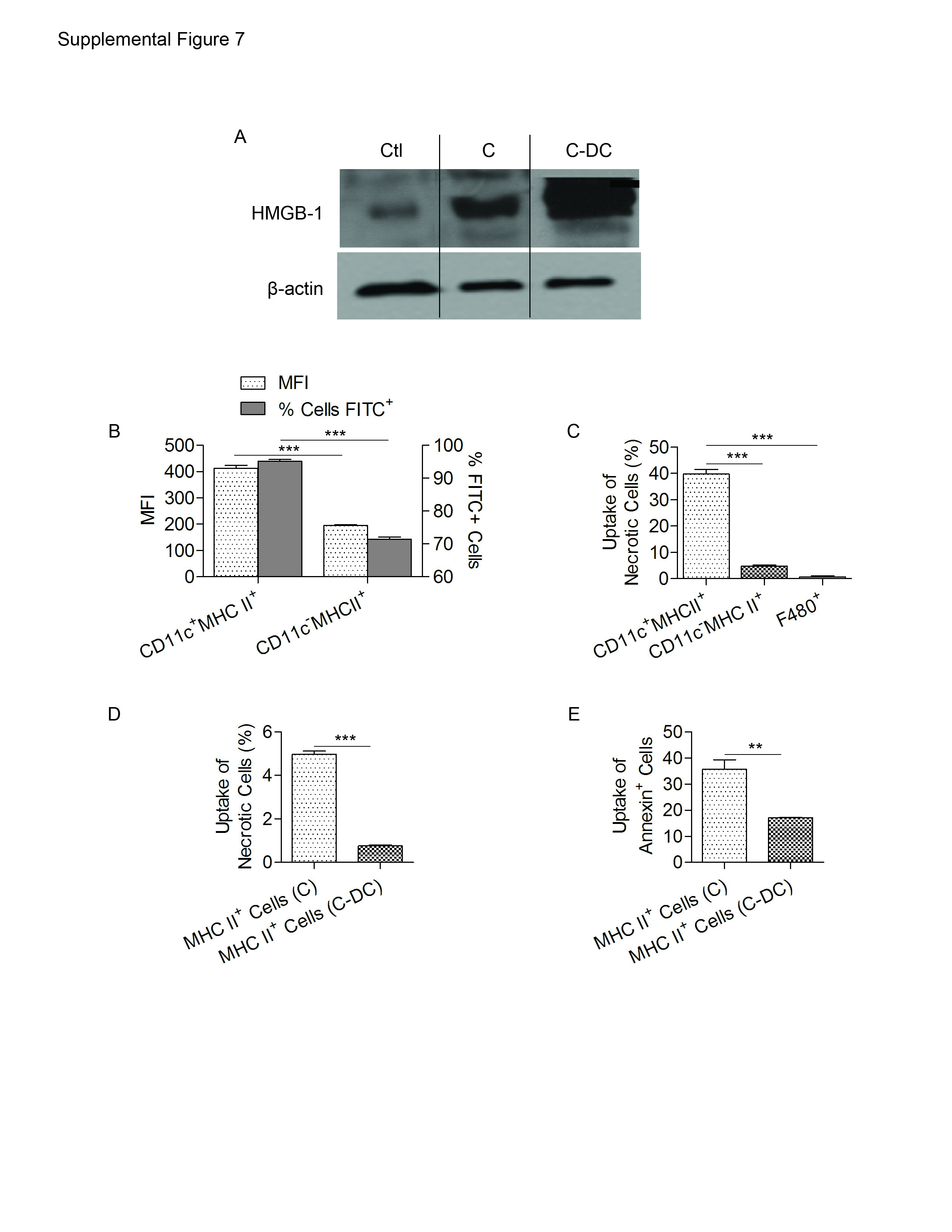
DC have recently been assigned an important role in the clearance of cellular debris in inflammatory disease (25, 26). Since DC expand most sharply during the regeneration phase of AP (Figure 1a), and DC depletion results in an marked increase in apoptotic cells and necrotic elements, we postulated that DC may have a primary role in clearance of cellular debris in pancreatitis, and, consequently, DC depletion may result in expansion of sterile inflammation in AP. In support of this notion, we found that C-DC pancreata have markedly higher levels of HMGB-1 (Supplemental Figure 7A) which is also consistent with our histologic findings showing increased apoptotic and necrotic acini. We tested the relative capacity of pancreatic DC to capture antigen as well as necrotic and apoptotic cells in AP. CD11c+MHCII+ DC from AP pancreata captured FITC-Dextran at a higher rate than CD11c-MHCII+ antigen presenting cells from the same pancreata (Supplemental Figure 7B). In addition, we found that pancreatic DC captured 7AAD+ necrotic cells at a far greater rate than other antigen presenting cells in AP (Supplemental Figure 7C). Moreover, the uptake of 7AAD+ necrotic cellular debris (Supplemental Figure 7D) and Annexin+ apoptotic bodies (Supplemental Figure 7E) by MHC II+ cells was severely deficient in pancreata depleted of DC compared with pancreata with a normal complement of DC. Taken together, these data suggest that DC may exert their protective effects by clearance of necrotic and apoptotic cells thereby limiting sterile inflammation.
