

Abstract
The synthesis and direct comparison of the chemical reactivity of the two highly oxidized bicyclic lactone fragments found in rearranged spongian diterpenes (8-substituted 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-one and 6-substituted 7-acetoxy-2,8-dioxabicyclo[3.3.0]octan-3-one) are reported. Details of the first synthesis of the 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-one ring system, including an examination of several possibilities for the key bridging cyclization reaction, are described (Schemes 2–5). In addition, the first synthesis of 7-acetoxy-2,8-dioxabicyclo[3.3.0]octanones containing quaternary carbon substituents at C6 is disclosed (Scheme 6). Aspects of the chemical reactivity and Golgi-modifying properties of these bicyclic lactone analogs of rearranged spongian diterpenes are also reported. Under both acidic and basic conditions, 8-substituted 2,7-dioxabicyclo[3.2.1]octanones are converted to 6-substituted-2,8-dioxabicyclo[3.3.0]octanones. Moreover, these dioxabicyclic lactones react with primary amines and lysine side chains of lysozyme to form substituted pyrroles, a conjugation that could be responsible for the unique biological properties of these compounds. These studies demonstrate that acetoxylation adjacent to the lactone carbonyl group—in either the bridged or fused series—is required to produce fragmented Golgi membranes in the pericentriolar region that is characteristic of macfarlandin E.
Keywords: chemical synthesis, natural product, reactivity, organelle organization
Introduction
Skeletal rearrangement and oxidation of spongian diterpene precursors of general structure 1 (Figure 1A) provides a structurally diverse group of marine-derived natural products referred to as rearranged spongian diterpenes.1 These natural products are isolated from sponges and dorid nudibranchs, the latter of which are believed to acquire these diterpenes from sponge sources as a chemical defense mechanism.1 Among the most structurally complex of the rearranged spongian diterpenes is a structurally unique group that contain a polycyclic hydrocarbon fragment joined to an oxidized lactone unit (Figure 1). The biological activity of this group of spongian-derived diterpenes has been characterized to only a limited extent. Antimicrobial,2,3,4 cytotoxic,4 and nematocidal4 activities have been reported, and norrisolide (9) has been shown to induce irreversible fragmentation and delocalization of Golgi membranes throughout the cytosol in human cell lines.5 In addition, we reported in 2010 that macfarlandin E (4, MacE) and a simplified analog 13 (t-Bu-MacE, see eq 1) induce a novel Golgi organization phenotype that is characterized by small, pericentriolar Golgi fragments and blockage of protein transport from the Golgi to the plasma membrane.6
Figure 1.

Representative rearranged spongian diterpene natural products containing lactone fragments.
A central feature of the group of rearranged spongian natural products depicted in Figure 1 is the presence of highly oxygenated and hydrophobic subunits. The oxygenated fragment is structurally diverse and includes monocyclic variants, such as that found in shahamin K (2),7 and more complex dioxabicyclic lactone fragments. The 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-one subunit is particularly rare; at the onset of our studies, it was known only in the diterpene macfarlandin E (4)2,8 and 13 additional rearranged spongian diterpenes such as aplyviolene (3),8 chromodorolide A (5),4a shahamin I (6),9 and norrlandin (7)10 (Figure 1B). The isomeric 7-acetoxy(or hydroxy)-2,8-dioxabicyclo[3.3.0]octan-3-one fragment is somewhat more abundant in rearranged spongian diterpenes,11 as exemplified by dendrillolide A (8),12 norrisolide (9),13 cheloviolenes A (10) and B (11),14 and omriolide A (12)15 (Figure 1C). The substituted dioxabicyclo[3.3.0]octanone ring system of these diterpenes has been the subject of limited synthetic efforts,16 highlighted by two total syntheses of norrisolide.17,18
Last year we disclosed the first synthesis of the 4,6-diacetoxy-2,7-dioxabicylo[3.2.1]octan-3-one moiety of MacE and evidence obtained by evaluation of related diterpenes and various analogs that the entire oxygenated subunit—including both acetoxy substituents—is required to elicit the MacE Golgi phenotype.6 Additionally, we showed that t-Bu-MacE (13) and its deacetoxy congener 14 are converted to substituted pyrroles such as 15 and 16 in the presence of primary amines under mild conditions (eq 1). This latter finding suggests a functional role for the oxygenated ring system of MacE—formation of a protein-bound pyrrole species—which could be responsible for the unique biological properties of these compounds.
 |
(1) |
Although both MacE and norrisolide have pronounced effects on the structure and function of the Golgi, their phenotypes are different: MacE causes the conversion of the Golgi ribbon into membrane fragments that remain in the pericentriolar region, whereas norrisolide induces fragmentation and dispersal of Golgi membranes throughout the cytoplasm.6,7 In addition, the structure-activity relationships reported to date for these two rearranged spongian natural products are quite distinct: the hydrophobic fragment is suggested to be essential for norrisolide's activity,7 whereas the full 4,6-diacetoxy-2,7-dioxabicylo[3.2.1]octan-3-one subunit, and not the hydroazulene fragment, is believed to be essential for eliciting the MacE-Golgi phenotype.6
This article reports the synthesis of simplified analogs of the oxygenated subunits of MacE (4) and dendrillolide A (8) and a survey of their chemical reactivity and Golgi-modifying properties. We find that both 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-ones and 7-acetoxy-2,8-dioxabicyclo[3.3.0]octan-3-ones react under mild conditions with benzylamine or lysine side chains of lysozyme to form substituted pyrrole products. Moreover, we show that the presence of acetoxy substitution adjacent to the lactone carbonyl in either the bridged or fused dioxabicyclooctanone ring system induces a nearly identical Golgi organization phenotype and greater reactivity with lysine side chains.
Results and Discussion
Enantioselective Synthesis of t-Bu-MacE
As an appropriate initial target for developing a chemical synthesis of the 4,6-diacetoxy-2,7-dioxabicyclo[3.2.1]octan-3-one ring system, we chose t-Bu-MacE (13) which possesses a tert-butyl group in place of the hydroazulene subunit of MacE. Our synthetic approach to t-Bu-MacE was based on the prospect that kinetically controlled cyclization of dialdehyde acid 17 would not form the desired dioxabicyclo[3.2.1]octanone but rather the 2,8-dioxabicyclo[3.3.0]octan-3-one isomer 19 (eq 2). Moreover, we anticipated, and later established experimentally (see below), that the fused isomer also would be favored thermodynamically. Bolstering our expectation that direct cyclization of 17 was unlikely to generate the 2,7-dioxabicyclo[3.2.1]octan-3-one ring system is the conversion outlined in equation 3. In this example, even enforced 1,3-diaxial proximity of the carboxyl nucleophile and the distal aldehyde of precursor 20 did not result in forming the bridged dioxabicyclo product, but rather fused isomer 21.19
 |
(2) |
 |
(3) |
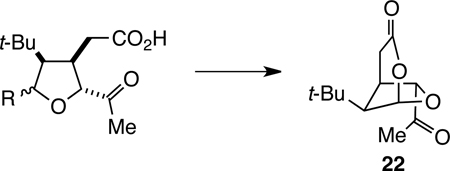
The analysis that guided our initial synthesis of t-Bu-MacE (13) is summarized in Scheme 1. Late stage Baeyer–Villiger oxidation was anticipated to form the C6-acetoxy substituent from an acetyl precursor.18,20 It was anticipated initially that the C4 acetate could be installed at a late stage as well. Bridged-bicyclic intermediate 22 was seen arising from the cyclic acetal 23, wherein X is a leaving group. Acyclic tricarbonyl intermediate 24 was seen resulting from oxidative cleavage of cyclopentyloxysilane 25, which in turn would arise from facial-selective conjugate addition of a tert-butylcuprate to cyclopentenone 26 in the presence of a silyl electrophile.
Scheme 1.

