

A comprehensive variation map of the human metabolome identifies genetic and stable-environmental sources as major drivers of metabolite concentrations. The data suggest that sample sizes of a few thousand are sufficient to detect metabolite biomarkers predictive of disease.
Keywords: biomarker, 1H nuclear magnetic resonance spectroscopy, metabolome-wide association study, top-down systems biology, variance decomposition
Abstract
1H Nuclear Magnetic Resonance spectroscopy (1H NMR) is increasingly used to measure metabolite concentrations in sets of biological samples for top-down systems biology and molecular epidemiology. For such purposes, knowledge of the sources of human variation in metabolite concentrations is valuable, but currently sparse. We conducted and analysed a study to create such a resource. In our unique design, identical and non-identical twin pairs donated plasma and urine samples longitudinally. We acquired 1H NMR spectra on the samples, and statistically decomposed variation in metabolite concentration into familial (genetic and common-environmental), individual-environmental, and longitudinally unstable components. We estimate that stable variation, comprising familial and individual-environmental factors, accounts on average for 60% (plasma) and 47% (urine) of biological variation in 1H NMR-detectable metabolite concentrations. Clinically predictive metabolic variation is likely nested within this stable component, so our results have implications for the effective design of biomarker-discovery studies. We provide a power-calculation method which reveals that sample sizes of a few thousand should offer sufficient statistical precision to detect 1H NMR-based biomarkers quantifying predisposition to disease.
Introduction
1H Nuclear Magnetic Resonance spectroscopy (1H NMR)-based metabolic profiling is a discovery-driven experimental technique that allows high-throughput quantification of small molecules, metabolites (Nicholson et al, 1999; Wishart et al, 2009), in biological samples. There has been a recent surge in the application of 1H NMR in biomedical research, with metabolic profiles being used to characterize, diagnose, and predict pathological states. The application of 1H NMR spectroscopy to urine and plasma samples is attractive from an experimental perspective, as the collection of such samples is minimally invasive, the sample-assay process is non-destructive, and 1H NMR-based quantification of metabolites in urine has been demonstrated to be highly reproducible (Keun et al, 2002; Dumas et al, 2006; Maher et al, 2007). 1H NMR metabonomics (Nicholson et al, 2002) has a substantial history of application in toxicology (Robertson, 2005), and promises to have an important biomedical role in drug-response characterization (Le Moyec et al, 2005; Holmes et al, 2006) and personalized medicine (Clayton et al, 2006; Qiu et al, 2008), as well as in human nutritional research (Gibney et al, 2005; Stella et al, 2006; Rezzi et al, 2007; Favé et al, 2009; Heinzmann et al, 2010). Furthermore, 1H NMR-based metabolic profiling has helped guide the search for diagnostic biomarkers for a number of diseases (Odunsi et al, 2005; Ala-Korpela, 2008; Saude et al, 2009; Williams et al, 2009; Zhou et al, 2009).
Metabolome-wide association studies (MWASs) have emerged as an interesting approach to explore systematically the statistical relationships between disease risk factors and metabolite concentrations, patterns, networks, or fluxes in human biological samples, in order to generate testable physiological hypotheses on disease aetiology (Nicholson et al, 2008; Chadeau-Hyam et al, 2010). MWASs provide a ‘top-down’ perspective on the physiology of complex organisms, usefully complementing other systems-biology approaches. The profiling of metabolite concentrations adds value by summarizing the global physiological impact of interacting multilevel biological systems (including genetic, epigenetic, transcriptomic, and proteomic) with environmental and lifestyle factors. A particular use of the MWAS is in prospective biomarker discovery, in which the goal is to find metabolites whose levels are predictive of disease development several years beyond the time of sample collection. Prospective biomarkers are much rarer in the literature than those simply offering diagnosis or interpretation of pre-existing disease states. We discuss the impact of our findings on the potential utility of the 1H NMR metabolome as a medium for biomarker discovery.
For a biomarker to be useful, its level across a population must clearly associate with disease risk or progression, while not varying too much over the short term within an individual, as that would undermine the predictive association from a single sample. Nor should it be completely heritable if disease risk is significantly influenced by environmental factors. Driven by these considerations, we set out to characterize systematically the sources of variation underpinning the 1H NMR metabolome, so as to inform the design and interpretation of MWASs in the future.
Analysis of a biofluid sample by 1H NMR spectroscopy provides a richly informative functional datum, a spectrum, in which the concentration of each detectable hydrogen-containing metabolite is represented quantitatively by the area under its specific profile. The full biofluid NMR spectrum is the sum of the intensities (i.e., a superposition) of the spectra of individual metabolites; a metabolite's spectrum is made up of peaks from each chemically distinct hydrogen atom in the molecule, with the peaks split into multiplets by inter-proton coupling interactions. The peak position of a given hydrogen on the frequency axis is known as a chemical shift and is quoted in parts per million (p.p.m., often termed a δ value) from that of a reference substance. Our study characterizes the variation landscape of the 1H NMR metabolome through the extraction and statistical analysis of a comprehensive set of 526 peaks.
In order to decompose peak-specific population variation into meaningful subcomponents, we designed a longitudinal twin study (Neale and Cardon, 1992; Martin et al, 1997); see also Materials and methods. The study was designed on the basis of statistical power considerations. Specifically, the ratio of identical to non-identical twin pairs and the longitudinal sampling scheme were chosen in such a way as to maximize information content on the variance parameters of interest, which are described in the following paragraph.
Familial variation comprised all heritable and common-environmental effects (i.e., arising from genetics or shared environment after conception). The current study incorporated a sufficient number of twin pairs to enable estimation of familial variation with useful precision, but not sufficient for the estimation of heritability, which would have required much larger sample sizes (Supplementary information). The incorporation of longitudinal sampling into the study design allowed the decomposition of the remaining non-familial variation into that which was stable over time (individual environmental) and that which was temporally dynamic. The temporally dynamic part of variation was modelled with two components—individual visit, capturing within-individual short-term fluctuations and common visit, allowing for the fact that each twin pair visited the clinic together. Finally, extensive technical replication within the current study's design allowed estimation and separation of non-biological variation (i.e., experimental random noise), so that it was not included in the primary decomposition of biological population variation. The current study was thus distinct from the majority of twin studies in which stable-environmental variation, short-term dynamic variation, and random experimental error are not separable.
The specifics of our study design were as follows. We analysed plasma and urine samples collected longitudinally from 154 female, post-menopausal twins. Of the 77 pairs of twins, 56 were identical (i.e., monozygotic, or MZ, genetically identical) and 21 were non-identical (i.e., dizygotic, or DZ, sharing half their genes as do ordinary siblings); 34 of the MZ twin pairs donated samples twice over the space of several months. We split each of the 222 samples into two aliquots, and analysed all aliquots by 1H NMR spectroscopy. We pre-processed and extracted peaks from each resulting spectrum, and fitted a robust variance-components model to each peak's intensity across spectra.
Our main result was the identification and quantification of a substantive proportion of stable variation in the 1H NMR plasma and urine metabolomes, where stable variation is defined as the sum of familial and individual-environmental components. The current paper lays out the nature and relevance of its results in three stages, by (a) summarizing the estimated variance decomposition across a comprehensive set of 526 peaks, (b) focusing in on the variability of 66 metabolites, whose peaks we annotated, and (c) demonstrating the relevance of its findings to study design in MWASs.
Results
Variation landscape of the 1H NMR metabolome
The 1H NMR acquisition process delivered a single, standard one-dimensional (1D) spectrum for each urine sample. For plasma samples, acquisition of the standard 1D spectrum was supplemented with acquisition of two other types of 1D spectrum, enabling quantification of a range of metabolites, extending from small molecules such as amino acids (targeted by the Carr-Purcell-Meiboom-Gill (CPMG) spin-echo pulse sequence; Nicholson et al, 1995) to large metabolites such as lipids and lipoproteins (targeted by the diffusion-edited pulse sequence; Liu et al, 1996). These (biofluid, pulse sequence) combinations produced four data sets (urine standard 1D, plasma standard 1D, plasma spin-echo, and plasma diffusion-edited); each such data set was analysed separately.
For each of 526 common peaks (a peak was defined to be common if it was present in >80% of spectra in its corresponding data set), we quantified its height—as a proxy for area—in each spectrum in its data set, and fitted a variance-components model to the resulting data (see Materials and methods for methodological details; Supplementary Table S1 shows peak-specific variance decompositions for all 526 common peaks). For the urine data, the mean (across all peaks) of the non-biological variance proportion was 10% (IQR: 2–13); for the combined plasma data, it was 36% (IQR: 16–53). All common peaks were included, irrespective of signal-to-noise ratio. The observation of a higher proportion of non-biological variation in plasma relative to urine was partially attributable to there being more variation across spectra in the spectral baseline (caused by a collection of broad peaks in plasma spectra arising from proteins), as well as to the presence of less population variation in (homeostatically controlled) plasma metabolite concentrations.