Systemic effects of DC depletion in acute pancreatitis
Consistent with our findings of pancreatic end organ destruction, by 48 hours, serum amylase levels were lower in C-DC mice than in animals challenged with caerulein alone (Figure 6A). Serum levels of inflammatory cytokines, including MCP-1, IL-6, and TNF-α were also elevated in C-DC mice, consistent with a severe inflammatory process (Figure 6B). Notably, however, tissue necrosis was specific to the pancreas as other organs were preserved when examined at 36 hours (Supplemental Figure 8) or 72 hours (not shown). Furthermore, cell death was limited to the exocrine pancreas. The endocrine pancreas was entirely protected in C-DC treated mice as there was no evident islet cell destruction (Figure 6C) and serum glucose levels were not elevated in C-DC mice (Figure 6D).
Figure 6. Effects of DC depletion in pancreatitis.

Mice were challenged for two days with caerulein after DC depletion or mock depletion. Serum levels of (A) amylase and (B) TNF-α, IL-6, and MCP-1 were measured at 48 hours. (C) The fraction of pancreatic area occupied by islets and (D) serum levels of glucose were also measured. (E, F) RNA was harvested from whole pancreata and analyzed for expression of (E) IL6, MIP1A, and (F) EGR1. Assays were performed in triplicate and repeated three times (*p<0.05; ***p<0.001).
Elevated expression of IL6, MIP1A, and EGR1in C-DC pancreata
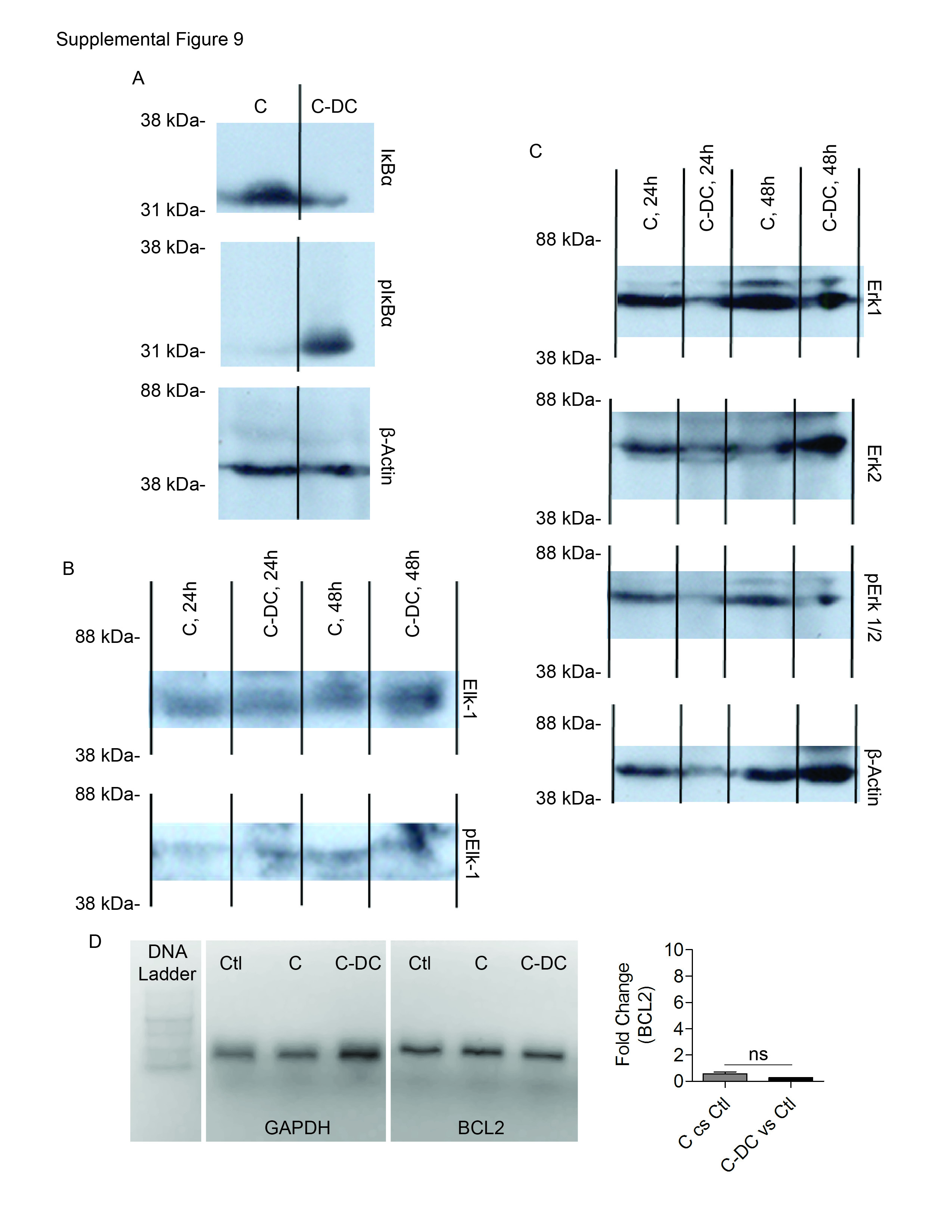