Retrosynthetic Analysis of t-Bu-MacE
Guided by these considerations, we initiated the synthesis of t-Bu-MacE by targeting cyclopentenone 26. Attempts to access enone 26 from known cyclopentanone 2721 were unsuccessful, as regioselective installation of the double bond proved problematic under a variety of conditions (Scheme 2).22 Consequently, enantiopure cyclopentenone 29, which is readily available in 4 steps from cyclopentadiene, was used as the Michael acceptor.23 The union of silyl ketene acetal 2824 and enone 29 to afford cyclopentenone 30 was realized in good yield in dry DMF in the presence of lithium acetate.25 Earlier attempts to promote this reaction with Lewis acids (SnCl4 or TiCl4 in CH2Cl2) or tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF) were less successful. After consumption of the starting materials at 0 °C, addition of a slight molar excess of water to the reaction mixture and allowing the reaction to warm to room temperature prior to aqueous work-up delivered the enone product in reproducibly good yield. If this step was omitted, conjugate addition product 31 was obtained in 30–60% yield.26 We speculate that the trimethylsilyl group is only partially transferred in the initial 1,4-addition; addition of water and warming to room temperature is believed to transform any enoxysilane intermediate to the corresponding ketone, allowing enolate equilibration and β-elimination to take place. Cyclopentenone 26 was then obtained in high yield by cleavage of the butane diacetal group with aqueous trifluoroacetic acid and methylation of the resulting carboxylic acid.27
Scheme 2.

Synthesis of Enantiopure Cyclopentenone 26
In five steps, enone 26 was elaborated in good overall yield to tricarbonyl intermediate 24 (Scheme 3). The tert-butyl substituent was first incorporated by stereoselective addition of the cuprate reagent generated from tert-butyllithium and CuCN (2:1 molar ratio) in the presence of tert-butyldimethylsilyl chloride (TBSCl) to provide exclusively trans-substituted cyclopentenyl silyl ether 32.28 At this stage, we needed to transform the methyl ester to an acetyl group without cleaving the enoxysilane. A two-step sequence, proceeding via methyl enol ether intermediate 33, was eventually developed. Although methylenation of 32 with the Tebbe reagent29 proceeded sluggishly, resulting in incomplete conversion, the Takai methylenation conditions developed by Rainer and coworkers produced methyl enol ether 33 in nearly quantitative yield.30 After examining several conditions for hydrolyzing the methyl enol ether, the use of 1.5 equiv of oxalic acid in aqueous i-PrOH at 0 °C was identified as particularly effective in achieving the conversion to methyl ketone 25 with minimal cleavage of the enoxysilane. We ascribe this rare, if not unprecedented, selective acidic cleavage of a methyl enol ether in the presence of a silyl enol ether to steric shielding of protonation of the cyclopentenyl double bond by the two bulky trans-oriented substituents.31 In our initial efforts, we cleaved the double bond of 25 by reaction with OsO4 and NaIO4, followed by addition of trimethylsilyldiazomethane32 to the crude product mixture to provide tricarbonyl product 24 in useful, albeit variable, yields (55–84%).6 To improve the reproducibility of this conversion and avoid the undesirable use of TMSCHN2, an alternate sequence was developed. In this improved procedure, enoxysilane 25 was oxidized with OsO4 (0.05 equiv) and N-methylmorpholine-N-oxide (NMO, 2.0 equiv), followed by cleavage of the resulting α-hydroxyketone with 1.3 equiv of methanolic Pb(OAc)4, a sequence that reproducibly delivered tricarbonyl intermediate 24 in 95% yield.
Scheme 3.