Then, after removal of the non-biological variation, the remaining biological variation was decomposed into two stable (familiality and individual-environment) and two unstable (individual-visit and common-visit) components. These biological variance components are summarized in Table I. The proportion of familial variation was found to be substantive in both biofluids, and somewhat higher in plasma (42% is the mean across all peaks) than in urine (30%). Finally, we aggregated the familial and individual-environment effects to estimate the total proportion of biological variation that was longitudinally stable. We found the inter-peak average percentage of stable variation to be 60% (IQR: 51–72) and 47% (IQR: 35–60) for plasma and urine, respectively.
Table 1. Percentage decomposition of biological population variation—summary of results.
| Plasma standard 1D (87 peaks) | Plasma spin-echo (87 peaks) | Plasma diffusion-edited (24 peaks) | Plasma all (198 peaks) | Urine standard 1D (328 peaks) | |
|---|---|---|---|---|---|
| aMean of estimates, across peaks. | |||||
| bInterquartile range of estimates, across peaks. | |||||
| (A) Familiality | 38a (28–48)b | 43 (33–56) | 49 (45–56) | 42 (32–52) | 30 (17–39) |
| (B) Individual environment | 17 (9–22) | 20 (10–26) | 22 (14–25) | 19 (10–25) | 18 (9–25) |
| (C) Individual visit | 35 (24–47) | 27 (14–39) | 20 (12–28) | 30 (17–39) | 45 (34–55) |
| (D) Common visit | 10 (4–15) | 10 (4–13) | 9 (5–13) | 10 (4–14) | 8 (4–10) |
| (A+B) Stable total | 55 (42–69) | 63 (54–73) | 71 (63–79) | 60 (51–72) | 47 (35–60) |
| (C+D) Unstable total | 45 (31–58) | 37 (27–46) | 29 (21–37) | 40 (28–49) | 53 (40–65) |
Variance decomposition for annotated metabolites
We assigned peaks to metabolites in each data set using a combination of the web-based human metabolome database (Wishart et al, 2009), an in-house developed database, statistical total correlation analysis (Cloarec et al, 2005), and other literature (Nicholson et al, 1995). We annotated a total of 38 metabolites in plasma and 27 in urine. Several metabolites were represented in the data with a degree of redundancy: a single metabolite can create multiple peaks within a single spectrum, and may also be represented in more than one plasma data set. We used this feature for model validation, and, with the exception of one metabolite (lactate in plasma), we were successfully able to verify the consistency of our findings across multiple peaks of the same metabolite (Supplementary Figure S1 and Supplementary information).
To summarize the results for each metabolite, a single representative peak was chosen on the basis of (a) being present in a high proportion of spectra, (b) having high signal-to-noise ratio, and (c) exhibiting limited overlap with other peaks (Supplementary Figure S2 displays these criteria, and details which peak was selected in each case). For plasma, the peak was drawn from across the three plasma data sets. The biological variance decomposition for each such representative peak is shown in Figure 1 (the underlying numbers are in a subset of the rows of Supplementary Table S1). The mean proportion of stable biological variation across annotated metabolites was 68% (IQR: 60–79) for plasma and 53% (IQR: 38–67) for urine. There was variation across metabolites in the statistical precision with which variance components could be estimated. We quantified this aspect of the results by providing Bayesian credible intervals (BCIs) for the variance parameters of each metabolite (Figure 1; Supplementary Table S1).
Figure 1.

Decomposition of biological variance for each annotated metabolite. The plot displays estimates (and measures of precision) for the proportion of biological variance explained by each of four components (familial, individual environmental, individual visit, and common visit). The central tick within each box marks the posterior mean, the box extends to the posterior quartiles, and the whiskers extend to the 2.5 and 97.5 posterior percentiles. Metabolites are ordered by estimated familiality.
Ten metabolites were annotated in both urine and plasma data sets (acetate, acetoacetate, alanine, citrate, creatine, creatinine, dimethylamine, glycine, lactate, and dimethylsulfone). For each of these, we compared the estimate of each biological variance proportion across biofluids, finding the 95% BCIs to overlap in all cases but two—dimethylamine and dimethylsulfone each exhibited higher individual-visit variance proportion in urine than in plasma (Figure 1; Supplementary Table S1).
Sample sizes for MWASs
The MWAS has emerged as an interesting ‘top-down’ approach for the characterization of disease-risk biomarkers (Nicholson et al, 2008; Chadeau-Hyam et al, 2010). Physiological concentrations of metabolites reflect both genetic and environmental risk factors, and can thus offer a relatively comprehensive and accurate assessment of complex-disease susceptibility, compared with molecular markers that are mechanistically closer to the genome (e.g., mRNA-transcript or protein levels). We examined the implications of our findings for the effective design of an MWAS in search of such disease-susceptibility metabolite biomarkers.
Let x denote a metabolite's concentration and y denote a quantitative disease-related phenotype. Consider, for example, a prospective MWAS, in which x is a subject's blood low-density lipoprotein cholesterol concentration (LDL) 10 years ago, and y quantifies the subject's cardiovascular disease status (CV) at the present time. Short-term variations in LDL are unlikely to provide useful predictive information about long-term CV risk, so CV-predictive variation in LDL is more likely to be nested within LDL's longitudinally stable component. This motivates a model under which the longitudinally stable variation in x contributes to the (x, y) association.
Suppose variation that is shared by x and y contributes a proportion p of the variance of x, and a proportion q of the variance of y (in the example, the biological processes underlying the association between LDL and CV explain a proportion p of variation in LDL and a proportion q of variation in CV). The underlying absolute correlation between x and y in such a scenario is 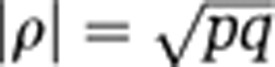 . We calculated the sample size of bivariate Gaussian observations
. We calculated the sample size of bivariate Gaussian observations 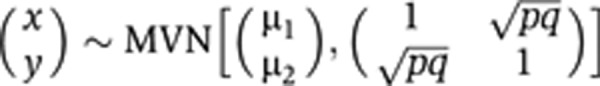 required to detect the (x, y) association with high power, as a function of p and q (Figure 2A). It is likely in practice that q will be small (explaining <10% of disease risk), while p is bounded above by the proportion of stable variation in the metabolite, which can be large (e.g., exceeding 50%), as the current study has demonstrated.
required to detect the (x, y) association with high power, as a function of p and q (Figure 2A). It is likely in practice that q will be small (explaining <10% of disease risk), while p is bounded above by the proportion of stable variation in the metabolite, which can be large (e.g., exceeding 50%), as the current study has demonstrated.
Figure 2.

Sample size calculations for 1H NMR-based MWASs. A hypothetical study was designed to detect an association between a metabolic phenotype, x, and a disease phenotype, y, when the biological processes linking x and y explain a proportion p of population variation in x, and a proportion q of population variation in y (so  . Calculations were based on the study attaining 80% power to reject H0:ρ=0 at a 10−4 level of significance (a Bonferroni-corrected significance level of 0.05, assuming that 500 metabolite peaks were tested for disease association). (A) Sample size as a function of p and q. Darker grey represents a larger required sample size (the colour scale is indicated by labelled contour lines on the plot). (B) Sample sizes for the discovery of 1H NMR-based urine biomarkers. Bottom panel (annotated ‘
. Calculations were based on the study attaining 80% power to reject H0:ρ=0 at a 10−4 level of significance (a Bonferroni-corrected significance level of 0.05, assuming that 500 metabolite peaks were tested for disease association). (A) Sample size as a function of p and q. Darker grey represents a larger required sample size (the colour scale is indicated by labelled contour lines on the plot). (B) Sample sizes for the discovery of 1H NMR-based urine biomarkers. Bottom panel (annotated ‘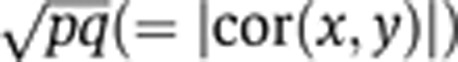 ’): Probability distributions on the magnitude of the underlying correlation (on logarithmic scale) between urinary metabolite concentration, x, and the disease phenotype, y. The probability distribution on p (not shown) was constructed using (for upper bounds) the current paper's estimates of the stable proportion of variation for peaks in the urine data (details are in Results). The proportion of disease risk explained, q, was fixed at four different values (annotated on plot). Main panel (annotated with four different values for q): Relationship between the underlying (x, y) correlation, and the sample size required for effect detection (both on logarithmic scale). Left panel (annotated ‘Sample Size for 80% Power’): Probability distribution on sample size (on logarithmic scale) required for effect detection, mapped from the correlation distributions in the bottom panel.
’): Probability distributions on the magnitude of the underlying correlation (on logarithmic scale) between urinary metabolite concentration, x, and the disease phenotype, y. The probability distribution on p (not shown) was constructed using (for upper bounds) the current paper's estimates of the stable proportion of variation for peaks in the urine data (details are in Results). The proportion of disease risk explained, q, was fixed at four different values (annotated on plot). Main panel (annotated with four different values for q): Relationship between the underlying (x, y) correlation, and the sample size required for effect detection (both on logarithmic scale). Left panel (annotated ‘Sample Size for 80% Power’): Probability distribution on sample size (on logarithmic scale) required for effect detection, mapped from the correlation distributions in the bottom panel.