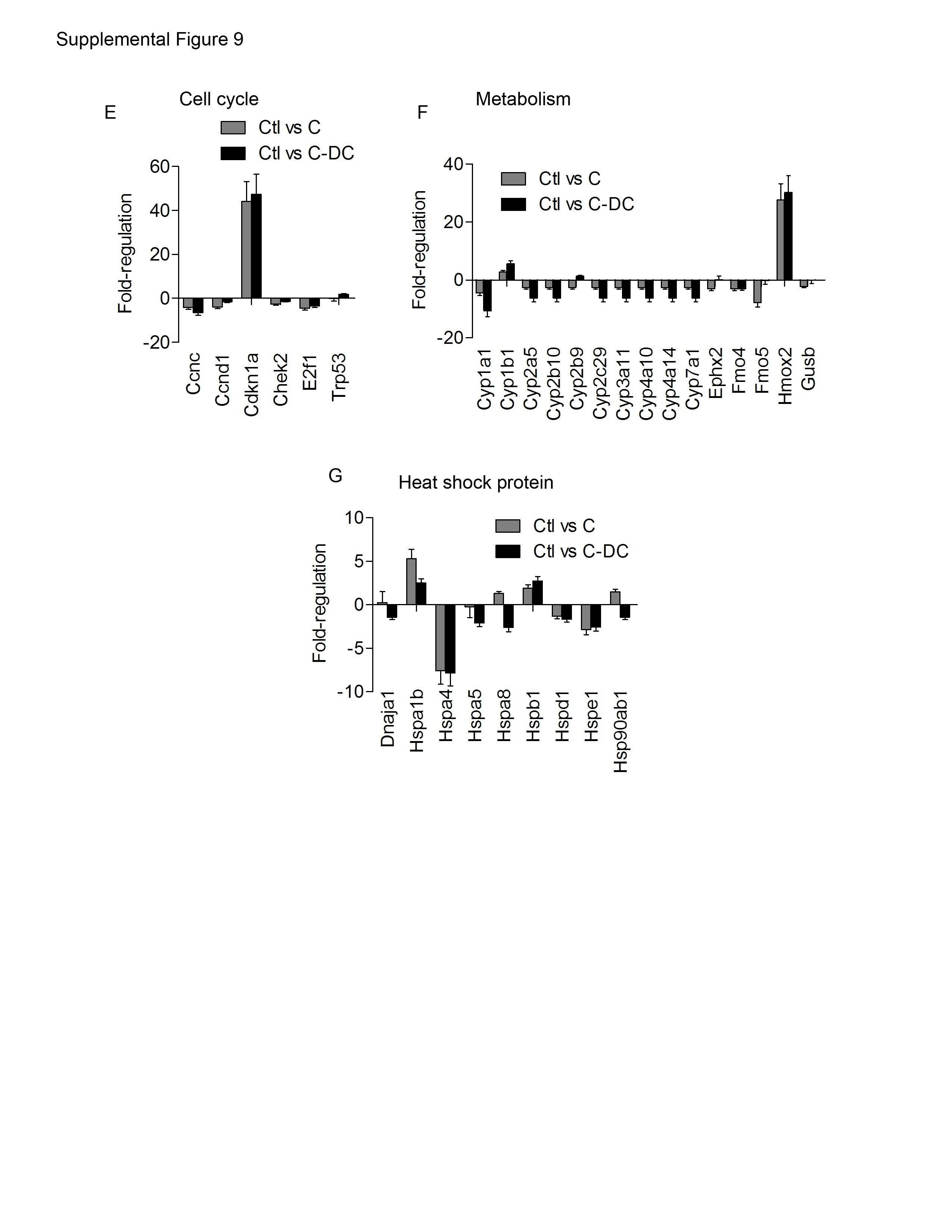
Since TNF-α was upregulated in C-DC mice, we postulated that there may be differences in NF-κB signaling in pancreatitis in the context of DC depletion. Consistent with this hypothesis, we found lower IκBα and elevated pIκBα in C-DC pancreata (Supplemental Figure 9A) which is consistent with increased signaling via NF-κB (27). We performed additional experiments investigating differential expression of genes that are known modulate pancreatic injury in C-DC pancreata compared with caerulein alone. We found that pancreatic expression of IL6 and EGR1, both positive regulators of pancreatic injury (28, 29), as well as MIP1A, were markedly increased in the pancreata of C–DC mice (Figure 6E, F). Since the MAPK pathway is known to regulate EGR-1 via the phosphorylation of the transcription factor Elk-1 we investigated MAPK signaling in C-DC mice. We found a small elevation in pElk-1 in C-DC pancreata at early time points; however, there was no detectable increase in Erk1/2 phosphorylation (Supplemental Figure 9B, C). Since there is increased acinar cell death in C-DC pancreata, we also examined the relative expression of cell cycle regulator, BCL2; however, there was no change (Supplemental Figure 9D). We similarly did not find differences in expression in additional genes related to cell cycle regulation, metabolism, or heat shock proteins which have been shown to regulate the severity of pancreatitis (30) (Supplemental Figure 9E-G).