Synthesis of Tricarbonyl Intermediate 24
As a prelude to examining the pivotal bridging reaction, acyclic intermediate 24 was transformed to several potential tetrahydrofuryl cyclization precursors (Scheme 4). Cleavage of the silyl ether by reaction of 24 with 1.5 equiv of TBAF provided tetrahydrofuryl lactol 34 in moderate yield, which upon careful saponification with 1.5 equiv of NaOH at 0 °C gave the crude carboxylic acid 35 in sufficient purity for subsequent cyclization studies. Attempts to access intermediate 35 more expeditiously from cyclopentenyl precursor 25 by sequential reaction with OsO4/NaIO4 and TBAF were unsuccessful, providing only complex mixtures of products. Reaction of tricarbonyl intermediate 24 with 1.6 equiv of camphorsulfonic acid (CSA) and trimethylorthoformate in methanol at room temperature generated diacetal 36 as a 1:1 mixture of separable methoxy anomers in 65% yield. The α epimer of 36 could be converted to the β epimer by equilibration in methanol in the presence of camphorsulfonic acid, allowing the β-36 to be obtained in 66% overall yield from acyclic precursor 24 after two recycles. Subsequent saponification of the ester and selective cleavage of the dimethyl acetal with aqueous HCl at 0 °C yielded crude tetrahydrofuryl ketoacid 37. As glycosyl fluorides are used extensively as glycosyl donors because of their stability and the mild, orthogonal methods available for their activation,33 hemiacetal 34 was transformed to anomeric fluoride 38 by reaction with 1.6 equiv of diethylaminosulfur trifluoride (DAST) at −78 °C in CH2Cl2. Saponification of this product with 1.5 equiv of 1 N NaOH in MeOH at room temperature provided carboxylic acid 39.
Scheme 4.

Formation of Tetrahydrofuryl Cyclization Precursors
With cyclization substrates 35, 37, and 39 in hand, their transformation to 2,7-dioxabicyclo[3.2.1]octan-3-one 22 was studied (Table 1). Mitsunobu conditions failed to promote cyclization of hemiacetal 35 (entry 1); however, in the presence of 1 equiv of camphorsulfonic acid (CSA) in chloroform, lactol 35 was converted at room temperature to dioxabicyclooctanone 22 in 54% overall yield from ester precursor 34 (entry 2). In a similar fashion, methoxy acetal 37 was transformed in the presence of 1 equiv of BF3•Et2O to dioxabicyclooctanone 22 in 66% yield (entry 3). Anomeric fluoride 39 cyclized with slightly enhanced efficiency when exposed to 2 equiv of SnCl2 in DMF at room temperature, generating bicyclic lactone 22 in 71% yield (entry 4).34
Table 1.
Cyclization Reactions to Form 22
 | |||
|---|---|---|---|
| Entry | R | Conditions | Yield 22a |
| 1 | OH (35) | DEAD, PPh3, CH2Cl2 | 0% |
| 2 | OH (35) | 0.2 equiv CSA, CHCl3, rt, 12 h | 54% |
| 3 | OMe (37) | 1.2 equiv BF3• OEt2, CH2Cl2, 0 °C, 1 h | 66% |
| 4 | F (39) | 2 equiv SnCl2, DMF, rt, 18 h | 71% |
Yield reported as conversion from methyl ester precursor (34, 36, and 38, respectively)
With several complementary methods for accomplishing the critical bridging lactonization reaction identified, all that remained was installing the additional oxygen functionality of t-Bu-MacE. The C6 acetoxy substituent was readily introduced by reaction of dioxabicyclooctanone 22 with 4 equiv of trifluoroperacetic acid at room temperature,35 providing the tert-butyl analog of aplyviolene, 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-one (14), in 88% yield. We had hoped that the enhanced acidity of lactones relative to esters would allow the remaining acetoxy substituent to be incorporated at this stage.36 However, all attempts to directly introduce a hydroxyl substituent adjacent to the lactone carbonyl group were unsuccessful. For example, enolization of 14 with 1–2 equiv of LDA or KHMDS, followed by oxidation (Davis oxaziridine37 or O2), silylation, or acylation resulted in either recovered starting material or extensive decomposition.38
 |
(4) |
As a result of our inability to selectively oxidize bicyclic lactone 14, we turned to examine introduction of the α-acetoxy substituent prior to forming the dioxabicyclo[3.2.1]octanone ring system. Hydroxylation of the sodium enolate of the β-methoxy epimer of tetrahydrofuryl acetal 36 with oxaziridine 4039 proceeded smoothly to generate α-hydroxy ester 41 as a single stereoisomer (Scheme 5). The stereoselectivity of this oxidation is rationalized by orientation of the enolate away from the tetrahydrofuran ring and delivery of the electrophile to the face opposite the tert-butyl group. As the hydroxyl substituent of 41 has the opposite relative configuration to the corresponding acetate of MacE, it was inverted by oxidation with Dess–Martin reagent40 and subsequent stereoselective reduction of the ketone product with sodium borohydride at −78 °C. This sequence provided alcohol 42 in 88% overall yield. Saponification of the ester and acidic hydrolysis of the ketal, followed by exhaustive acylation and anhydride methanolysis yielded α-acetoxycarboxylic acid 43. We were delighted to find that tetrahydrofuryl acetal 43, when exposed to 1.1 equiv of BF3•OEt2 at 0 °C, gave rise to 2,7-dioxabicyclo[3.2.1]octan-3-one 44 in 71% overall yield from tetrahydrofuran 42. Baeyer–Villiger oxidation of this product with trifluoroperacetic acid then provided t-Bu-MacE (13) in 90% yield.41 The synthetic sequence outlined in Schemes 2–5 allowed 0.5 g of t-Bu-MacE to be synthesized, enabling the chemical reactivity and biological profile of this compound to be studied in detail.
Scheme 5.

Completion of the Synthesis of t-Bu-MacE (13)
Enantioselective Synthesis of 7-Acetoxy-2,8-Dioxabicyclo[3.3.0]octan-3-ones
Several intermediates prepared during the synthesis of t-Bu-MacE provide potential access to related structures in the 2,8-dioxabicyclo[3.3.0]octan-3-one series. We demonstrated this chemistry with the α-methoxy epimer of intermediate 36, thus allowing both epimers of acetal 36 to be utilized (Scheme 6). Sequential ester saponification and ketal hydrolysis cleanly provided tetrahydrofuran α-37 from acetal precursor α-36. Baeyer–Villiger oxidation of this intermediate took place with concomitant cyclization to provide 7-methoxy-2,8-dioxabicyclo[3.3.0]octanone 45 in 82% overall yield for the three steps. Hydrolysis of 45 with dilute HCl yielded a mixture of bicyclic lactols, which upon acetylation delivered crystalline 7-acetoxy-2,8-dioxabicyclo[3.3.0]octanone 46 as a single stereoisomer. The structure and relative configuration of this product was initially secured by 1H NMR NOE analysis and subsequently confirmed by single crystal X-ray diffraction.42
Scheme 6.