We created a distribution for p that quantified the stability of common 1H NMR-detectable urine metabolites. The probability distribution on p was constructed using (for upper bounds) the current paper's estimates of the stable proportion of variation for peaks in the urine data. Specifically, we defined the distribution on p to be a non-weighted mixture of the set of uniform densities {Uniform(0, pi): i=1,…,328}, where pi denotes the estimate of the stable proportion of total phenotypic variance for the ith peak. We combined this distribution on p with various fixed values of the explained proportion of disease risk, q, to give corresponding distributions on underlying correlations, via
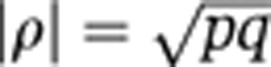 (Figure 2B, bottom panel). We then translated this uncertainty in the underlying correlation into uncertainty in the sample size required to detect the effect (Figure 2B, left-hand panel). The plot indicates that a sample size of 5000 would be sufficient to detect associations explaining 10% of disease risk (q=0.1), should they exist, but would be insufficient to detect most associations explaining just 1%. Supplementary Figure S3 is the corresponding plot based on the plasma data, showing that estimated sample sizes for plasma are similar to, but very slightly higher than, those for urine.
(Figure 2B, bottom panel). We then translated this uncertainty in the underlying correlation into uncertainty in the sample size required to detect the effect (Figure 2B, left-hand panel). The plot indicates that a sample size of 5000 would be sufficient to detect associations explaining 10% of disease risk (q=0.1), should they exist, but would be insufficient to detect most associations explaining just 1%. Supplementary Figure S3 is the corresponding plot based on the plasma data, showing that estimated sample sizes for plasma are similar to, but very slightly higher than, those for urine.
It is important to note that the underlying result shown in Figure 2A is applicable to other metabolic phenotypes (e.g., metabolite concentrations measurable by mass spectrometry—MS), and also to other ‘omics’ platforms (e.g., transcriptomic and proteomic). Figure 2B and Supplementary Figure S3 are specific to the 1H NMR urine and plasma metabolomes, respectively; they depend on the stability of the constituent metabolites' concentrations and the precision of the measurements. The sample-size calculations are applicable to molecular epidemiological studies (not necessarily involving twins) in which the underlying disease model is assumed to be one where persistent overexpression or underexpression of an individual's baseline molecular level, relative to that of the general population, is associated with an increase or decrease in disease susceptibility relative to the background disease prevalence. We further assume that each participant donates a sample at a single time point. In this situation, variation due to longitudinal instability will reduce the precision in the estimate of the true baseline level and hence affect power to detect systematic differences between baseline measurements in cases versus controls. Studies with repeated longitudinal sampling of individuals could estimate the within-individual baseline level with greater precision, by averaging over the longitudinal variation. Such studies could thereby increase power to detect disease associations by increasing the numbers of samples and assays, without increasing the number of participants.
Discussion
Our study has substantively extended pre-existing knowledge of the sources of variation in the human 1H NMR metabolome. We extracted peak heights to quantify concentrations of 1H NMR-detectable metabolites in human urine and plasma. We decomposed population variation in the concentration of common metabolites—those found to be present in >80% of samples. Rare metabolites, such as exogenous medications, were intentionally excluded, and this should be a consideration in the interpretation of our results. We employed a longitudinal twin-based design, allowing a relatively detailed decomposition of variation. Pre-existing research into metabolomic variation had focused on the heritability, or the longitudinal fluctuation, or the experimental variation, of a metabolite's concentration. The current study simultaneously estimated familial, individual-environmental, short-term dynamic (visit), and non-biological variation.
The current study included the first systematic quantification of the familiality and stability of urine metabolite levels in humans. Previous work had identified some examples of instability in the urine 1H NMR metabolome (Saude et al, 2007), raising concerns that urine metabolites might have limited utility as predictive biomarkers. Here, we have quantified the relative contributions of stable and unstable sources to population variation in urine metabolite concentration, and identified a substantive average level of stability (47%). We have demonstrated the important implications of this finding on the design of MWASs. We identified higher levels of stability in the plasma 1H NMR metabolome (60%) than in urine (47%), consistent with plasma homeostasis being largely controlled through urinary excretion (Simpson, 1983). We further contextualize our findings around pre-existing work later in Discussion.
1H NMR and MS measure different subsets of the metabolome. 1H NMR spectroscopy quantifies the most abundant 50–100 metabolites in a biofluid, typically those above 10 micromolar in concentration. 1H NMR covers many important substances involved in major biochemical functions and key intermediary processes. In contrast to 1H NMR, MS-based methods can detect molecules at lower concentrations, but are hindered by additional experimental variability, since they typically rely on a preliminary chromatographic separation stage. Furthermore, different chromatographic methods have to be used for different classes of compounds, and so MS approaches are usually applied in a more targeted manner (e.g., to specifically quantify bile acids or phospholipids). The two approaches can be considered complementary, but 1H NMR is typically used before MS to provide an extensive overview of the metabolic profile. Furthermore, the majority of publications in mammalian metabonomics use NMR rather than MS. Thus, in our 1H NMR-based study, we have addressed an important, representative, and interesting subset of the human metabolome (Lindon and Nicholson, 2008).
We incorporated a number of safeguards into our analysis to prevent our findings being influenced by the use of concomitant medications by members of our study group (see Materials and methods for full details). We explicitly removed peaks that we annotated as exogenous metabolites. We only retained peaks that were present in at least 80% of spectra, thus eliminating peaks arising directly from rare exogenous metabolites. Finally, we implemented a robust variance-components model that automatically down-weighted anomalous observations (such as might be induced indirectly in peaks adjoining the peak of an exogenous metabolite).
We addressed longitudinal variation by sampling individuals twice, with the two visits separated by several months. This provided a decomposition of population phenotypic diversity into variation that persisted for at least several months and variation that did not. The rationale for this study design was that stability over long time scales implied stability over shorter time scales: variation that persisted for several months also persisted over days or weeks (with the caveat that the current study's design did not address the dynamics of those metabolites that varied diurnally about a relatively stable baseline). While the current study's design did not directly address long-term stability beyond ∼4 months, it is reasonable to expect a gradual, smooth decay in stable behaviour as the time scale increases from months to years. The rate and nature of the decay in metabolic stability is an interesting topic for further research, and will be facilitated as biobanks mature, fuelling cohort studies capable of characterizing very long-term molecular variation.
Several aspects of longitudinal variation in metabolic profiles have been characterized previously (Lenz et al, 2003; Bollard et al, 2005; Saude et al, 2007; Slupsky et al, 2007; Assfalg et al, 2008; Lewis et al, 2010). This previous work has focused on low-dimensional subspaces of the metabolome defined by pattern recognition methods (Lenz et al, 2003; Bollard et al, 2005), or on a restricted subset of metabolites, as did Saude et al (2007), who measured daily concentrations of 10 urine metabolites in 6 subjects over 30 days. Saude et al reported results for 6 randomly selected metabolites (they omitted results for 4 of the 10 metabolites). Of these, 5 are annotated and analysed in the current study—alanine (54%), citrate (76%), creatine (70%), hippurate (57%), and lactate (35%); parenthesized percentages are our estimates of the stable proportion of biological variation. We are unable to make a direct quantitative comparison between our results and those of Saude et al due to fundamental differences between the two studies in design and data analysis. Instead, we describe how our results develop knowledge of longitudinal stability of urine metabolites against the background of Saude et al's study. Saude et al reported some instances of within-individual longitudinal fluctuations (specifically, for citrate and tyrosine in a subset of individuals) that were of the same magnitude as one to two times the inter-individual standard deviation (i.e., the standard deviation, across individuals in the population, of the within-individual baseline mean concentration). They thereby demonstrated the existence of substantive within-individual longitudinal variation (relative to population variation) in the concentrations of some urine metabolites in some individuals. Against this background created by the results of Saude et al, an important next goal was to quantify the relative contributions of stable and unstable variation to population variation in urine metabolite concentrations. Our research has done this, providing a formal and comprehensive treatment of longitudinal variation in the urine and plasma 1H NMR metabolomes. In contrast to previous work, we have explicitly modelled and estimated the proportional contribution of longitudinally fluctuating variation to population variance in metabolite concentration. We have demonstrated the importance of such results to the design and interpretation of MWASs.