Effects of DC depletion are not contingent on secondary changes in cellular immunity
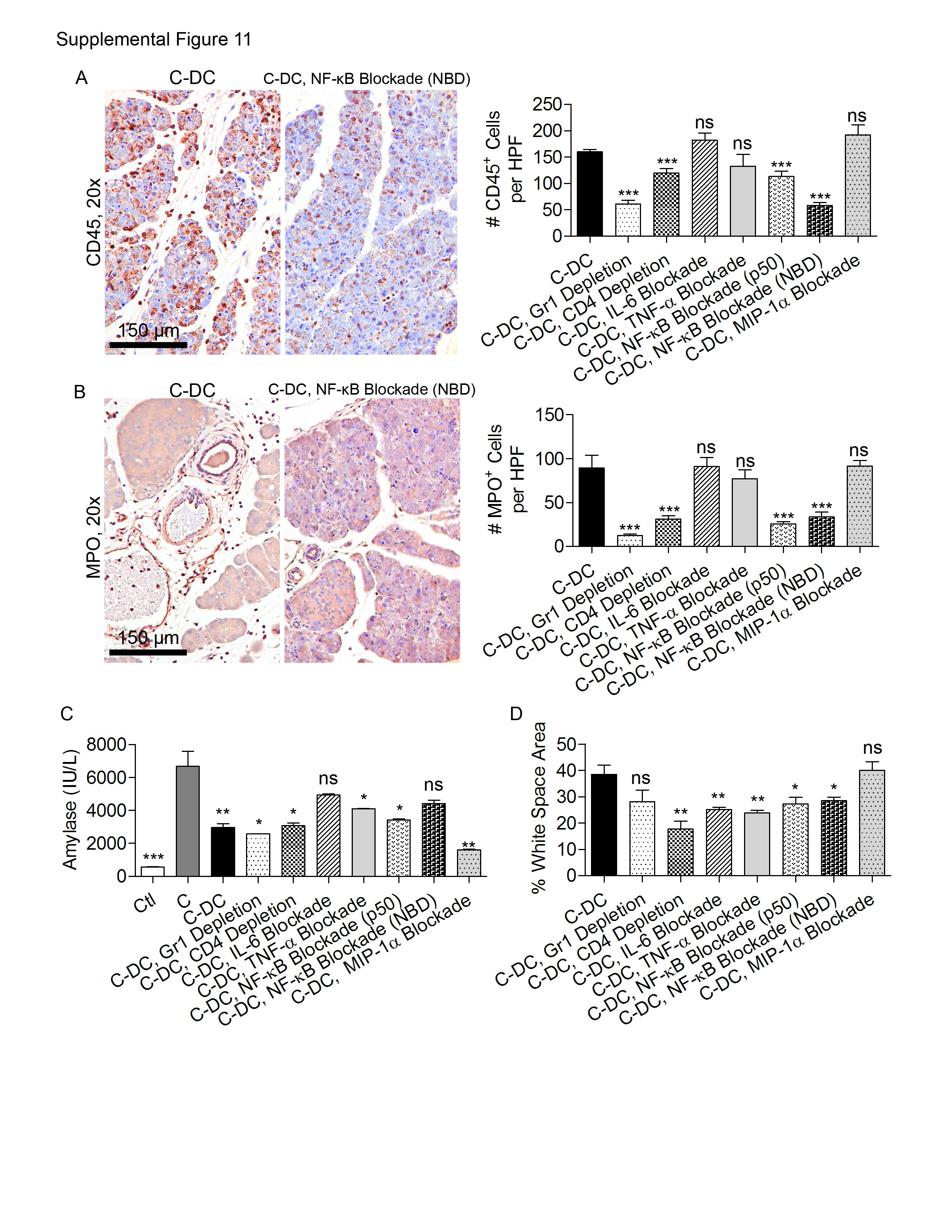
To determine whether a baseline level of endogenous DC directly protects the stressed pancreas from necrosis or whether DC depletion results in a secondary sequence of events leading to organ necrosis, we investigated the alterations in cellular immunity in acute pancreatitis upon DC depletion. We found that in C-DC mice, the total number of CD45+ pancreatic leukocytes was elevated (Figure 7A). Moreover, this infiltrate was attributable to a disproportionate increase in pancreatic Gr1+ neutrophils which increased in step with progressive acinar necrosis (Figure 7B, C). Nevertheless, whereas neutrophil depletion partially protected caerulein treated and C-DC mice from edema and inflammation (Supplemental Figures 10, 11) as expected (31), it did not protect C-DC pancreata from cellular death (Figure 7D), suggesting that the neutrophil infiltration was a consequence of the exacerbated injury associated with DC depletion rather than a causative factor. Furthermore, since CD4+ T cells have been implicated in exacerbating acute pancreatitis (8), we postulated that CD4+ T cell depletion, in association with DC depletion, may protect pancreata from necrosis. However, whereas depletion of CD4+ T cells alone improved pancreatic edema in non-DC-depleted mice (Supplemental Figure 10) and mitigated inflammation in C-DC treated mice (Supplemental Figure 11), it did not protect against complete exocrine destruction (Figure 7D).
Figure 7. Additional cellular depletion or cytokine blockade does not mitigate the effects of DC depletion on pancreas viability.