Synthesis of 2,8-Dioxabicyclo[3.3.0]octanones 46 and 49
Analog 49, which possesses an acetoxy group adjacent to the lactone carbonyl, was also prepared. The enolate of bicyclic lactone 45 was generated with NaHMDS in THF at −78 °C and hydroxylated to afford 47 (Scheme 6). Subsequent acetylation of the secondary alcohol gave acetate 48 in low (unoptimized) yield over two steps.43 The methoxy acetal functional group of 48 was hydrolyzed with dilute HCl, followed by acetylation of the hemiacetal product with acetic anhydride and pyridine to provide an inseparable mixture of 4,7-diacetoxy-2,8-dioxabicyclo[3.3.0]octan-3-one 49 and an uncharacterized aldehyde byproduct. Pure dioxabicyclo[3.3.0]octanone 49 was obtained in modest yield from this crude product mixture by sodium chlorite oxidation,44 which allowed for easy removal of carboxylic acid impurities by chromatography. Of note, the syntheses summarized in Scheme 6 are the first of 7-acetoxy-2,8-dioxabicyclo[3.3.0]octan-3-ones possessing a quaternary-carbon substituent at C6, a structural feature found in many rearranged spongian diterpene natural products (see Figure 1).
Chemical Reactivity of the 6-Acetoxy-2,7-dioxabicylo[3.2.1]octan-3-one and 7-Acetoxy-2,8-dioxabicylo[3.3.0]octan-3-one Ring Systems
Our expectation that the 2,7-dioxabicyclo[3.2.1]octan-3-one ring system would be less stable than the isomeric 2,8-dioxabicyclo[3.3.0]octan-3-one ring system was readily confirmed by exposure of dioxabicyclo[3.2.1]octanone 14 to BF3•OEt2 in acetic acid at 0 °C to generate dioxabicyclo[3.3.0]octanone isomer 46 as 3.5:1 mixture of separable acetal epimers, favoring the β-acetoxy isomer (Scheme 7).45 Alternatively, hydrolysis of 14 at room temperature with 1 N HCl in THF gave a mixture of dioxabicyclo[3.3.0]octanone lactol epimers 50,46 which upon acetylation provided the α epimer of dioxabicyclo[3.3.0]octanone 46 exclusively. This sample was identical to the material prepared by the approach outlined in Scheme 6. In a similar fashion, acidic hydrolysis of t-Bu-MacE (13) provided a lactol intermediate, which was identical to the product formed by acidic hydrolysis of 48. Ensuing acetylation of this lactol intermediate gave α-acetoxylactone 49 in 36% overall yield from t-Bu-MacE.
Scheme 7.

Conversion of Bridged Dioxabicyclic Lactones 13 and 14 to Fused Isomers 46 and 49
The transformation of the 2,7-dioxabicyclo[3.2.1]octan-3-one ring system to fused bicyclic lactone products could also be accomplished under basic conditions. In the presence of sodium hydroxide at room temperature, dioxabicylo[3.2.1]octanone 14 yielded a single 2,8-dioxabicylo[3.3.0]octan-3-one lactol product. However, in this case, 1H NMR NOE analysis showed that the tert-butyl group of product 51 resides on the convex face of the 2,8-dioxabicyclo[3.3.0]octanone ring system (Scheme 8). Confirmation that this product resulted from epimerization of the carbon bearing the tert-butyl substituent, and not the methine hydrogens of the ring junction, was obtained by conversion of 51 to the R and S Mosher esters 52. Enhanced Mosher analysis established that 52 possesses the 1R,5R,6S,7R absolute configuration with a slightly eroded enantiomeric purity of 78% ee.47
Scheme 8.

Synthesis of 51 and its Mosher Ester
The formation of product 51 from dioxabicyclo[3.2.1]octanone precursor 14 requires further comment. Certainly isomer 51 having the tert-butyl group on the convex face of the cis-dioxabicyclo[ 3.3.0]octanone ring system should be more stable than stereoisomer 50. Under the basic reaction conditions (pH ~14), the predominant aldehyde intermediates generated from hydroxide opening of the lactone (or cleavage of the acetate substituent) of precursor 14 would be expected to be 53 and 54 (eq 5). Base-promoted epimerization of the likely predominant species, uncharged 54, would lead to the formation of the observed product 51. The partial erosion of enantiomeric purity observed in the conversion of 14 to 51 indicates that there is some epimerization under these conditions of the central methine carbon of intermediate 53.48
 |
(5) |
To gain further insight into the reactivity of these dioxabicyclic lactone ring systems, we examined the rate of hydrolysis of compounds 13, 14, 46, and 49. Hydrolytic rate was measured by NMR observation of the disappearance of the dioxabicyclic ring system in a pD 8.3 phosphate buffer containing 10% DMSO at 37 °C (Table 2). The presence of an acetoxy group adjacent to the lactone carbonyl results in an enhanced hydrolysis rate of both the substituted dioxabicyclo[3.2.1]octanone and dioxabicyclo[3.3.0]octanone ring systems, with the latter ring system hydrolyzing more rapidly than the former.
Table 2.
Half-life of Hydrolysis at pD 8.3
| Compound | t1/2 (h)a |
|---|---|
| 13 | 10.2 |
| 14 | 91.9 |
| 46 | 9.7 |
| 49 | 1.5 |
Incubated at 37 °C in 50 mM phosphate buffer; disappearance of substrate (5 mM) was monitored by 1H NMR.
In our initial report, we demonstrated that the reaction of substituted dioxabicyclo[3.2.1]octanones 13 and 14 with benzylamine leads to substituted pyrroles.6 Specifically, we showed that t-Bu-MacE (13) was converted to pyrrole 15 in a mixture of perdeutero-benzylamine (2.5 equiv) and THF-d8, and that dioxabicyclooctanone 14 is transformed to pyrrole 16 in high yield in the presence of 5 equiv of benzylamine in DMSO and water (eq 1). Under these conditions, fragmentation of these precursors undoubtedly generates transient 1,4-dialdehyde intermediates, 17, which undergo Paal–Knorr pyrrole formation (Scheme 9).6 In the case of the pyrrole derived from t-Bu-MacE (13), the oxygen substituent of the acetic acid side chain is exchanged for a benzylamino substituent, likely after formation of the pyrrole by a gramine-type fragmentation/addition pathway.
Scheme 9.