The most extensive prior work on the heritability of metabolite levels in human plasma was conducted by Shah et al (2009) using MS. They estimated heritabilities for >60 targeted metabolites using samples from families at increased risk of premature cardiovascular disease. Some of the metabolites in our study overlapped with those examined by Shah et al, and hence we were able to check the consistency of a number of our findings against pre-existing work. To this end, we compared our plasma familiality estimates with Shah et al's heritability estimates for the subset of metabolites appearing in both studies (i.e., for alanine, glutamine/glutamate, glycine, leucine/isoleucine, tyrosine, and valine). Shah et al's heritability estimates all fell within our corresponding 95% credible intervals for familiality, with the exception of their glutamate/glutamine heritability estimate, which, while consistent with our familiality estimate for glutamate (59%), was higher than our estimate for glutamine (24%); these metabolites are discussed in greater detail below. It is reassuring that our plasma familiality findings are consistent with previous work.
An estimate of heritability or familiality draws on variation from a potentially large number of genetic loci. Contrastingly, Illig et al (2010) searched for single-locus genetic drivers of metabolite levels. They quantified the strength of association in a human population between serum metabolite concentration and genetic variation at each of many single-nucleotide polymorphisms spanning the genome (see also Gieger et al, 2008). They reported nine loci, each of which exhibited a significant, replicable association either with a metabolite's concentration or with a concentration ratio (i.e., the ratio of one metabolite's concentration to another's), with the loci explaining between 5.6 and 36.3% of the observed variance in concentration ratios. The MS-based Biocrates platform used by these authors was largely non-overlapping with 1H NMR in the subset of the metabolome it targeted (it targeted mostly amino acids and lipids). Some of the strongly familial 1H NMR-detectable metabolites of our study may also be driven substantively by single-locus variation.
Our sample of individuals comprised only post-menopausal females, and so our results are not immediately transferable to males and younger females. Some studies have reported association of metabolite concentrations with age or gender (Bollard et al, 2005; Kochhar et al, 2006; Saude et al, 2007; Slupsky et al, 2007). We note, though, that inter-gender differences in the mean concentration of a metabolite do not imply inter-gender differences in variance components (including longitudinal stability). We are unaware of work comparing longitudinal stability across genders or other strata, and so further research will be necessary to determine the extent of transferability of our findings to other contexts.
Analyses of 1H NMR metabolic profiles between and within heterogeneous populations have revealed striking systematic differences in metabolite concentration between geographic regions (Holmes et al, 2008; Yap et al, 2010). Our study design takes the opposite sampling approach, drawing its subjects from a single, homogeneous population. We observe a stable component of metabolite variation arising from the genetic and environmental diversity within our Northern European panel. A multipopulation cohort with greater genetic and/or environmental heterogeneity than ours would exhibit a correspondingly greater proportion of stable variation than we observe (assuming levels of intra-individual longitudinal variation are consistent with those observed in our study). An interesting question, beyond the scope of our study, but potentially addressable in broader cohorts, is: ‘What are the relative contributions of genetics and environment to worldwide metabolic diversity?’ Initial studies suggest that environmental influences may have the major role (Holmes et al, 2008; Yap et al, 2010).
We have performed a separate variance decomposition on each metabolite's concentration. An interesting extension to our work is to analyse the data in such a way as to acknowledge the biological relationships between metabolites (Wheelock et al, 2009; Pontoizeau et al, 2011). We mapped 36 of the annotated metabolites in our study to KEGG compound identifiers, and then to KEGG pathways (Xia et al, 2009); the mapping is shown in Supplementary Table S2. We performed a hyper-geometric test for overrepresentation of highly familial (>50% familiality) or highly stable (>60% stability) metabolites within each KEGG pathway (Xia et al, 2009). After correction for multiple testing, we discovered no instances of significant overrepresentation. An alternative, empirical approach is to develop network models of partial correlation that are appropriate in the current longitudinal, twin-based data setting. Of particular interest would be models that allow inter-metabolite correlations to be driven by separately parameterized genetic, environmental, and short-term dynamic influences. Though beyond the scope of the current paper, we identify this as an interesting avenue of future research.
The variability results for a number of annotated metabolites are worthy of particular discussion in their own right. Glutamate is a major excitatory neurotransmitter, but also has an important role as an inter-organ carrier of nitrogen. Most dietary ammonia is converted to urea in periportal hepatocytes, but some escapes detoxification and is converted to glutamine in perivenous hepatocytes. This residual glutamine is converted to urea on the next visit to the periportal cells after conversion to glutamate by glutaminase. This has been termed the ‘Intercellular Glutamine Cycle,’ and is under regulation by factors which increase glutaminase activity, such as plasma ammonia concentration, plasma pH, and hormones (McGivan, 1998). Phosphate-dependent glutaminase is responsible for 90% of the glutamine hydrolyzing activity of the liver (Horowitz and Knox, 1968), and this enzyme is also found in blood platelets (Sahai, 1983). A previous twin study, (Sahai and Vogel, 1983), found the activity of this enzyme to be highly heritable, with an intra-class correlation of 0.96 for MZ twins, compared with 0.53 for DZ twins. Thus, our finding of high familiality for glutamate but not glutamine may be suggestive of mediation by glutaminase.
The plasma metabolite with the highest familiality was creatinine (77%). Formed from muscle creatine at a steady rate of ∼2% per day, creatinine production is dependent on total muscle mass, while its clearance is determined by the glomerular filtration rate (Perrone et al, 1992). The high stability of plasma creatinine in our cohort of healthy individuals was consistent with the well-established clinical utility of blood creatinine levels as a measure of renal function. Blood creatine, however, had a much lower familiality (37%), and high visit effect (40%). Biosynthesis of creatine takes place in the liver, but it can also be absorbed from the gut after ingestion of creatine-rich foods (Wyss and Kaddurah-Daouk, 2000); thus, the high visit effect of blood creatine levels was likely due to variations in dietary consumption before collection. We found that urinary creatinine had a familiality of 58%, within the heritability confidence intervals previously estimated in a study of older female twins (Bathum et al, 2004).
3-hydroxybutyrate (3-HB) is a ketone body produced by the liver as metabolic fuel for peripheral tissues, including heart and skeletal muscle, and is elevated during starvation to provide additional fuel for the brain (Voet and Voet, 1995). In our study, plasma 3-HB had a moderate familiality (41%) but a high visit effect (51%). This probably reflected variations in total fasting time before collection of samples. Since this molecule is used as a marker in metabonomic studies of diabetes (Griffin, 2006), caution should be exercised in interpreting changes in plasma 3-HB levels, as fasting time might have a strong influence on levels of this biomarker.
In conclusion, we have designed and conducted a study of human variation in 1H NMR-based metabolic profiles. We collected plasma and urine samples longitudinally from healthy, post-menopausal twins, and analysed each sample using 1H NMR spectroscopy. From each resulting spectrum, we extracted a comprehensive set of peaks, arising from common metabolites, and robustly decomposed the population variation underlying each peak. Our results show that a human's genetic and long-term environmental background exerts a stable and pervasive influence on the concentration of 1H NMR-detectable metabolites. Predictive biomarkers are likely to be nested within this stable component of variation, so our analysis maps out a substantial biomarker-harbouring zone within the 1H NMR metabolome. Our results will act as a resource to aid the future design and interpretation of 1H NMR-based epidemiological studies.
Materials and methods
Recruitment and sample collection
A total of 154 twins, comprising 21 DZ and 56 MZ pairs, were ascertained from the Twins UK database at St Thomas Hospital (http://www.twinsUK.ac.uk) and recruited to participate in this study. Eligible volunteers were healthy, Caucasian, post-menopausal females of Northern European descent, aged between 45 and 76 years old.
Eligible twins were sent an information sheet containing details of the study, as well as two consent forms. After they had returned a completed consent form, twins were contacted by letter and phone to book their appointment.
Fasting blood and urine samples were collected at all visits of each twin. Twins who visited in the morning (scheduled at 1000 h) fasted overnight from midnight. Twins who visited in the afternoon (scheduled at 1400 h) fasted from 0600 h on the day of the visit. Spot urine samples from the twin volunteers were centrifuged (16 060 g) at 4°C for 10 min before being stored at −80°C. Fresh blood was collected in a 9-ml heparin tube from each twin through venepuncture. The blood samples were kept on ice for 20 min before centrifugation (16 060 g) at 4°C for 10 min, and subsequent storage at −80°C.
Thirty-four of the MZ twin pairs donated samples twice; the median inter-visit time across all such pairs was 118 days (IQR: 96–134). Both twins in a pair always visited on the same day, and each visit was scheduled at either 1000 or 1400 h (with repeated visits of each individual not necessarily scheduled at the same time of day). The study was approved by St Thomas' Hospital Research Ethics Committee (EC04/015 Twins UK).
Sample preparation and 1H NMR data acquisition
Thawed samples were centrifuged at 16 060 g for 10 min. Samples were aliquotted into two technical replicates before sample preparation. Plasma was diluted 1:4 in physiological saline prepared in 20% D2O supplemented with 0.1% (w/v) sodium azide as a bacteriostatic agent and 1.5 mM sodium formate as a chemical-shift reference (δ8.452). Urine was diluted 2:1 in phosphate buffer (20% D2O, pH 7.4) supplemented with 1 mM trimethylsilyl-2,2,3,3-tetradeuteropropionic acid (TSP; δ0.00) and 0.1% (w/v) sodium azide. Sample aliquots were allocated to 96-well plates (and wells thereon) in a randomized design.