Pancreata were harvested from C-DC mice and controls and (A) the number of intra-pancreatic CD45+ leukocytes and (B) the number and fraction of pancreas infiltrating neutrophils were examined by both immunohistochemistry and flow cytometry. (C) The time course of neutrophil recruitment was determined by immunohistochemistry. Mice were sacrificed either one hour after the final dose of caerulein or after a 12 hour delay. (D) Acinar death in C-DC mice was studied in mice concurrently depleted of neutrophils, CD4+ T cells, mice with blockaded NF-κB signaling, or treated with neutralizing antibodies targeting IL-6, MIP-1α, or TNF-α (mean 4 mice/group; *p<0.05; **p<0.01; ***p<0.001).
To further investigate whether DC depletion in pancreatitic animals results in necrosis as a result of secondary effects of DC depletion, rather than the specific absence of DC, we selectively blocked TNF-α, IL-6, and MIP-1α, each of which is markedly elevated in C-DC treated animals (Figures 4B and 6E), or NF-κB, a key regulator in pancreatitis (9, 11). Blockade of TNF-α was not protective (Figure 7D and Supplemental Figures 10, 11). IL-6 and MIP-1α blockade had variable effects on pancreatitis in absence of DC depletion (Supplemental Figure 10); however, selective cytokine or chemokine blockade offered no protection against acute pancreatitis in the context of DC depletion (Figure 7D and Supplemental Figure 11). Inhibition of NF-κB using either an NBD inhibitor or a p50 inhibitor mitigated pancreatic edema and inflammation as expected (32) (Supplemental Figures 10, 11). However, mice were not protected from acinar death in the context of DC depletion despite abatement in inflammation (Figure 7D and Supplemental Figure 11). Similarly, MAPK blockade in vivo partially improved pancreatic edema and inflammation but did not protect C-DC treated mice from organ destruction (Supplemental Figure 12).
Discussion
Pancreatitis is a disease with significant medical and economic impact, yet one whose specific immunological pathogenesis is uncertain. We have shown here that DC, principal modulators of the immune response, play a key role in the progression of acute pancreatitis and the extent of organ-specific injury. Using mouse models of acute pancreatitis which mimic human disease, we revealed an expansion of the DC population specific to the pancreas. DC also expressed a mature phenotype and produced high levels of pro-inflammatory cytokines without a commensurate increase in the regulatory cytokine IL-10. Again, these changes were noted to be pancreas-specific; cytokine levels of spleen DC remained constant in pancreatitic mice. The recognition of a discrete, mature and immunogenic body of DC in the pancreas led us to investigate whether DC contribute to the destructive inflammatory response in acute pancreatitis.
A very surprising and novel finding was that depletion of DC using CD11c.DTR mice treated with caerulein or L-arginine resulted in a massive increase in the severity of pancreatitis, with near-complete pancreatic exocrine cell death and subsequent mortality of all animals treated in this manner. Markers of injury and inflammation, including IL-6, Egr1, MCP-1, and TNF-α, were elevated in pancreatitic mice depleted of DC. Tissue necrosis was limited to the exocrine pancreas and other organs were preserved. This was in considerable contrast to our previous work in chronic liver disease, in which DC depletion abrogated inflammatory markers in the fibrotic liver (14), suggesting that distinct regulatory mechanisms govern DC function in acute pancreatitis.
We next investigated the pancreas' immunological profile to shed light on the deleterious effects of DC depletion on acute pancreatitis. The severe necrosis observed in caerulein-treated, DC-depleted mice could be attributable to either a protective influence of endogenous DC on the injured/inflamed pancreas, or a secondary destructive pathway in which DC depletion promotes other events culminating in organ necrosis. In the DC-depleted acute pancreatitis model, there was a disproportionate increase in Gr1+ neutrophils among the pancreatic leukocyte population with progressive acinar necrosis. However, this neutrophil infiltration, which was confirmed by myeloperoxidase staining, appeared to be merely a sequela of DC depletion-induced necrosis, as neutrophil depletion had no protective effect on necrosis in DC-depleted pancreatitic mice. Demols et al. (8) established that CD4+ T lymphocytes are key players in tissue injury in rodent models of acute pancreatitis. However, in the present study, CD4+ T cell depletion did not mitigate the damage seen in DC-depleted pancreatitic mice. Selective blockade of the pro-inflammatory mediators IL-6, MIP-1α and TNF-α, and NF-κB and MAPK - all known to play a role in pancreatitis and showing differential activity in C-DC mice - did not protect from severe organ destruction.