Mechanistic Outline for Pyrrole Formation from 13 and 14
The conversion of t-Bu-MacE (13) and analog 14 to pyrrole products was examined in CD3OD to gain insight into the initial step of the pyrrole-forming process under protic conditions (Scheme 10).49 NMR analysis of the reaction of 13, perdeutero-benzylamine (5 equiv), and CD3OD at room temperature indicated that pyrrole 57 was formed along with equimolar quantities of acetic acid and methyl acetate.50 Similarly, when 14 was converted to 58 under identical conditions, methyl acetate was observed.49 The formation of methyl acetate, as well as the exclusive formation of the carboxylates 57 and 58, demonstrates that fragmentation of the 6-acetoxy-2,7-dioxabicyclo[3.2.1]octanones 13 and 14 is initiated by initial reaction of the protic solvent at the anomeric acetoxy substituent.
Scheme 10.

In Situ Observation of the Formation of 57 and 58 and Reaction Byproducts
Pyrroles are also formed from the reaction of 7-acetoxy-2,8-dioxabicyclo[3.3.0]octan-3-ones with primary amines. For example, 7-acetoxydioxabicyclo[3.3.0]octanone 46 was converted to pyrrole carboxylic acid 59 when exposed at room temperature to 2 equiv of benzylamine in DMSO/H2O (Scheme 11). When this reaction was conducted in methanol, pyrrole methyl ester 60 was observed as the major product. The formation of methyl ester 60 suggests, in contrast to bridged dioxabicyclo[3.2.1]octanone compounds 13 and 14, that fragmentation in this series is initiated by reaction of the protic solvent at the lactone carbonyl group.
Scheme 11.

Conversion of Dioxabicyclo[3.3.0]octanones 46 to Pyrroles 59 and 60
The divergence in fragmentation pathways of the two isomeric bicyclic lactone ring systems (summarized in Scheme 12) is consistent with the reactivity of related simple lactones. Although sixmembered lactones are typically more reactive than their five-membered ring counterparts,51 oxabicyclo[3.2.1]octanone 61 is saponified at a dramatically reduced rate (Figure 2).52 The reduced rate of saponification of lactone 61 is readily ascribed to developing destabilizing syn-pentane interactions during axial approach of hydroxide to the lactone carbonyl group. Similar destabilizing interactions would be involved in the tetrahedral intermediate generated from the addition of nucleophiles to the lactone carbonyl of 6-acetoxy-2,7-dioxabicyclo[3.2.1]octanones 14.
Scheme 12.

Regioselectivity of Initial Nucleophilic Attack on Bicyclic Lactones 14 and 46 Leading to 1,4-Dialdehyde Intermediates
Figure 2.

The saponification rates of select lactones.53
To ascertain whether the oxygenated bicyclic lactones would react with lysine residues of proteins under biologically relevant conditions, we examined the reactivity of these molecules with hen egg white lysozyme (HEWL).53 In initial experiments, we found that dioxabicyclo[3.2.1]octanones 13 and 14 converted lysine residues of HEWL to pyrrole adducts at room temperature in pH 7 phosphate buffer (Figure 3 and Table 3, entries 1 and 3). Analog 14 provided the pyrrole-3-acetic acid modification (+164 mu), whereas t-Bu-MacE (13) led to the pyrrole-3-hydroxyacetic acid adduct (+180 mu) resulting from solvolytic incorporation of a hydroxyl substituent at the heterobenzylic site (Figure 3). The reaction of dioxabicyclo[3.3.0]octanones 46 and 49 with HEWL at pH 7 and 8 was also examined. As expected, 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-ones and isomeric 7-acetoxy-2,8-dioxabicyclo[3.2.1]octan-3-ones provided identical pyrrole adducts: 14 and 46 (+164 mu), 13 and 49 (+180 mu).54 t-Bu-MacE (13) and 49, which possess an acetoxy substituent adjacent to the lactone carbonyl group, modified lysozyme to a greater extent than the corresponding desacetoxy congeners 14 and 46. Subjecting the modified lysozymes to trypsin digestion, followed by standard MALDI-MS-MS peptide sequencing, revealed that the two most surface-accessible lysines (K-33 and K-97) were the predominant sites of covalent modification.55
Figure 3.