Each spectrum was acquired on a Bruker advanced DRX 600 MHz spectrometer (Rheinstetten, Germany) operating at 600 MHz (for 1H) using a 5-mm TXI flow-injection probe equipped with a z-gradient coil, at 300 K, at a spectral width of 12 019 Hz, with 96 transients being collected with 8 dummy scans using 64k time domain data points. For both plasma and urine samples, a standard 1D spectrum (RD–90°–3 μs–90°–tm–90°–acquire) with selective irradiation of the water resonance during the relaxation delay (RD, 2 s) and during the mixing time (tm, 0.1 s) was acquired. Additionally, for the plasma samples, a spin-echo (CPMG) spectrum (RD–90°–(τ/2–180°–τ/2)n–acquire) with a total echo time of 608 ms (n=304, τ=2000 μs) and a diffusion-edited spectrum made using a bipolar pulse-pair longitudinal eddy current delay pulse sequence with spoil gradients immediately following the 90° pulses after the bipolar gradient pulse pairs were acquired. Continuous wave irradiation was applied during the relaxation delay at the frequency of the water (or HOD) resonance. Eddy current recovery time (Te) was 5 ms, and the time interval between the bipolar gradients was 0.5 ms. Further details may be found in Nicholson et al (1983, 1984, 1995).
Pre-processing and feature extraction
Each of four data sets was passed independently through a semiautomated pre-processing pipeline: phasing, alignment, denoising, baseline correction, manual bin selection, normalization, quality control, peak extraction, and logarithmic transformation.
Spectra were phased using in-house software (NMRProc, Doctors Tim Ebbels and Hector Keun, Imperial College London). All other data analysis was performed in R (R Development Core Team, 2010). Spectra were zero-filled to 216 points. Urine spectra were aligned to TSP; plasma spectra to formate (peak centres were defined by the position of the local maximum).
The spectra were denoised in the frequency domain using wavelet-based methodology similar to that described by Johnstone and Silverman (2005). For baseline correction, we initially fitted a constant baseline to each spectrum; however, visual inspection revealed that, for a number of spectra, the fit was better on one side of the water peak than on the other; imperfect phasing might contribute such an effect. Hence, a two-piece piecewise-constant baseline was fitted to and subtracted from each spectrum; specifically, the baseline on each side of the water peak was estimated by the fifth percentile of the spectral points in the corresponding interval (a robust estimator of baseline location).
We plotted each peak, and for those that visually displayed consistent presence across spectra, we manually created a bin, and that bin was used to extract the peak's data across all spectra. The datum extracted from a bin was the intensity of the highest local maximum, or was coded as a missing value if no local maximum was present. This approach used peak height as a proxy for peak area. We note that if the width (at half height) of a peak varies substantially across spectra then peak height may be less precise than area at quantifying concentration. Plots of peaks did not reveal substantial peak-width variation in our data sets (Supplementary Figure S2).
Only common peaks (present in at least 80% of spectra in their corresponding data set) were included in downstream statistical analysis, and only a peak's non-missing data were included in the variance decomposition of that peak. Before fitting the variance-components model, we discarded any peaks that were annotated to an exogenous metabolite (ibuprofen or acetaminophen), to a spike-in compound (TSP in urine and formate in plasma), or to urea. Across the three plasma data sets, 104 peaks were annotated to glucose. In order to prevent the analysis of the plasma data from being dominated by a single metabolite, we retained just one representative glucose peak in each plasma data set (the parts of the analysis to which the glucose peak-omission is relevant are the normalization of each of the three plasma data sets; the summary of variance-decomposition results for all metabolite peaks in Table I; and the calculation of sample sizes for biomarker discovery presented in Supplementary Figure S3).
The spectra were normalized using probabilistic quotient normalization (Dieterle et al, 2006). The normalization was performed using data from the retained peaks only; spectra were normalized to a reference spectrum comprising median peak heights; missing values were excluded from the calculation of medians. After quality control, each of the four data sets comprised spectra from a total of 152 twins.
A logarithmic transformation was applied to make the peak height distributions more symmetric—the entire spectrum-wide set of peak heights were collectively shifted and scaled to lie between 0 and 100 and then transformed y ↦ log(1+y).
The data have been uploaded to an FTP server, from which they can be freely downloaded (host: svilpaste.mii.lu.lv; login: Moltwin_NMR; password: Moltwin_NMR1; path: /home/George/MSB_NMR_data). For each of the four data sets analysed in the current paper, the following data formats are available for download: (a) raw frequency domain spectral data; (b) pre-processed spectral data (denoised, baseline corrected, and normalized); (c) extracted peak heights (logarithmically transformed, as described above). Sample metadata are also available.
Statistical model for twin data
The analysis of twin data typically proceeds by estimation of (functions of) the additive-genetic, dominant-genetic, common-environment, and individual-environment variance components—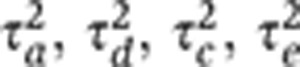 , respectively. The structural-equation model (SEM) for the classical twin study (e.g., Rijsdijk and Sham, 2002) provides a model for the covariance structure in phenotypic data obtained from MZ and DZ twin pairs. The covariance matrix of the phenotype measurements, x1 and x2, from a pair of MZ twins is
, respectively. The structural-equation model (SEM) for the classical twin study (e.g., Rijsdijk and Sham, 2002) provides a model for the covariance structure in phenotypic data obtained from MZ and DZ twin pairs. The covariance matrix of the phenotype measurements, x1 and x2, from a pair of MZ twins is
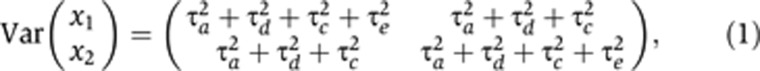 |
while the corresponding matrix for a pair of DZ twins is
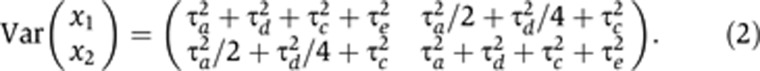 |
A common approach to fitting an SEM proceeds by assuming a multivariate Gaussian model for the phenotype data, and finding maximum likelihood estimates of the variance parameters (Neale, 2001; Rijsdijk and Sham, 2002).
It is not possible within the standard twin-study design to estimate all of 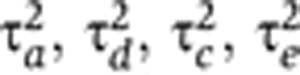 identifiably. One approach that is commonly taken to address this non-identifiability issue is to constrain to zero either the dominant-genetic variance (
identifiably. One approach that is commonly taken to address this non-identifiability issue is to constrain to zero either the dominant-genetic variance (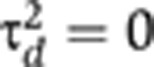 , giving the ACE model) or the common-environment variance (
, giving the ACE model) or the common-environment variance (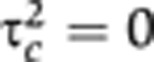 , giving the ADE model), and then to estimate the remaining unconstrained parameters (Neale, 2001; Posthuma et al, 2003).
, giving the ADE model), and then to estimate the remaining unconstrained parameters (Neale, 2001; Posthuma et al, 2003).
The mixed-effects model of the current paper creates the same covariance structure (and hence the same likelihood) as the SEM-induced covariance described in Equations (1) and (2). We addressed non-identifiability by re-parameterizing the four non-identifiable variance parameters, 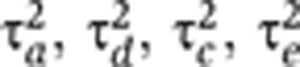 , in terms of three identifiable parameters:
, in terms of three identifiable parameters: 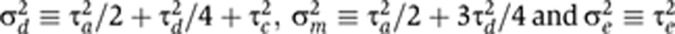 . Visscher et al (2004) used an analogous parameterization under the ACE model. By direct substitution into Equations (1) and (2), it can be seen that, for MZ twins, the covariance structure of the σ2-parameterized model is
. Visscher et al (2004) used an analogous parameterization under the ACE model. By direct substitution into Equations (1) and (2), it can be seen that, for MZ twins, the covariance structure of the σ2-parameterized model is
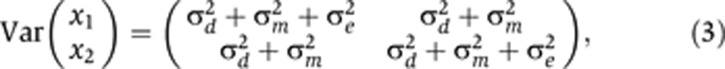 |
while for DZ twins it is
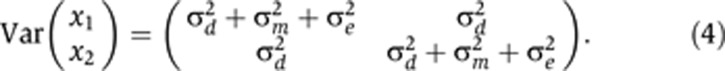 |
The three parameters σd2, σm2, σe2 are all identifiable in the standard twin design, though relatively large sample sizes are required to separate σd2 from σm2. In the current paper, the familial variance (i.e., σd2+σm2≡τa2+τd2+τc2) is estimated, but is not further decomposed into genetic and non-genetic components, because our study's sample size is insufficient for this purpose (see ‘Sample Size for Heritability Estimation’ section in Supplementary information, and Supplementary Figure S4).