Another notable finding was that macrophage-depleted mice treated with caerulein did not exhibit the adverse effects associated with DC depletion (Figure 3D, E). Like DC, macrophages are antigen presenting cells with the capability of mobilizing other leukocytes against pathogenic insults. However, macrophages and DC appear to have contrasting roles in acute pancreatitis. Pancreatic and peritoneal resident macrophages release TNF-α and IL-1β during the initial pancreatic insult (33, 34), and thus propagate local and systemic inflammation, as well as organ damage by recruitment of monocytes and neutrophils and positive regulation of other pro-inflammatory chemokines, including MIP-1α and RANTES (35). In this manner, peritoneal macrophages can measurably expand the inflammatory response to pancreatic injury and contribute to pancreatic necrosis. Administration of the macrophage-pacifying compound CNI-1493 prior to the induction of severe pancreatitis in rats resulted in an increased survival and a reduction in the severity of pancreatitis as exemplified by lower levels of circulating pancreatic enzymes and pro-inflammatory cytokines (36, 37). A protective effect was also observed by depleting macrophages with the injection of liposome-encapsulated dichloromethylenediphosphonate in a mouse model of virus-induced pancreatitis (38). In the context of our work, we have found that, like their macrophage ‘cousins’, DC are pro-inflammatory in acute pancreatitis by releasing TNF-α, IL-6, and MCP-1. However, DC also protect the pancreas from severe injury. The mechanism underlying these paradoxical effects is unclear. It can be speculated that DC depletion in acute pancreatitis prevents the induction of Tregs, allowing inflammation to go unchecked. Recent work by Tiemessen et al. (39) has shown that CD4+CD25+Foxp3+ Tregs can both inhibit the pro-inflammatory function of macrophages and direct their differentiation toward the “alternatively activated”, or M2, anti-inflammatory phenotype. However, we did not appreciate a diminution in the intra-pancreatic Treg population upon DC depletion (not shown). Furthermore, the fact that depletion of CD4+ T cells did not exacerbate pancreatitis suggests that the DC-Treg axis is not a central modulator in acute pancreatitis.
Our findings implicating endogenous pancreatic DC in an innate protective mechanism against pancreatic necrosis are, in part, analogous to the role of DC in sepsis (40). The development of shock and multiple organ system failure in sepsis has been attributed not to the inciting infectious agent or the hyper-inflammatory response, but to progressive immunosuppression (41). In consort with these findings, studies in rodents have identified a marked depletion in splenic DC 12-36 hours after initiation of septic insult (42, 43). This immune depletion results from apoptosis of dendritic cells, seen with increased active caspase 3 and Annexin V staining in lymph nodes of mice undergoing cecal ligation and puncture, simulating polymicrobial sepsis (44). Similarly, our study showed that a lack of DC severely worsened organ injury in murine acute pancreatitis and resulted in frank pancreatic necrosis. Thus, in acute pancreatitis, DC appear to both galvanize the inflammatory response to injury, by producing high levels of pro-inflammatory cytokines, but also protect the pancreas upon cellular stress. The physiologic or molecular switch that governs DC functionality has yet to be determined in acute pancreatitis. However, given the apparent increase in sterile inflammation in acute pancreatitis associated with DC depletion (Supplemental Figure 7A), the emerging role of DC in clearance of the byproducts of injury (25), as well as our findings that DC are uniquely capable of clearance of antigen as well as apoptotic and necrotic debris (Supplemental Figure 7B, C) our work suggest that DC may limit sterile inflammation in pancreatitis via this exact role. In support of this hypothesis, we show that, in the context of DC depletion, intra-pancreatic antigen presenting cells have poor capabilities in the clearance of the byproducts of pancreatic injury (Supplemental Figure 7D, E) suggesting that DC are required for optimal clearance of cellular debris. Further work to elucidate the regulatory function of DC in pancreatitis may be a key step in immune-directed therapy against acute pancreatitis and its sequelae.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Grant Support: This work was supported in-part by grants from National Pancreas Foundation (AM), the Society of University Surgeons (GM), and National Institute of Health Awards CA108573 (ABF), DK085278 (GM), CA155649 (GM).
Abbreviations
- DC
Dendritic cells
- BM-DC
Bone marrow derived dendritic cells
- DAMP
Damage-associated molecular patterns
- DPT
Diphtheria toxin
- Ctl
Saline
- C
Caerulein
- MPO
Myeloperoxidase
- NBD
NEMO Binding Domain inhibitor
- PAMP
Pathogen-associated molecular patterns
- pDC
Plasmacytoid DC
Footnotes
Disclosures: None
Writing Assistance: None