Modification of lysozyme under the conditions shown with 13 and 14 to form lysine adducts (a) and the distribution of alkylated products obtained from 13 (b) and 14 (c). The ESI-MS spectra were reconstructed from charge ladders.
Table 3.
Modification of HEWL by Dioxabicyclolactones 13, 14, 46, and 49a
| Entry | Substrate | pH | Unmod. (%) |
+1 (%) |
+2 (%) |
+3 (%) |
+4 (%) |
|---|---|---|---|---|---|---|---|
| 1 | 13 | 7 | 9 | 54 | 33 | 4 | 0 |
| 2 | 13 | 8 | 0 | 14 | 49 | 32 | 5 |
| 3 | 14 | 7 | 88 | 12 | 0 | 0 | 0 |
| 4 | 14 | 8 | 43 | 47 | 2 | 0 | 0 |
| 5 | 46 | 7 | 61 | 35 | 4 | 0 | 0 |
| 6 | 46 | 8 | 7 | 48 | 38 | 7 | 0 |
| 7 | 49 | 7 | 13 | 47 | 28 | 12 | 0 |
| 8b | 49 | 8 | 0 | 4 | 19 | 43 | 7 |
Conditions: 50 µM substrate, 10 µM lysozyme, 50 mM K2HPO4 buffer incubated at 22 °C for 20 h. Product distributions were determined from ESI-MS analyses. With 13 and 46, the addition of +180 mu was observed per modification and with 14 and 49, +164 mu was observed.
In addition +5 (7%).
Effects of 6-Acetoxy-2,7-dioxabicylo[3.2.1]octan-3-ones and 7-Acetoxy-2,8-dioxabicylo[3.2.1]octan-3-ones on Golgi Organization
Synthetic access to 2,8-dioxabicyclo[3.3.0]octan-3-ones 46 and 49 allowed us to compare the Golgi-modifying properties of these structures with those of isomers in the 2,7-dioxabicyclo[3.2.1]octan-3-one series. Normal Rat Kidney (NRK) cells grown on coverslips were incubated with analogs 46 and 49 for 60 min at 37 °C, followed by examination of the Golgi for the MacE-induced reorganization phenotype by immunofluorescence analysis with antibodies to the known Golgi resident protein, mannosidase-II.56 The 4,7-diacetoxy-2,8-dioxabicyclo[3.3.0]octanone 49 produced a Golgi phenotype indistinguishable from that of MacE or t-Bu-MacE (13), with the conversion of the Golgi ribbon into small fragments that remained localized adjacent to the centrosome (Figure 4). By contrast, 7-acetoxy-2,8-dioxabicyclo[3.3.0]octanone 46, which lacks an acetoxy substituent adjacent to the lactone carbonyl group, did not affect Golgi structure at concentrations of up to 80 µg/mL. As 6-acetoxy-2,7-dioxabicyclo[3.2.1]octanone 14 also did not impact Golgi structure,6 these findings indicate that the formation of pericentrosomal Golgi fragments characteristic of MacE depends on the presence of oxygenation adjacent to the lactone carbonyl group, either in the substituted dioxabicyclo[3.2.1]octanone or dioxabicyclo[3.3.0]octanone ring systems.
Figure 4.