The current paper's parameterization approach to non-identifiability was preferable to the use of the ACE or ADE model in the current context in which the familial variance (i.e., σd2+σm2≡τa2+τd2+τc2) was estimated, but was not further decomposed. This was because our parameterization provided direct estimates of the familial variance under the full, ‘true’ model defined by Equations (1) and (2). In contrast, the ACE or ADE approach would have first approximated this model (by setting τd2=0 or τc2=0 for the ACE and ADE models, respectively). Hence, for example, under the ACE parameterization the resulting estimates of τa2, τc2, τe2 can no longer be interpreted as estimates of additive-genetic, common-environment, and individual-environment variance components, since these estimates are conditional on τd2 being zero, and therefore will be biased if the unknown true dominant-genetic effect, τd2, is non-zero. In contrast, the σ2 parameterization used in the current paper provides interpretable estimates of familial and individual-environment variance components irrespective of the unknown actual values of τa2, τd2, τc2, τe2.
The current study was complex in its design, in that it included multiple longitudinal measurements on participants, and also incorporated technical replication. Within this relatively complex twin-study design, the standard SEM approach would still have employed the identical likelihood to the current paper's mixed-model approach, and would have differed only in the aforementioned approach to parameterization. An additional, practical, reason for our use of mixed models rather than SEMs was that it was considerably simpler to specify and fit the complex covariance structure directly in R, than it was to do so using SEM software such as Mx.
Full variance-components model
At each metabolite peak, we fitted the linear mixed-effects model (Searle et al, 2006):
The ‘fixed-effect’ parameters {βb: b=1,…,5} controlled for experimental inter-plate effects—b(·) maps spectra to plates. The ‘fixed-effect’ coefficient τ was included to control for sampling time-related effects—t(·) maps visits to sample-collection times in 24-h format, with times being mostly 10 or 14. The other terms on the right-hand side of the equation are ‘random effects’ that model the covariance structure across observations induced by familial (d, m), individual-environmental (e), temporally dynamic (w, v), and non-biological (ε) effects. In the formula, i∈{1,…,77} indexes twin pairs, j∈{1,2} indexes twins within a pair, k∈{1,2} indexes the visits of a twin pair, and l∈{1,2} indexes the two aliquots of a sample. The variances of the ‘random effects’ (d, m, e, w, v, ε) are, respectively, represented by the elements of (σd2, σm2, σe2, σw2, σv2, σε2)′≡σ2.
The subscript in the mz(i,j) term on the right-hand side of Equation (5) was defined conditionally on the zygosity of pair i. Specifically, z(i, j)=i if i was an MZ pair, and z(i, j)=(i, j) if i was a DZ pair (Visscher et al, 2004). This allocated one such term (mi) to each MZ pair, and two such terms (mi1, mi2) to each DZ pair. The terms di+mz(i,j)+eij on the right-hand side of Equation (5) thereby created the covariance structure described in Equations (3) and (4).
The familial variance (σd2+σm2) represented the combined effects of genetics and common environment. The individual-environmental variance (σe2) captured non-familial variation that was stable over time within an individual. The longitudinal design of our study allowed short-term (temporal) phenotypic variation to be quantified—the common-visit (σw2) and individual-visit (σv2) variance terms captured inter-visit variation that was (respectively) shared and non-shared by twins in a pair. The residual or non-biological variance component (σε2) represented variation that could not be explained by the biological model, and which corresponded to variation between pairs of aliquots of the same biological sample. Table II relates the mathematical notation used for variance parameters (and functions thereof) to the descriptions used in the text. Supplementary Table S3 relates variance components to real-world sources of variation.
Table 2. Variance parameters—textual description and mathematical notation.
| Familial variance | σd2+σm2 |
| Individual-environment variance | σe2 |
| Common-visit variance | σw2 |
| Individual-visit variance | σv2 |
| Non-biological (residual) variance | σε2 |
| Total phenotypic variance | σd2+σm2+σe2+σw2+σv2+σε2 |
| Total biological variance | σd2+σm2+σe2+σw2+σv2 |
| Non-biological proportion of total phenotypic variance |
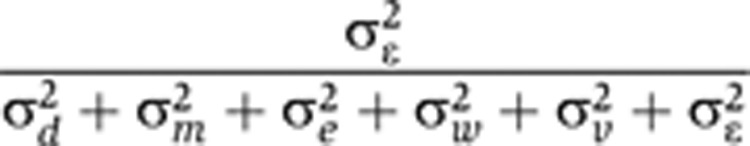
|
| Familiality (familial proportion of biological variance) |
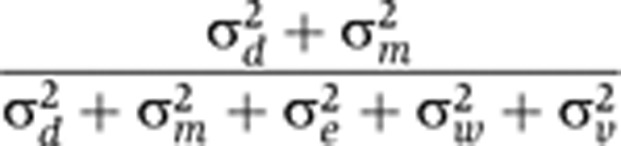
|
| Stable proportion of biological variance |
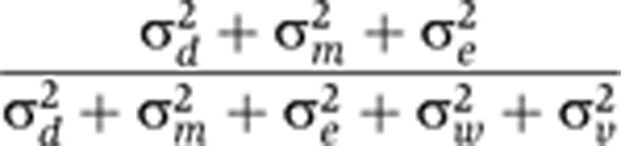
|
| Unstable proportion of biological variance |
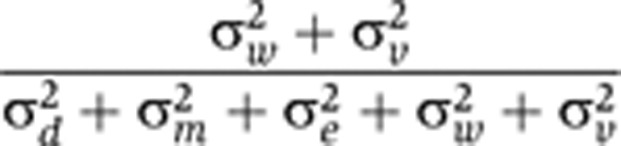
|
Robust Bayesian implementation
Under the model described in Equation (5), we estimated σ2 within a Bayesian hierarchical framework, in which the conventional Gaussian distribution on the ‘random effects’ was replaced by a heavy-tailed distribution, in order to prevent extreme observations from exerting undue influence on inference. Specifically, we defined the heavy-tailed probability density function (pdf) q(·) to be a Gauss–Student mixture:
where δ defines the mixture proportions, tv(·∣μ, σ2) is the pdf of Student's t-distribution with v degrees of freedom, and N(·∣μ, σ2) is the pdf of a Gaussian distribution (in both cases with mean μ and scale parameter σ). The conditional density function of each random effect (denoted by u), conditional on the corresponding variance parameter (denoted by σu2), was defined to be:
Independent Uniform priors were placed on the standard deviation parameters (Gelman, 2006), p(σ)∼Uniform(σ∣0, 10 × sy), where sy denotes the sample standard deviation of the data, y. The prior on the ‘fixed effects’ vector, α≡(β′, τ)′, was a diffuse multivariate Gaussian distribution, with mean at the least-squares estimates, 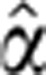 , and diagonal covariance matrix with entries
, and diagonal covariance matrix with entries  . Samples were drawn from the posterior distribution of α and σ2, that is p(α, σ2∣y), using Gibbs sampling, with a burn-in of 10 000 updates followed by the collection of 50 000 samples from the joint posterior.
. Samples were drawn from the posterior distribution of α and σ2, that is p(α, σ2∣y), using Gibbs sampling, with a burn-in of 10 000 updates followed by the collection of 50 000 samples from the joint posterior.
To check the qualitative robustness of our findings to the statistical method used, we compared the results of the robust Bayesian analysis with the results obtained by a distinct but parallel non-Bayesian approach (Supplementary Figure S5; Supplementary information). There was a high level of consistency across the two approaches.
Supplementary Material
Acknowledgments
We thank the editor and the anonymous reviewers for their careful and helpful comments. This work was supported by funding from the European Commission to the MolPAGE Consortium (LSHGCT-2004-512066). GN and CCH acknowledge funding from MRC Harwell, UK. MIM, TDS, and CCH acknowledge funding from the Wellcome Trust. KTZ is supported by a Wellcome Trust Career Development Fellowship (085235/Z/08/Z). Twins UK receives funding from the Wellcome Trust and the NIHR/NHS via a BRC grant to Guy′s and St Thomas' Hospitals and King's College London. MR is an MRC biomedical informatics fellow (Medical Research Council, fellowship G0802460). PD acknowledges funding from the Wellcome Trust (grants 084575/Z/08/Z and 075491/Z/04/B), as well as support from a Wolfson Royal Society Merit Award.
Author contributions: GN, ADM, DM, JHF, IBH, HT, M-ED, BWS, PD, JKN, MA, KTZ, JCL, TDS, MIM, EH, DB, and CCH designed the research. ADM, JHF, AB, and HT carried out experimental work. MK, JV, SGN, and US designed the database. GN, MR, ADM, JVL, and DM analysed data and annotated peaks. GN, MR, ADM, KRA, JCL, and CCH wrote the paper.