Author Contributions: Andrea S. Bedrosian (acquisition of data, drafting of manuscript), Andrew H. Nguyen (acquisition of data, analysis and interpretation of data, drafting of manuscript), Michael Hackman (acquisition of data), Michael K. Connolly (acquisition of data), Ashim Malhotra (acquisition of data, critical revision), Napoleon E. Cieza-Rubio (acquisition of data), Justin R. Henning (acquisition of data, critical revision), Junaid Ibrahim (acquisition of data), Rocky Barilla (acquisition of data), Adeel Rehman (acquisition of data), H. Leon Pachter (critical revision), Marco V. Medina-Zea (immunoblotting), Steven M. Cohen (critical revision), Alan B. Frey (critical revision; study design), Devrim Acehan (immunoblotting), George Miller (study concept and design, analysis and interpretation of data, drafting of manuscript)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fagenholz PJ, Fernandez-del Castillo C, Harris NS, et al. Direct medical costs of acute pancreatitis hospitalizations in the United States. Pancreas. 2007;35:302–7. doi: 10.1097/MPA.0b013e3180cac24b. [DOI] [PubMed] [Google Scholar]
- 2.Bradley EL, 3rd, Dexter ND. Management of severe acute pancreatitis: a surgical odyssey. Ann Surg. 2010;251:6–17. doi: 10.1097/SLA.0b013e3181c72b79. [DOI] [PubMed] [Google Scholar]
- 3.Hegyi P, Rakonczay Z, Jr, Sari R, et al. L-arginine-induced experimental pancreatitis. World J Gastroenterol. 2004;10:2003–9. doi: 10.3748/wjg.v10.i14.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willemer S, Elsasser HP, Adler G. Hormone-induced pancreatitis. Eur Surg Res. 1992;24 1:29–39. doi: 10.1159/000129237. [DOI] [PubMed] [Google Scholar]
- 5.Glasbrenner B, Adler G. Pathophysiology of acute pancreatitis. Hepatogastroenterology. 1993;40:517–21. [PubMed] [Google Scholar]
- 6.Sandoval D, Gukovskaya A, Reavey P, et al. The role of neutrophils and platelet-activating factor in mediating experimental pancreatitis. Gastroenterology. 1996;111:1081–91. doi: 10.1016/s0016-5085(96)70077-x. [DOI] [PubMed] [Google Scholar]
- 7.Bhatia M, Saluja AK, Hofbauer B, et al. The effects of neutrophil depletion on a completely noninvasive model of acute pancreatitis-associated lung injury. Int J Pancreatol. 1998;24:77–83. doi: 10.1007/BF02788564. [DOI] [PubMed] [Google Scholar]
- 8.Demols A, Le Moine O, Desalle F, et al. CD4(+) T cells play an important role in acute experimental pancreatitis in mice. Gastroenterology. 2000;118:582–90. doi: 10.1016/s0016-5085(00)70265-4. [DOI] [PubMed] [Google Scholar]
- 9.Gukovsky I, Gukovskaya AS, Blinman TA, et al. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol. 1998;275:G1402–14. doi: 10.1152/ajpgi.1998.275.6.G1402. [DOI] [PubMed] [Google Scholar]
- 10.Vaquero E, Gukovsky I, Zaninovic V, et al. Localized pancreatic NF-kappaB activation and inflammatory response in taurocholate-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1197–208. doi: 10.1152/ajpgi.2001.280.6.G1197. [DOI] [PubMed] [Google Scholar]
- 11.Orlichenko LS, Behari J, Yeh TH, et al. Transcriptional regulation of CXC-ELR chemokines KC and MIP-2 in mouse pancreatic acini. Am J Physiol Gastrointest Liver Physiol. 2010;299:G867–76. doi: 10.1152/ajpgi.00177.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dominguez PM, Ardavin C. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol Rev. 2010;234:90–104. doi: 10.1111/j.0105-2896.2009.00876.x. [DOI] [PubMed] [Google Scholar]
- 13.Zeytun A, Chaudhary A, Pardington P, et al. Induction of cytokines and chemokines by Toll-like receptor signaling: strategies for control of inflammation. Crit Rev Immunol. 2010;30:53–67. doi: 10.1615/critrevimmunol.v30.i1.40. [DOI] [PubMed] [Google Scholar]
- 14.Connolly MK, Bedrosian AS, Mallen-St Clair J, et al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J Clin Invest. 2009;119:3213–25. doi: 10.1172/JCI37581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bamboat ZM, Ocuin LM, Balachandran VP, Obaid H, Plitas G, DeMatteo RP. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J Clin Invest. 2010;120:559–69. doi: 10.1172/JCI40008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kerschen E, Hernandez I, Zogg M, et al. Activated protein C targets CD8+ dendritic cells to reduce the mortality of endotoxemia in mice. J Clin Invest. 2010;120:3167–78. doi: 10.1172/JCI42629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pene F, Zuber B, Courtine E, Rousseau C, Ouaaz F, Toubiana J, Tazi A, Mira JP, Chiche JD. Dendritic cells modulate lung response to Pseudomonas aeruginosa in a murine model of sepsis-induced immune dysfunction. J Immunol. 2008;181:8513–20. doi: 10.4049/jimmunol.181.12.8513. [DOI] [PubMed] [Google Scholar]
- 18.Cailhier JF, Partolina M, Vuthoori S, et al. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J Immunol. 2005;174:2336–42. doi: 10.4049/jimmunol.174.4.2336. [DOI] [PubMed] [Google Scholar]
- 19.Jung S, Unutmaz D, Wong P, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–20. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller G, Lahrs S, Pillarisetty VG, et al. Adenovirus infection enhances dendritic cell immunostimulatory properties and induces natural killer and T-cell-mediated tumor protection. Cancer Res. 2002;62:5260–6. [PubMed] [Google Scholar]