NRK cells were treated with either control (DMSO), MacE (4) at 20 µg/mL, 49 at 40 µg/mL, or 46 at 80 µg/mL for one hour at 37 °C. MacE and 49 are shown at the minimal active concentration, and 46 is shown at the maximal concentration tested. Cells were fixed and stained with an antibody to the Golgi resident protein mannosidase II (green) and the DNA dye Hoechst 33342 (blue). Area demarcated by the white square is enlarged in the insets to show detail of Golgi organization following each treatment. Scale bar is 10 microns.
Conclusions
The synthesis and initial comparison of the chemical reactivity of two highly oxidized bicyclic lactone fragments found in rearranged spongian diterpenes—8-substituted 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-one and 6-substituted 7-acetoxy-2,8-dioxabicyclo[3.3.0]octan-3-one—are reported. The syntheses of t-Bu-MacE (13) and congener 14 are the first syntheses of the 8-substituted 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-one ring system found in rearranged spongian diterpenes such as macfarlandin E (4) and aplyviolene (3). A late-stage intermediate, 36, was diverted to provide the first synthetic entry to isomeric structures (46 and 49) in the 6-acetoxy-2,8-dioxabicyclo[3.3.0]octanone series.
Access to these highly oxidized bicyclic lactones allowed the chemical reactivity and Golgi-modifying activity of these ring systems to be studied. Under both acidic and basic conditions, 8-substituted 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-ones are converted to 6-substituted 7-acetoxy-2,8-dioxabicyclo[3.3.0]octan-3-one products. Both dioxabicyclooctan-3-one ring systems are found to react readily with primary amines to form pyrrole products. Of particular significance, lysine side chains of hen egg white lysozyme are converted under physiologically relevant conditions to substituted pyrroles upon exposure to dioxabicyclic lactones 13, 14, 46 and 49. The presence of an acetoxy substituent adjacent to the lactone carbonyl group, in either the bridged or fused dioxabicyclooctanone series, increases the extent of the lysine to pyrrole conversion and is essential for induction of the macfarlandin E Golgi phenotype. These investigations provide a basis for future studies aimed at identifying the biological target(s) of these Golgi-modifying natural products, as well as initial insight into the reactivity of the family of structurally distinctive rearranged spongian diterpenes depicted in Figure 1.
Supplementary Material
Acknowledgement
We gratefully acknowledge the late Professor John Faulkner, Scripps Institution of Oceanography, for authentic macfarlandin E. Professor Gregory Weiss, University of California, Irvine, is acknowledged for helpful discussions and Issa Moody, University of California, Irvine, for initial protein-labeling studies. We thank Dr. Joe Ziller, University of California, Irvine, for the single-crystal X-ray analyses and Dr. John Greaves, University of California, Irvine, for mass spectrometric analyses. This research was supported by the NIH Neurological Disorders & Stroke Institute (Grant NS-12389), the NIH National Institutes of General Medical Sciences (Grant GM098601), and by NIH postdoctoral fellowships for M.J.S. (CA-138084) and C.M.B. (GM-079937). B.D.W.K. was supported by a postdoctoral fellowship from the UC Irvine Training Program in Cancer Biology (NCI grant 5 T32 CA009054-34). NMR spectra, mass spectra, and the X-ray analyses were obtained at UC Irvine using instrumentation acquired with the assistance of NSF and NIH Shared Instrumentation grants. Unrestricted funds from Amgen and Merck are also gratefully acknowledged.
Footnotes
Supporting Information Available Experimental details and copies of 1H and 13C NMR spectra of new compounds; CIF files for compounds 46 and 51. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews see Keyzers RA, Northcote PT, Davies-Coleman MT. Nat. Prod. Rep. 2006;23:321. doi: 10.1039/b503531g. González M. Cur. Bioact. Comp. 2007;3:1. Faulkner DJ. Nat. Prod. Rep. 2001;18:1. doi: 10.1039/b006897g.
- 2.Molinski TF, Faulkner DJ, He CH, Van Duyne GD, Clardy J. J. Org. Chem. 1986;51:4564. [Google Scholar]
- 3.Bobzin SC, Faulkner DJ. J. Nat. Prod. 1991;54:225. doi: 10.1021/np50073a023. [DOI] [PubMed] [Google Scholar]
- 4.(a) Dumdei EJ, Dilip de Silva E, Andersen RJ, Choudhary MI, Clardy J. J. Am. Chem. Soc. 1989;111:2712. [Google Scholar]; (b) Morris SA, Dilip de Silva E, Andersen RJ. Can. J. Chem. 1991;69:768. [Google Scholar]; (c) Rungprom W, Chavasiri W, Kokpol U, Kotze A, Garson MJ. Marine Drugs. 2004;2:101. [Google Scholar]
- 5.(a) Guizzunti G, Brady TP, Malhotra V, Theodorakis EA. J. Am. Chem. Soc. 2006;128:4190. doi: 10.1021/ja058259a. [DOI] [PubMed] [Google Scholar]; (b) Guizzunti G, Brady TP, Fischer D, Malhotra V, Theodorakis EA. Bioorg. Med. Chem. 2010;18:2115. doi: 10.1016/j.bmc.2010.02.007. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schnermann MJ, Beaudry C, Egovora AV, Polishchuk RS, Sütterlin C, Overman LE. Proc. Nat. Acad. Sci. USA. 2010;107:6158. doi: 10.1073/pnas.1001421107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Isolation: Dilip de Silva E, Morris SA, Miao SC, Dumdei E, Andersen RJ. J. Nat. Prod. 1991;54:993. (b) Total synthesis: Lebsack AD, Overman LE, Valentekovich RJ. J. Am. Chem. Soc. 2001;123:4851. doi: 10.1021/ja015802o.
- 8.Hambley TW, Poiner A, Taylor WC. Tetrahedron Lett. 1986;27:3281. [Google Scholar]
- 9.Carmely S, Cojocaru M, Loya Y, Kashman Y. J. Org. Chem. 1988;53:4801. [Google Scholar]
- 10.Rudi A, Kashman Y. Tetrahedron. 1990;46:4019. [Google Scholar]
- 11.This ring system is also referred to as oxytetrahydro[2,3-b]furan-2(3H)one.
- 12.Sullivan B, Faulkner DJ. J. Org. Chem. 1984;49:3204. [Google Scholar]
- 13.Hochlowski JE, Faulkner DJ, Matsumoto GK, Clardy J. J. Org. Chem. 1983;48:1141. [Google Scholar]
- 14.Bergquist PR, Bowden BF, Cambie RC, Craw PA, Karuso P, Poiner A, Taylor WC. Aust. J. Chem. 1993;46:623. [Google Scholar]
- 15.Rudi A, Erez Y, Benayahu Y, Kashman Y. Tetrahedron Lett. 2005;46:8613. [Google Scholar]
- 16.Select preparative approaches Corey EJ, Su WG. J. Am. Chem. Soc. 1987;109:7534. Petit R, Furstoss R. Synthesis. 1995:1517. Corey EJ, Letavic MA. J. Am. Chem. Soc. 1995;117:9616. Weisser R, Yue W, Reiser O. Org. Lett. 2005;7:5353. doi: 10.1021/ol051457m.
- 17.Brady TP, Kim SH, Wen K, Theodorakis EA. Angew. Chem., Int. Ed. 2004;43:739. doi: 10.1002/anie.200352868. [DOI] [PubMed] [Google Scholar]
- 18.Granger KE. Ph.D. Thesis. Boston, MA: Boston College; 2009. Norrisolide: Convergent Total Synthesis and Preliminary Biological Investigation. [Google Scholar]
- 19.(a) Arnó M, González MA, Zaragozá RJ. J. Org. Chem. 2003;68:1242. doi: 10.1021/jo026536f. [DOI] [PubMed] [Google Scholar]; (b) Arnó M, Gonzélez MA, Marin ML, Zaragozá RJ. Tetrahedron Lett. 2001;42:1669. [Google Scholar]