MolPAGE Consortium Partners (In no particular order) John Bell1, Mark McCarthy1, Lon Cardon1, Peter Donnelly1, Edwin Southern2, John Anson2, Mark Lathrop3, Ivo Gut3, Matthias Schuster4, Kurt Berlin4, Esper Boel5, Jan Fleckner5, Mathias Uhlén6, Thomas Bergman7, Vladimir Stich8, Alvis Brazma9, Ugis Sarkans9, Juris Viksna10, Jeremy Nicholson11, Luisa Bernardinelli12, Stephen Hoffmann13, Mats Inganas13, Tim Spector14, Dominique Langin15, Fredrik Ponten16, Hanno Langen17, Stefan Evers17 1University of Oxford, Oxford, UK 2Oxford Gene Technology, Oxford, UK 3Centre National de Génotypage, Evry, France 4Epigenomics AG, Berlin, Germany 5Novo Nordisk, Copenhagen, Denmark 6Royal Institute of Technology, Stockholm, Sweden 7Affibody AB, Bromma, Sweden 8Charles University Prague, Czech Republic 9European Bioinformatics Institute, Cambridge, UK 10Institute of Mathematics and Computer Science, Riga, Latvia 11Imperial College London, London, UK 12University of Pavia, Pavia, Italy 13Gyros AB, Uppsala, Sweden 14Guy's and St Thomas' Hospital NHS Trust, London, UK 15Obesity Research Unit, INSERM, Toulouse, France 16University of Uppsala, Uppsala, Sweden 17Roche, Basel, Switzerland
Footnotes
The authors declare that they have no conflict of interest.
References
- Ala-Korpela M (2008) Critical evaluation of 1H NMR metabonomics of serum as a methodology for disease risk assessment and diagnostics. Clin Chem Lab Med 46: 27–42 [DOI] [PubMed] [Google Scholar]
- Assfalg M, Bertini I, Colangiuli D, Luchinat C, Schäfer H, Schütz B, Spraul M (2008) Evidence of different metabolic phenotypes in humans. Proc Natl Acad Sci USA 105: 1420–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathum L, Fagnani C, Christiansen L, Christensen K (2004) Heritability of biochemical kidney markers and relation to survival in the elderly—results from a Danish population-based twin study. Clin Chim Acta 349: 143–150 [DOI] [PubMed] [Google Scholar]
- Bollard M, Stanley E, Lindon J, Nicholson J, Holmes E (2005) NMR-based metabonomic approaches for evaluating physiological influences on biofluid composition. NMR Biomed 18: 143–162 [DOI] [PubMed] [Google Scholar]
- Chadeau-Hyam M, Ebbels T, Brown I, Chan Q, Stamler J, Huang CC, Daviglus M, Ueshima H, Zhao L, Holmes E, Nicholson J, Elliott P, De Iorio M (2010) Metabolic profiling and the metabolome-wide association study: significance level for biomarker identification. J Proteome Res 9: 4620–4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton A, Lindon J, Cloarec O, Antti H, Charuel C, Hanton G, Provost J-P, Le Net J-L, Baker D, Walley R, Everett J, Nicholson J (2006) Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature 440: 1073–1077 [DOI] [PubMed] [Google Scholar]
- Cloarec O, Dumas M-E, Craig A, Barton R, Trygg J, Hudson J, Blancher C, Gauguier D, Lindon J, Holmes E, Nicholson J (2005) Statistical total correlation spectroscopy: an exploratory approach for latent biomarker identification from metabolic 1H NMR data sets. Anal Chem 77: 1282–1289 [DOI] [PubMed] [Google Scholar]
- Dieterle F, Ross A, Schlotterbeck G, Senn H (2006) Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal Chem 78: 4281–4290 [DOI] [PubMed] [Google Scholar]
- Dumas M-E, Maibaum E, Teague C, Ueshima H, Zhou B, Lindon J, Nicholson J, Stamler J, Elliott P, Chan Q, Holmes E (2006) Assessment of analytical reproducibility of 1H NMR spectroscopy based metabonomics for large-scale epidemiological research: the INTERMAP Study. Anal Chem 78: 2199–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favé G, Beckmann ME, Draper JH, Mathers JC (2009) Measurement of dietary exposure: a challenging problem which may be overcome thanks to metabolomics? Genes Nutr 4: 135–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman A (2006) Prior distributions for variance parameters in hierarchical models. Bayesian Anal 1: 515–533 [Google Scholar]
- Gibney MJ, Walsh M, Brennan L, Roche HM, German B, van Ommen B (2005) Metabolomics in human nutrition: opportunities and challenges. Am J Clin Nutr 82: 497–503 [DOI] [PubMed] [Google Scholar]
- Gieger C, Geistlinger L, Altmaier E, Hrabé de Angelis M, Kronenberg F, Meitinger T, Mewes H-W, Wichmann HE, Weinberger K, Adamski J, Illig T, Suhre K (2008) Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet 4: e1000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J (2006) The Cinderella story of metabolic profiling: does metabolomics get to go to the functional genomics ball? Philos Trans R Soc Lond B Biol Sci 361: 147–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzmann S, Brown I, Chan Q, Bictash M, Dumas M-E, Kochhar S, Stamler J, Holmes E, Elliott P, Nicholson J (2010) Metabolic profiling strategy for discovery of nutritional biomarkers: proline betaine as a marker of citrus consumption. Am J Clin Nutr 92: 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E, Loo RL, Stamler J, Bictash M, Yap I, Chan Q, Ebbels T, De Iorio M, Brown I, Veselkov K, Daviglus M, Kesteloot H, Ueshima H, Zhao L, Nicholson J, Elliott P (2008) Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 453: 396–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E, Tsang T, Jeffrey TJ, Leweke M, Koethe D, Gerth C, Nolden B, Gross S, Schreiber D, Nicholson J, Bahn S (2006) Metabolic profiling of CSF: evidence that early intervention may impact on disease progression and outcome in schizophrenia. PLoS Med 3: e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz ML, Knox WE (1968) A phosphate activated glutaminase in rat liver different from that in kidney and other tissues. Enzymol Biol Clin (Basel) 9: 241–255 [DOI] [PubMed] [Google Scholar]
- Illig T, Gieger C, Zhai G, Romisch-Margl W, Wang-Sattler R, Prehn C, Altmaier E, Kastenmuller G, Kato B, Mewes H-W, Meitinger T, de Angelis M, Kronenberg F, Soranzo N, Wichmann HE, Spector T, Adamski J, Suhre K (2010) A genome-wide perspective of genetic variation in human metabolism. Nat Genet 42: 137–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone I, Silverman B (2005) Empirical Bayes selection of wavelet thresholds. Ann Stat 33: 1700–1752 [Google Scholar]
- Keun H, Ebbels T, Antti H, Bollard M, Beckonert O, Schlotterbeck G, Senn H, Niederhauser U, Holmes E, Lindon J, Nicholson J (2002) Analytical reproducibility in (1)H NMR-based metabonomic urinalysis. Chem Res Toxicol 15: 1380–1386 [DOI] [PubMed] [Google Scholar]
- Kochhar S, Jacobs D, Ramadan Z, Berruex F, Fuerholz A, Fay L (2006) Probing gender-specific metabolism differences in humans by nuclear magnetic resonance-based metabonomics. Anal Biochem 352: 274–281 [DOI] [PubMed] [Google Scholar]
- Le Moyec L, Valensi P, Charniot J-C, Hantz E, Albertini J-P (2005) Serum 1H-nuclear magnetic spectroscopy followed by principal component analysis and hierarchical cluster analysis to demonstrate effects of statins on hyperlipidemic patients. NMR Biomed 18: 421–429 [DOI] [PubMed] [Google Scholar]
- Lenz EM, Bright J, Wilson ID, Morgan SR, Nash AF (2003) A 1H NMR-based metabonomic study of urine and plasma samples obtained from healthy human subjects. J Pharm Biomed Anal 33: 1103–1115 [DOI] [PubMed] [Google Scholar]
- Lewis G, Farrell L, Wood M, Martinovic M, Arany Z, Rowe G, Souza A, Cheng S, McCabe E, Yang E, Shi X, Deo R, Roth F, Asnani A, Rhee E, Systrom D, Semigran M, Vasan R, Carr S, Wang T et al. (2010) Metabolic signatures of exercise in human plasma. Sci Transl Med 2: 33ra37–33ra37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindon J, Nicholson J (2008) Spectroscopic and statistical techniques for information recovery in metabonomics and metabolomics. Annu Rev Anal Chem (Palo Alto, Calif) 1: 45–69 [DOI] [PubMed] [Google Scholar]
- Liu M, Nicholson JK, Lindon JC (1996) High-resolution diffusion and relaxation edited one- and two-dimensional 1H NMR spectroscopy of biological fluids. Anal Chem 68: 3370–3376 [DOI] [PubMed] [Google Scholar]