- 21.Connolly MK, Mallen-St Clair J, Bedrosian AS, et al. Distinct populations of metastases-enabling myeloid cells expand in the liver of mice harboring invasive and preinvasive intra-abdominal tumor. J Leukoc Biol. 2010;87:713–25. doi: 10.1189/jlb.0909607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoneyama H, Matsuno K, Toda E, et al. Plasmacytoid DCs help lymph node DCs to induce anti-HSV CTLs. J Exp Med. 2005;202:425–35. doi: 10.1084/jem.20041961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asselin-Paturel C, Brizard G, Pin JJ, et al. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol. 2003;171:6466–77. doi: 10.4049/jimmunol.171.12.6466. [DOI] [PubMed] [Google Scholar]
- 24.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–26. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 25.Sancho D, Joffre OP, Keller AM, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458:899–903. doi: 10.1038/nature07750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fransen JH, Hilbrands LB, Ruben J, et al. Mouse dendritic cells matured by ingestion of apoptotic blebs induce T cells to produce interleukin-17. Arthritis Rheum. 2009;60:2304–13. doi: 10.1002/art.24719. [DOI] [PubMed] [Google Scholar]
- 27.Liu S, Chen ZJ. Expanding role of ubiquitination in NF-kappaB signaling. Cell Res. 2011;21:6–21. doi: 10.1038/cr.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki S, Miyasaka K, Jimi A, Funakoshi A. Induction of acute pancreatitis by cerulein in human IL-6 gene transgenic mice. Pancreas. 2000;21:86–92. doi: 10.1097/00006676-200007000-00056. [DOI] [PubMed] [Google Scholar]
- 29.Ji B, Chen XQ, Misek DE, et al. Pancreatic gene expression during the initiation of acute pancreatitis: identification of EGR-1 as a key regulator. Physiological Genomics. 2003;14:59–72. doi: 10.1152/physiolgenomics.00174.2002. [DOI] [PubMed] [Google Scholar]
- 30.Kubisch C, Dimagno MJ, Tietz AB, et al. Overexpression of heat shock protein Hsp27 protects against cerulein-induced pancreatitis. Gastroenterology. 2004;127:275–86. doi: 10.1053/j.gastro.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Sandoval D, Gukovskaya A, Reavey P, et al. The role of neutrophils and platelet-activating factor in mediating experimental pancreatitis. Gastroenterology. 1996;111:1081–91. doi: 10.1016/s0016-5085(96)70077-x. [DOI] [PubMed] [Google Scholar]
- 32.Ethridge RT, Hashimoto K, Chung DH, et al. Selective inhibition of NF-kappaB attenuates the severity of cerulein-induced acute pancreatitis. J Am Coll Surg. 2002;195:497–505. doi: 10.1016/s1072-7515(02)01222-x. [DOI] [PubMed] [Google Scholar]
- 33.Sameshima H, Ikei S, Mori K, et al. The role of tumor necrosis factor-alpha in the aggravation of cerulein-induced pancreatitis in rats. Int J Pancreatol. 1993;14:107–15. doi: 10.1007/BF02786116. [DOI] [PubMed] [Google Scholar]
- 34.Lundberg AH, Eubanks JW, 3rd, Henry J, et al. Trypsin stimulates production of cytokines from peritoneal macrophages in vi- tro and in vivo. Pancreas. 2000;21:41–51. doi: 10.1097/00006676-200007000-00050. [DOI] [PubMed] [Google Scholar]
- 35.Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Denham W, Tracey KJ, et al. The physiologic consequences of macrophage pacification during severe acute pancreatitis. Shock. 1998;10:169–175. doi: 10.1097/00024382-199809000-00004. [DOI] [PubMed] [Google Scholar]
- 37.Yang J, Denham W, Carter G, et al. Macrophage pacification reduces rodent pancreatitis-induced hepatocellular injury through down-regulation of hepatic tumor necrosis factor alpha and interleukin-1 beta. Hepatology. 1998;28:1282–1288. doi: 10.1002/hep.510280517. [DOI] [PubMed] [Google Scholar]
- 38.Shifrin AL, Chirmule N, Gao GP, et al. Innate immune responses to adenoviral vector-mediated acute pancreatitis. Pancreas. 2005;30:122–129. doi: 10.1097/01.mpa.0000151578.99413.88. [DOI] [PubMed] [Google Scholar]
- 39.Tiemessen MM, Jagger AL, Evans HG, et al. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. PNAS. 2007;104(49):19446–51. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shields CJ, Winter DC, Redmond HP. Lung injury in acute pancreatitis: mechanisms, prevention, and therapy. Curr Opin Crit Care. 2002;8:158–63. doi: 10.1097/00075198-200204000-00012. [DOI] [PubMed] [Google Scholar]
- 41.Hotchkiss RS, Tinsley KW, Swanson PE, et al. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol. 2002;168:2493–500. doi: 10.4049/jimmunol.168.5.2493. [DOI] [PubMed] [Google Scholar]
- 42.Tinsley KW, Grayson MH, Swanson PE, et al. Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol. 2003;171:909–14. doi: 10.4049/jimmunol.171.2.909. [DOI] [PubMed] [Google Scholar]
- 43.Flohe SB, Agrawal H, Schmitz D, et al. Dendritic cells during polymicrobial sepsis rapidly mature but fail to initiate a protective Th1-type immune response. J Leukoc Biol. 2006;79:473–81. doi: 10.1189/jlb.0705413. [DOI] [PubMed] [Google Scholar]
- 44.Efron PA, Martins A, Minnich D, et al. Characterization of the systemic loss of dendritic cells in murine lymph nodes during polymicrobial sepsis. J Immunol. 2004;173:3035–43. doi: 10.4049/jimmunol.173.5.3035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.