- 20.Krow GR. Org. React. 1993;43:251. [Google Scholar]
- 21.Díez E, Dixon DJ, Ley SV. Angew. Chem., Int. Ed. 2001;40:2906. [PubMed] [Google Scholar]
- 22.(a) For an example of a similar functionalization, see reference 7b. (b) IBX in DMSO/toluene generated regioisomeric enone products as a 1:1 mixture. Similarly, reaction conditions such as PhSCl in MeCN or LDA, PhSSPh in THF provided unselective introduction of the sulfur unit.
- 23.Tietze LF, Stadler C, Böhnke N, Brasche G, Grube A. Synlett. 2007:485. doi: 10.1002/chem.200700464. [DOI] [PubMed] [Google Scholar]
- 24.(a) Dixon DJ, Ley SV, Rodriguez F. Org. Lett. 2001;3:3753. doi: 10.1021/ol016708f. [DOI] [PubMed] [Google Scholar]; (b) Ley SV, Dixon DJ, Guy RT, Rodriguez F, Sheppard TD. Org. Biomol. Chem. 2005;3:4095. doi: 10.1039/b512410g. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa T, Fujisawa H, Nagata Y, Mukaiyama T. Chem. Lett. 2004;33:1016. [Google Scholar]
-
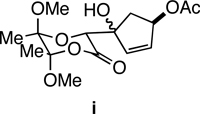
26.The lithium enolate (generated with LiHMDS at −78 °C in THF) of the butane-2,3-diacetal glycolic acid derivative (the precursor to 28) also reacted with 29 to afford 30; however, competing formation of aldol product i (~1:1 with 30) significantly reduced the yield of 30.
- 27.Ley SV, Humphries AC, Eick H, Downham R, Ross AR, Boyce RJ, Pavey JBJ, Pietruszka J. J. Chem. Soc. Perkin Trans. 1998;1:3907. [Google Scholar]
- 28.(a) Corey EJ, Boaz NW. Tetrahedron Lett. 1985;26:6019. [Google Scholar]; (b) Corey EJ, Kang M, Desai MC, Ghosh AK, Houpis JN. J. Am. Chem. Soc. 1988;110:649. doi: 10.1021/ja00210a083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tebbe FN, Parshall GW, Reddy GS. J. Am. Chem. Soc. 1978;100:3611. [Google Scholar]
- 30.(a) Roberts SW, Rainier JD. Org. Lett. 2007;9:2227. doi: 10.1021/ol0707970. [DOI] [PubMed] [Google Scholar]; (b) Takai K, Kataoka Y, Miyai J, Okazoe T, Oshima K, Utimoto K. Org Synth. 1996;73:73. [Google Scholar]
- 31.Steric shielding must be involved, as 1-(trimethylsiloxy)cyclopentene is a much stronger nucleophile than ethyl vinyl ether, see: Mayr H, Kempf H, Ofial AR. Acc. Chem. Res. 2003;36:66. doi: 10.1021/ar020094c. Mayr H, Bug T, Gotta MF, Hering N, Irrgang B, Janker B, Kempf B, Loos R, Ofial AR, Remennikov G, Schimmel H. J. Am. Chem. Soc. 2001;123:9500. doi: 10.1021/ja010890y.
- 32.Diazomethane could be employed, but the yield was reduced by 10–20%.
- 33.Shoda S. Glycoside Synthesis from Anomeric Halides. In: Demchenko AV, editor. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance. Weinheim: Wiley-VCH; 2008. pp. 29–94. [Google Scholar]
- 34.The use of dry DMF was critical for efficient cyclization. For a discussion of the role of DMF in glycosylation chemistry, see: Lu S, Lai Y, Chen J, Liu C, Mong KT. Angew. Chem., Int. Ed. 2011;50:7315. doi: 10.1002/anie.201100076.
- 35.Cooper MS, Heaney H, Newbold AJ, Sanderson WR. Synlett. 1990:533. [Google Scholar]
- 36.Arnett EM, Harrelson JA. J. Am. Chem. Soc. 1987;109:809. [Google Scholar]
- 37.Davis FA, Stringer OD. J. Org. Chem. 1982;47:1774. [Google Scholar]
- 38.It is likely that the acetate was competitively enolized under many of these conditions. We were able to generate the vinyl triflate of the ketone 22 in modest yield (~35%) with KHMDS and the Comins reagent in THF at −78 °C. Although the subsequent α-oxidation reaction with KHMDS and the Davis oxaziridine in THF at −78 °C provided the α-hydroxy lactone in useful yield (~60%), initial attempts to selectively cleave the vinyl triflate were unsuccessful.
- 39.Vishwakarma LC, Stringer OD, Davis FA. Org. Synth. 1988;66:203. [Google Scholar]
- 40.Dess DB, Martin JC. J. Am. Chem. Soc. 1991;113:7277. [Google Scholar]
- 41.The chemical structures of 13 and 30 were confirmed by X-ray crystallography: CDCC 762682 and 762683.6
- 42.Crystallographic data for this compound were deposited at the Cambridge Crystallographic Data Centre: CCDC 839175.
- 43.The low yield observed in this sequence is likely a result of the sensitivity of the α-hydroxylated intermediate 47 to silica gel.
- 44.(a) Bal BS, Childers WE, Pinnick HW. Tetrahedron. 1981;37:2091. [Google Scholar]; (b) Kraus GK, Taschner MJ. J. Org. Chem. 1980;45:1175. [Google Scholar]; (c) Lindgren BO, Nilsson T, Husebye S, Mikalsen Ø, Leander K, Swahn C. Acta Chem. Scand. 1973;27:888. [Google Scholar]
- 45.That the acetal mixture reflected a thermodynamic ratio was confirmed by individually exposing the separable acetal diastereomers of 46 to the reaction conditions (BF3•OEt2, AcOH, CH2Cl2, 0 °C) to provide an identical 3.5:1 α:βmixture of diastereomers.
- 46.The ratio of epimeric lactols was somewhat variable (between 1:1 and 3:1).
- 47.Further confirmation was obtained by chromatographic separation of the major diastereomer of 52 (possessing the S-Mosher-ester) and single crystal X-ray diffraction. Crystallographic data for this compound were deposited at the Cambridge Crystallographic Data Centre: CCDC 839176.
-
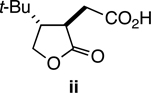
48.Longer reaction times do not afford fully equilibrated, racemic 51, as significant quantities of Cannizzaro product ii are formed. For example, after 72 h in 1 N NaOH at room temperature 54% of ii is isolated from the reaction mixture.
- 49.The initial site of nucleophilic attack in protic conditions was not established in ref 6 in which either aprotic (BnNH2, THF) or aqueous (BnNH2, DMSO, H2O) conditions were used.
- 50.Compound 57 was identified by NMR and mass spectroscopic observation. Efforts to isolate 57 were complicated by the formation of additional products during purification. Compound 58 was also identified by NMR and mass spectroscopic observation, but could be isolated in pure form. See supporting information for details.
- 51.For a discussion of the differences in reactivity between 5 and 6 membered lactones, see: Brown HC, Brewster JH, Shechter H. J. Am. Chem. Soc. 1954;76:467.
- 52.Hall HK. J. Org. Chem. 1963;28:2027. [Google Scholar]
- 53.Lundblad RL, editor. The Modification of Amino Groups. Chemical Reagents for Protein Modification. 3rd Ed. Washington, DC: CRC Press; 2005. pp. 31–67. [Google Scholar]
- 54.No products resulting from an acyl transfer reaction to HEWL appeared to be formed by ESI-MS analysis, suggesting that initial hydrolysis, and not protein side chain reactivity, provides the reactive 1,4-dialdehyde intermediate.
- 55.A similar lysine modification pattern is observed in the acylation of HEWL by acetic anhydride, see: Suckau D, Mak M, Przybylski M. Proc. Natl. Acad. Sci. USA. 1992;89:5630. doi: 10.1073/pnas.89.12.5630.
- 56.The Golgi staining was also carried out with an antibody to the known Golgi protein giantin, which led to nearly indistinguishable staining patterns (see supporting information)


Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.