- Maher A, Zirah S, Holmes E, Nicholson J (2007) Experimental and analytical variation in human urine in 1H NMR spectroscopy-based metabolic phenotyping studies. Anal Chem 79: 5204–5211 [DOI] [PubMed] [Google Scholar]
- Martin N, Boomsma D, Machin G (1997) A twin-pronged attack on complex traits. Nat Genet 17: 387–392 [DOI] [PubMed] [Google Scholar]
- McGivan JD (1998) Metabolism of glutamine and glutamate in the liver—regulation and physiological significance. In Glutamine and Glutamate in Mammals, Kvamme E (ed), Vol. 1. Boca Raton, FL: CRC Press, Inc. [Google Scholar]
- Neale M (2001) Twin Studies: Software and Algorithms. In Encyclopedia of the Human Genome, Cooper DN (ed). London: Macmillan Publishers Ltd, Nature Publishing Group [Google Scholar]
- Neale MC, Cardon LR (1992) Methodology for Genetic Studies of Twins and Families. Dordrecht: Kluwer Academic Publishers [Google Scholar]
- Nicholson J, Holmes E, Elliott P (2008) The metabolome-wide association study: a new look at human disease risk factors. J Proteome Res 7: 3637–3638 [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Buckingham MJ, Sadler PJ (1983) High resolution 1H n.m.r. studies of vertebrate blood and plasma. Biochem J 211: 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson JK, Connelly J, Lindon JC, Holmes E (2002) Metabonomics: a platform for studying drug toxicity and gene function. Nat Rev Drug Discov 1: 153–161 [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Foxall PJ, Spraul M, Farrant RD, Lindon JC (1995) 750 MHz 1H and 1H-13C NMR spectroscopy of human blood plasma. Anal Chem 67: 793–811 [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Lindon JC, Holmes E (1999) ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 29: 1181–1189 [DOI] [PubMed] [Google Scholar]
- Nicholson JK, O'Flynn MP, Sadler PJ, Macleod AF, Juul SM, Sönksen PH (1984) Proton-nuclear-magnetic-resonance studies of serum, plasma and urine from fasting normal and diabetic subjects. Biochem J 217: 365–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odunsi K, Wollman R, Ambrosone C, Hutson A, McCann S, Tammela J, Geisler J, Miller G, Sellers T, Cliby W, Qian F, Keitz B, Intengan M, Lele S, Alderfer J (2005) Detection of epithelial ovarian cancer using 1H-NMR-based metabonomics. Int J Cancer 113: 782–788 [DOI] [PubMed] [Google Scholar]
- Perrone RD, Madias NE, Levey AS (1992) Serum creatinine as an index of renal function: new insights into old concepts. Clin Chem 38: 1933–1953 [PubMed] [Google Scholar]
- Pontoizeau C, Fearnside J, Navratil V, Domange C, Cazier J-B, Fernández-Santamaría C, Kaisaki P, Emsley L, Toulhoat P, Bihoreau MT, Nicholson J, Gauguier D, Dumas M (2011) Broad-ranging natural metabotype variation drives physiological plasticity in healthy control inbred rat strains. J Proteome Res 10: 1675–1689 [DOI] [PubMed] [Google Scholar]
- Posthuma D, Beem L, de Geus E, van Baal C, von Hjelmborg J, Iachine I, Boomsma D (2003) Theory and practice in quantitative genetics. Twin Res 6: 361–376 [DOI] [PubMed] [Google Scholar]
- Qiu Y, Rajagopalan D, Connor S, Damian D, Zhu L, Handzel A, Hu G, Amanullah A, Bao S, Woody N, MacLean D, Lee K, Vanderwall D, Ryan T (2008) Multivariate classification analysis of metabolomic data for candidate biomarker discovery in type 2 diabetes mellitus. Metabolomics 4: 337–346 [Google Scholar]
- R Development Core Team (2010) R: A Language and Environment for Statistical Computing [Google Scholar]
- Rezzi S, Ramadan Z, Fay L, Kochhar S (2007) Nutritional metabonomics: applications and perspectives. J Proteome Res 6: 513–525 [DOI] [PubMed] [Google Scholar]
- Rijsdijk F, Sham P (2002) Analytic approaches to twin data using structural equation models. Brief Bioinform 3: 119–133 [DOI] [PubMed] [Google Scholar]
- Robertson DG (2005) Metabonomics in toxicology: a review. Toxicol Sci 85: 809–822 [DOI] [PubMed] [Google Scholar]
- Sahai S (1983) Glutaminase in human platelets. Clin Chim Acta 127: 197–203 [DOI] [PubMed] [Google Scholar]
- Sahai S, Vogel F (1983) Genetic control of platelet glutaminase: a twin study. Hum Genet 63: 292–293 [DOI] [PubMed] [Google Scholar]
- Saude E, Adamko D, Rowe B, Marrie T, Sykes B (2007) Variation of metabolites in normal human urine. Metabolomics 3: 439–451 [Google Scholar]
- Saude E, Obiefuna I, Somorjai R, Ajamian F, Skappak C, Ahmad T, Dolenko B, Sykes B, Moqbel R, Adamko D (2009) Metabolomic biomarkers in a model of asthma exacerbation: urine nuclear magnetic resonance. Am J Respir Crit Care Med 179: 25–34 [DOI] [PubMed] [Google Scholar]
- Searle SR, Casella G, McCulloch CE (2006) Variance Components. John Wiley & Sons, Inc., Hoboken, NJ, USA [Google Scholar]
- Shah S, Hauser E, Bain J, Muehlbauer M, Haynes C, Stevens R, Wenner B, Dowdy E, Granger C, Ginsburg G, Newgard C, Kraus W (2009) High heritability of metabolomic profiles in families burdened with premature cardiovascular disease. Mol Syst Biol 5: 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson DP (1983) Citrate excretion: a window on renal metabolism. Am J Physiol 244: F223–F234 [DOI] [PubMed] [Google Scholar]
- Slupsky CM, Rankin KN, Wagner J, Fu H, Chang D, Weljie AM, Saude EJ, Lix B, Adamko DJ, Shah S, Greiner R, Sykes BD, Marrie TJ (2007) Investigations of the effects of gender, diurnal variation, and age in human urinary metabolomic profiles. Anal Chem 79: 6995–7004 [DOI] [PubMed] [Google Scholar]
- Stella C, Beckwith-Hall B, Cloarec O, Holmes E, Lindon J, Powell J, van der Ouderaa F, Bingham S, Cross A, Nicholson J (2006) Susceptibility of human metabolic phenotypes to dietary modulation. J Proteome Res 5: 2780–2788 [DOI] [PubMed] [Google Scholar]
- Visscher P, Benyamin B, White I (2004) The use of linear mixed models to estimate variance components from data on twin pairs by maximum likelihood. Twin Res 7: 670–674 [DOI] [PubMed] [Google Scholar]
- Voet D, Voet JG (1995) Biochemistry. John Wiley and Sons, Inc., New York, NY, USA [Google Scholar]
- Wheelock C, Wheelock A, Kawashima S, Diez D, Kanehisa M, van Erk M, Kleemann R, Haeggström J, Goto S (2009) Systems biology approaches and pathway tools for investigating cardiovascular disease. Mol BioSyst 5: 588–602 [DOI] [PubMed] [Google Scholar]
- Williams H, Cox J, Walker D, North B, Patel V, Marshall S, Jewell D, Ghosh S, Thomas H, Teare J, Jakobovits S, Zeki S, Welsh K, Taylor-Robinson S, Orchard T (2009) Characterization of inflammatory bowel disease with urinary metabolic profiling. Am J Gastroenterol 104: 1435–1444 [DOI] [PubMed] [Google Scholar]
- Wishart D, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau D, Psychogios N, Dong E, Bouatra S, Mandal R, Sinelnikov I, Xia J, Jia L, Cruz J, Lim E, Sobsey C, Shrivastava S, Huang P, Liu P et al. (2009) HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res 37: D603–D610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss M, Kaddurah-Daouk R (2000) Creatine and creatinine metabolism. Physiol Rev 80: 1107–1213 [DOI] [PubMed] [Google Scholar]
- Xia J, Psychogios N, Young N, Wishart D (2009) MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res 37: W652–W660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap I, Brown I, Chan Q, Wijeyesekera A, Garcia-Perez I, Bictash M, Loo RL, Chadeau-Hyam M, Ebbels T, Iorio MD, Maibaum E, Zhao L, Kesteloot H, Daviglus M, Stamler J, Nicholson J, Elliott P, Holmes E (2010) Metabolome-wide association study identifies multiple biomarkers that discriminate North and South Chinese populations at differing risks of cardiovascular disease: INTERMAP Study. J Proteome Res 9: 6647–6654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Xu B, Huang J, Jia X, Xue J, Shi X, Xiao L, Li W (2009) 1H NMR-based metabonomic and pattern recognition analysis for detection of oral squamous cell carcinoma. Clin Chim Acta 401: 8–13 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.